Article Text

Abstract
Primary erythermalgia is a rare autosomal dominant disease characterised by intermittent burning pain with redness and heat in the extremities. A previous study established the linkage of primary erythermalgia to a 7.94 cM interval on chromosome 2q, but the causative gene was not identified. We performed linkage analysis in a Chinese family with primary erythermalgia, and screened the mutations in the two candidate genes, SCN9A and GCA, in the family and a sporadic patient. Linkage analysis yielded a maximum lod score of 2.11 for both markers D2S2370 and D2S2330. Based on critical recombination events in two patients in the family, we further limited the genetic region to 5.98 cM between D2S2370 and D2S2345. We then identified two missense mutations in SCN9A in the family (T2573A) and the sporadic patient (T2543C). Our data suggest that mutations in SCN9A cause primary erythermalgia. SCN9A, encoding a voltage-gated sodium channel alpha subunit predominantly expressed in sensory and sympathetic neurones, may play an important role in nociception and vasomotor regulation.
- mutation
- pain
- primary erythermalgia
- SCN9A
- sodium channel
Statistics from Altmetric.com
Primary erythermalgia (MIM 133020) is a rare disorder characterised by recurrent episodes of severe pain associated with redness and warmth in the feet or hands.1 In patients, standing, exercise, or local exposure to heat can induce the symptoms, and keeping the involved extremities at an icy cold temperature is the most effective way to relieve pain. Symptoms of primary erythermalgia arise in childhood or adolescence; with age, the symptoms may progress and become constant. Histopathological examination reveals a reduced density of skin autonomic nerve plexuses.2 Many therapeutic options have been tested with variable alleviatory results.3 This disorder may be inherited as an autosomal dominant trait.4–6 A previous study7 established the linkage of primary erythermalgia to a 7.94 cM interval on chromosome 2q. We report here the linkage of primary erythermalgia in a Chinese family to chromosome 2q24.2–q24.3. Critical recombination events in two patients in this family further defined the genetic region to 5.98 cM between D2S2370 and D2S2345. This genomic interval contains a cluster of sodium channel genes. We then identified two missense mutations in SCN9A in the family and a sporadic patient with primary erythermalgia. SCN9A, encoding a voltage-gated sodium channel alpha subunit mainly expressed in sensory and sympathetic neurones, may play an important role in nociception and vasomotor regulation. Our data suggest that mutations in SCN9A cause primary erythermalgia.
MATERIALS AND METHODS
Patients (fig 1)
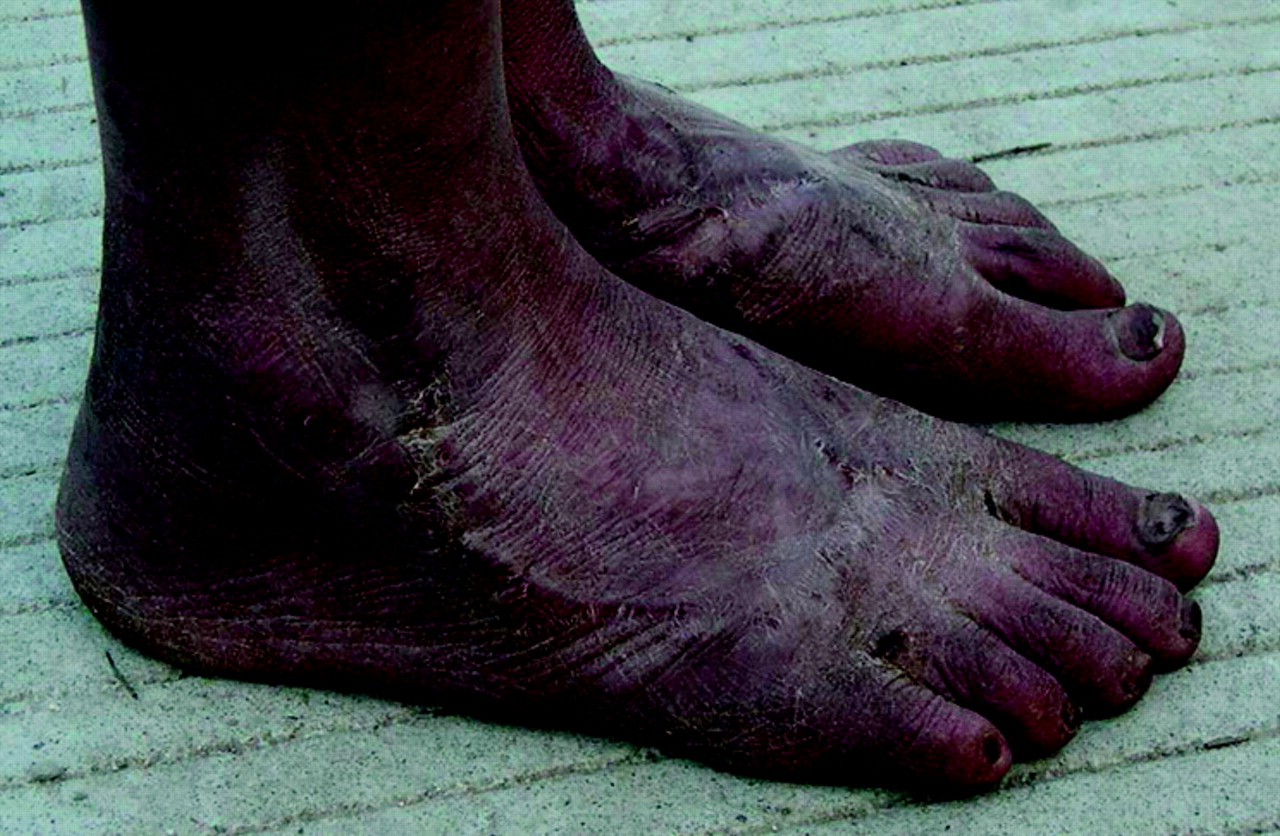
Clinical features of the proband in the family. Diffuse redness is seen on the feet. Scars due to injury from repeatedly excessive cooling to relieve the symptoms are also shown.
We studied a Chinese family of three generations with autosomal dominant hereditary primary erythermalgia, and a sporadic patient. The two probands first showed attacks of bilateral burning pain with redness and warmth in the hands and feet at 4–8 years old. Symptoms were evoked by exercise or exposure to heat and relieved by elevation of the extremities and cold. The disorder was refractory to treatment and persistent during life. The diagnosis was made independently by at least two dermatologists based on the diagnostic criteria of primary erythermalgia.8 Blood samples were collected after informed consent was obtained.
Haplotype and linkage analysis
Five polymorphic microsatellite markers (D2S2370, D2S382, D2S2363, D2S2330, and D2S2345) in the region of interest on chromosome 2 were used to perform linkage analysis (fig 2) in the family with primary erythermalgia. We also used AmpFLSTR Identifiler PCR Amplification Kit (ABI, USA) to confirm the paternity and maternity of the sporadic case.

Pedigree of the family and haplotype analysis. Females are indicated by circles and males by squares. Blackened figures denote affected individuals. Recombinations occurred at marker D2S2345 in II-3 and III-1.
DNA sequencing
Genomic DNA samples from patients, their family members, and normal control subjects were screened for mutations in SCN9A (GenBank accession number, NM_002977) and GCA (GenBank accession number, NM_012198). PCR primers were used to amplify all of the exons and their flanking sequences of SCN9A and GCA (primer sequences are available upon request). PCR products were purified and sequenced on ABI PRISM 377 automated sequencer (PE Applied Biosystems, USA).
RESULTS
Two point linkage analysis yielded a maximum lod score (Zmax) of 2.11 at θ = 0 for both markers D2S2370 and D2S2330. Critical recombination events occurring in individual II-3 and III-1 indicate that marker D2S2345 defines the telomeric end. This marker and marker D2S2370, which is reported by the literature7 as the centromeric boundary, limit the responsible gene to a region of 5.98 cM (fig 2). Individual III-5 (3 years old) carried the risk haplotype but was clinically unaffected, possibly due to the late onset of the disease, because all of the affected individuals in this family first showed symptoms at 7–15 years old, and we have not found patients less than 5 years old in the literature.3,8
This genomic interval contains a cluster of sodium channel genes including SCN1A, SCN2A, SCN3A, SCN7A, and SCN9A. Primary erythermalgia can be evoked by various stimulations, comparable to sodium channel diseases such as severe myoclonic epilepsy in infancy (MIM 607208), which is caused by a SCN1A mutation,9 and hyperkalemic periodic paralysis (MIM 170500) and congenital paramyotonia (MIM 168300), which are caused by a SCN4A mutation.10–12 Furthermore, the rat homologue of SCN9A, peripheral nerve type 1 sodium channel alpha subunit, is expressed at high levels throughout the peripheral nervous system but not in the skeletal muscle, myocardium, or brain, and is targeted from an intracellular pool to neurite terminals.13 Other sodium channel genes in this interval, including SCN1A, SCN2A, SCN3A, and SCN7A, are mainly expressed in the central nervous system, heart, and muscles. SCN9A was therefore an intriguing candidate gene, likely to be responsible for the disease. By analysing the sequences of the PCR products amplified from exons and their flanking sequences of SCN9A, we identified a heterozygous T to A transversion (T2573A; fig 3A) (EMBL accession number: AJ580919) in exon 15, which results in the protein change of L858H, from the proband in the family. Results of DNA sequencing and restriction endonuclease Hph I digestion also showed that this transversion was found in all patients in the family, but not in the unaffected family members (except individual III-5) (fig 3B) and 400 alleles from normal Chinese controls. Results from the normal controls indicate that this transversion is unlikely to be a polymorphism in the normal population.

Mutations in SCN9A in patients with primary erythermalgia. (A) Affected members in the family carry the heterozygous mutation of T2573A. (B) Restriction endonuclease Hph I digestion for the identification of the T2573A mutation in the family. Mutation T2573A abolishes a recognition site for restriction endonuclease Hph I. Three fragments of 470 bp, 303 bp, and 167 bp are found in patients, and two fragments of 303 bp and 167 bp fragments are found in unaffected members. (C) The sporadic case carries a heterozygous mutation of T2543C. (D) Restriction endonuclease BsrS I digestion for the identification of the T2543C mutation in the family of the sporadic case. Mutation T2543C creates a recognition site for restriction endonuclease BsrS I. Three fragments of 470 bp, 320 bp, and 150 bp fragments are observed in the patient, and one 470 bp fragment is observed in his unaffected parents.
In a further study, we identified a de novo heterozygous mutation of T2543C (EMBL accession number, AJ580918), resulting in the protein change of I848T, in a sporadic patient (fig 3C). Restriction endonuclease BsrS I digestion of the PCR products confirmed the T2543C mutation (fig 3D). This transition was not found in his unaffected parents (paternity and maternity were confirmed) and 400 alleles from normal Chinese controls.
Another candidate gene GCA, encoding grancalcin, was also screened, but no mutation was found in the two probands.
DISCUSSION
SCN9A encodes Nav1.7, a voltage gated sodium channel alpha subunit expressed primarily in sensory and sympathetic ganglia,14 probably involved in nociception. Nav1.7 shares significant similarity with other isoforms of voltage gated sodium channel alpha subunit in the brain, peripheral nerve, muscle, and myocardium, including Nav1.1 (encoded by SCN1A), Nav1.2 (encoded by SCN2A), Nav1.3 (encoded by SCN3A), Nav1.4 (encoded by SCN4A), Nav1.5 (encoded by SCN5A), Nav1.6 (encoded by SCN8A), and Nav1.8 (encoded by SCN10A). These channels on the cell membrane are involved in the generation of action potentials. They have a very similar structure. Each of them contains four highly homologous domains (I-IV) and each domain has six transmembrane segments (S1-S6).13–15 Mutations in these ion channels have been related to epilepsy, periodic paralysis, and long Q-T syndrome. Although several sodium channel isoforms are preferentially expressed in dorsal root sensory neurones and may exert specific roles in pain sensation, no mutation relating in pain has been reported in these genes to date.16–18
The mutations we identified are located in the II/S5 segment (L858H) and the loop region between II/S4 and II/S5 (I848T) of Nav1.7. The leucine and isoleucine at the two sites are highly conserved in most voltage gated sodium channel alpha subunit isoforms (fig 4). The I848T mutation in Nav1.7 found in our sporadic case is comparable to the I693T mutation in Nav1.4 previously reported in a family congenital for paramyotonia,19 suggesting the important function of this isoleucine in the loop (fig 4). Since the mutations we identified in our cases are located in the critical sites in polypeptide Nav1.7, these mutations are expected to cause abnormal excitability of sensory and sympathetic neurones. As a result, mild stimulation may evoke episodes of sensory and vasomotor symptoms in patients with primary erythermalgia. Thus, mutations in SCN9A may cause primary erythermalgia. Our findings also provide a clue for the further study of pain and peripheral vasomotor regulation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Multiple alignment of the region from the loop II/S4-II/S5 to II/S5 of the human Nav1.7 protein with the corresponding segments of its homologues. Multiple alignment of the region was performed using the ClustalW program, using its default parameters. Numbers denote the positions of amino acids in the polypeptide. Nav1.7 mutations of L858H in the family and I848T in the sporadic case are aligned. Nav1.4 mutation of I693T, which was reported in a family congenital for paramyotonia19, is also shown for comparison.
Acknowledgments
We thank the family members for their participation. We also thank China Tianjin public security criminal investigative bureau for the confirmation of paternity and maternity of the sporadic case.
REFERENCES
Footnotes
-
↵* These authors contributed equally to this work
-
This work was supported by “985” Foundation of Peking University Health Science Centre, the National High Technology Research and Development Programme of China (Grant 2002BA711A07), and Xian-Janssen Dermatology Foundation.