


Abstract
Epipodophyllotoxin derivatives, such as etoposide (VP-16), constitute an important class of anticancer agents, the major cytotoxic effects of which are associated with trapping of the topoisomerase II/DNA cleavable complex and formation of protein-DNA cross-links and nicked DNA. VP-16, however, can be metabolized to several highly reactive products, including an ortho-quinone (VPQ). The inhibitory activity of VPQ against purified human topoisomerase II processing of supercoiled DNA was studied and compared with that of the parent compound, VP-16. Our results show that VPQ is a powerful inhibitor of topoisomerase II, which prevents DNA strand passage in the presence of ATP. As with VP-16, trapping of the cleavable complex is highly reversible upon removal of divalent ions, which indicating that VPQ alters the cleavage-reunion equilibrium of topoisomerase II and DNA mainly by noncovalent interactions, as does the parent compound. However, we observed several differences between the effects induced by VP-16 and VPQ, including a strong inhibition of the second DNA strand religation, which implies the involvement of additional (asymmetric) mode(s) of interactions of the VPQ, possibly by interference with ATP binding by the homodimeric enzyme, and/or involving covalent interactions. Reduced or oxidized glutathione prevented trapping of the topoisomerase/DNA cleavable complex by VPQ, but not by VP-16, probably by forming covalent adducts with the former.
Type II topoisomerases alter DNA topology by the formation of a double-stranded break, followed by DNA strand passage through an ATP-dependent protein clamp (referred to as a two-gate mechanism) (Liuet al., 1983; Hsieh, 1983; Roca and Wang, 1994). The eukaryotic enzymes function as homodimers and exist in two forms: p170 and p180. Both forms catalyze DNA strand passage, but the reaction mechanisms are different: the p170 form relaxes SC DNA in a highly distributive manner, whereas the p180 form changes the DNA linking number in a processive way, although the exact mechanistic differences remain obscure (Drake et al., 1989). DNA strand passage is preceded by the formation of a transient DNA-protein (cleavable) complex, anchored by covalent phosphotyrosil bonds between the active site of the homodimeric enzyme and the 5′-DNA ends of the cleaved strands of the gate-segment of DNA (Hsieh, 1990; Wigley, 1995). The final stages of DNA processing by topoisomerases are the religation of the cleaved strands, followed by release of the DNA. Most of the topoisomerase-targeting anticancer drugs (e.g., epipodophyllotoxins) exert their therapeutic action by a specific and reversible block of the DNA-strand rejoining steps, resulting in trapping of the covalent topoisomerase II/DNA cleavable complex (Chen et al., 1984), accumulation of protein-linked DNA breaks, late S and/or G2-phase cell-cycle arrest and, ultimately, cell death (Wozniak and Ross, 1983; Ross et al., 1984;Markovits et al., 1987; Gantchev et al., 1996). One of these topoisomerase poisons, used in the treatment of a number of neoplastic disorders, is the epipodophyllotoxin VP-16 [4′- demethyl-epipodophyllotoxin-9-(4, 6-O-ethylidene-β-d-glucopyranoside)]. VP-16, however, can undergo oxido-reductive transformations in cells. Two major pathways have been identified: O-demethylation catalyzed by cytochrome P450-dependent monooxygenases (van Maanenet al., 1987), and one- or two-electron oxidation mediated by some peroxidases (Haim et al., 1986) and tyrosinase (Gantchev et al., 1994). These enzymatic transformations affect the pendant dimethoxyphenolic group (E-ring) of the drug, leading to several products, the major one being theortho-quinone derivative VPQ, as summarized in Fig.1.

Major oxido-reductive pathways of metabolic transformations of etoposide (VP-16) mediated by P450 monooxygenases and/or peroxidases. These transformations alter the pendant dimethoxyphenolic group (E-ring): (1) VP-16;(2) phenoxyl free radical; (3) catechol;(4) semiquinone free radical; (5) VPQ.
In addition to any inhibitory effects resulting from noncovalent interactions, the cytotoxicity of the VP-16 ortho-quinone might also result from its nucleophilic character (i.e., its ability to form covalent adducts with -SH and -NH2 groups of enzymes) and/or its conversion to a toxic semiquinone radical (Kalyanaraman et al., 1987). Earlier studies have also shown that VPQ can directly induce damage to DNA (e.g., by forming adducts in alkaline solutions) and that glutathione protected DNA from inactivation (Mans et al., 1991; Mans et al., 1992). Other studies have correlated the antioxidant potential of cells (i.e. suppression/enhancement of VP-16 metabolic transformations) with VP-16 toxicity (Gantchev et al., 1994; Yokomizo et al., 1995; Yalowich et al., 1996; Gantchev and Hunting, 1997a). Analysis of the effects of the ortho-quinone VP-16 derivative on topoisomerase II processing of SC DNA is the subject of the present study.
Materials and Methods
Reagents.
Purified human type II topoisomerase (p170 form, 2 units/μl) was obtained from TopoGene (Columbus, OH). Activity was measured according to the unit definition of the supplier: 1 unit of topoisomerase II decatenates 0.2 μg of catenated DNA in 30 min at 37°. No nuclease or topoisomerase I contamination was detected. VP-16 was purchased from Sigma (St. Louis, MO) and stock solutions in dimethylsulfoxide were kept at −20°. The ortho-quinone derivative VPQ was synthesized by controlled potential electrolysis of VP-16 at a Pt-gauze electrode under nitrogen atmosphere as described previously (Holthuis et al., 1985). Reaction rates were followed by optical spectroscopy (broad absorption band between 300 and 400 nm, characteristic for the quinone and centered at λ = 356 nm). Purification of the product from unreacted starting material and impurities (mainly the aromatized derivative of VP-16) was performed by chloroform extraction followed by semipreparative liquid chromatography on LH-50 Sephadex (Pharmacia, Uppsala, Sweden). The structure and purity of VPQ were confirmed by high performance liquid chromatography, IR spectroscopy (carbonyl bands at 1627, 1661, and 1709 cm−1) and mass spectrometry. In all experiments, SC DNA, pBR 322 (∼30 superhelical turns per molecule) was used as a topoisomerase II substrate. Stock DNA was purchased from Pharmacia Biotech (Uppsala, Sweden), contained an average of 15–17% nicked circle DNA and was used as supplied after suitable dilutions.
Topoisomerase II reactions.
Topoisomerase II processing of SC DNA includes the following events: binding to DNA, strand-cleavage, strand-passage, strand-religation, and enzyme turnover (ATP-dependent). To monitor the effects of VP-16 and VPQ on the overall reaction of topoisomerase-catalyzed relaxation of SC DNA, standard assays were performed in 10 mm Tris, pH 7.7, 50 mm NaCl, 50 mm KCl, 0.1 mm EDTA, 5 mmMgCl2 in the presence, or absence of 0.5 mm ATP. Unless otherwise stated, reduced thiols and bovine serum albumin, which were expected to affect VPQ activity, were avoided. The topoisomerase/DNA complex was formed by mixing the enzyme and DNA on ice and the reactions were incubated for different times at 37° (unless otherwise stated). Typically, reactions contained 6 units of the enzyme (∼13 nm) and 100 ng of pBR 322 per 20-μl sample. Special experiments were performed to evaluate the effects of incubation times and addition order of reagents (topoisomerase, DNA, and drugs) on the inhibitory efficiency of VP-16 and VPQ. Cleavage products were trapped with 2 μl of 10% SDS, followed by an additional 5 min incubation at 37° in the presence of 10 mm EDTA and 200 mm NaCl. Thereafter, samples were treated with proteinase K (0.8 mg/ml final concentration at 55° for 2 hr). The reversibility of the drug-trapped cleavable complex was assayed after chelation of divalent metal ions by the addition of 2 μl of 100 mm EDTA before addition of SDS, while keeping samples at 37°, or at elevated temperature (56–58°) for 5 min. Kinetics of DNA strand religation reaction after topoisomerase-mediated DNA cleavage in the presence of Ca2+ ions and in the presence or absence of drugs were evaluated essentially as described (Osheroff, 1989).
Samples were subjected to electrophoresis in 1.3% agarose in the presence of 0.7 μg/ml ethidium bromide at 2 V/cm for 18 hr. In the presence of ethidium bromide, all DNA forms were separated and migrated as follows: RLX (fastest band), SC, LNR, and finally NC. Under these conditions, topoisomers of intermediate superhelicity (PRLX, observed also in the absence of ethidium) were also resolved when the average change of the linking number was between 10 and 20%. Negatives of gel photographs (Polaroid type 55 film; Polaroid, Cambridge, MA) were scanned and DNA bands were quantified using NIH Image (version 1.58) together with appropriate calibration curves. Numerical data for drug induced effects were expressed as percent difference from control: % DNA form = 100 × (C/T −C o/T o)/(C o/T o ), where C is the quantity of a given DNA form and Tis the total DNA in a sample, and C o andT o are the corresponding values for the control sample.
Results
Effect of drug concentration and addition order of reagents.
The gel photographs presented in Fig. 2show the effect of different concentrations of VP-16 and VPQ on topoisomerase II processing of SC pBR 322 DNA. As is well known for VP-16 (Chen et al., 1984; Ross et al., 1984), and demonstrated here for VPQ, the two drugs have a strong effect on the topoisomerase II/DNA cleavage/religation equilibrium, as shown by the increased formation of nicked DNA forms (LNR and NC). Quantitative analysis performed by measuring band intensities on photographic negatives reveals the rapid accumulation of linear DNA with drug doses up to about 25 μm for both VP-16 and VPQ (Fig.3). Thereafter, the amounts of linear DNA change only slightly with increasing concentration of VP-16 or VPQ (Fig. 3). Formation of linear DNA was accompanied by a monotonous increase in the levels of NC DNA within the entire range of drug concentrations used (not shown). Trapping of the topoisomerase/DNA cleavable complex in the presence of either VP-16 or VPQ resulted in a decreased formation of RLX DNA (Fig. 3). However, there was a marked difference in the rate of this conversion in the presence of VP-16 versus VPQ as seen in Figs. 2 and 3. In the case of VP-16, the concentration dependence of the formation of LNR DNA and the inhibition of DNA relaxation followed similar trends [e.g., steep slopes below 20 μm and gentle slopes (plateau) up to 125 μm]. In contrast, inhibition of DNA relaxation by VPQ was much stronger and at concentrations ⪞25 μm, no RLX DNA was formed; instead, PRLX (topoisomers with intermediate superhelicity, Fig. 2B) were observed, and at higher VPQ concentrations, neither PRLX nor RLX DNA were formed (Fig. 2B and Fig.3). The above results indicate that although VP-16 and VPQ exhibit similarities in their mode of action as inhibitors of the topoisomerase/DNA cleavable complex, the mechanism(s) may be different and/or VPQ may possess more than one mode of action. In addition, our previous results showed that VPQ does not inhibit the enzyme before its binding to DNA; rather, it suppresses topoisomerase processing of DNA after the complex has been formed (Gantchev and Hunting, 1997b).

Topoisomerase II-catalyzed reaction products during pBR322 relaxation in the absence or in the presence of different concentrations of VP-16 (A) and VPQ (B). Lane 1, 100 ng of pBR 322; lane 2, 6 units of topoisomerase/100 ng of pBR 322 (no drugs, control); lanes 3–9, topoisomerase II/DNA in the presence of 0.5 μm (lane 3), 1.25 μm (lane 4), 2.5 μm(lane 5), 12.5 μm (lane 6), 25 μm (lane 7), 62.5 μm(lane 8), and 125 μm (lane 9) drugs. Reaction conditions: drugs were mixed with the enzyme in cleavage buffer containing 0.5 mm ATP and incubated on ice for 10 min; 10 μl of the enzyme solutions were mixed with 10 μl DNA in the same buffer and further incubated for 8 min at 37°; the reactions were terminated by SDS. Positions of RLX, SC, NC, and PRLX are indicated.

Accumulation of linear (▴, ▵) and relaxed (•, ○) DNA forms at the end of the reaction in the presence of different concentrations of VP-16 (filled symbols) and VPQ (open symbols). Reaction conditions as in Fig. 2. Averaged data (mean ± standard deviation) from 3 independent experiments. ∗, DNA is only partially relaxed in the presence of increasing VPQ concentrations.
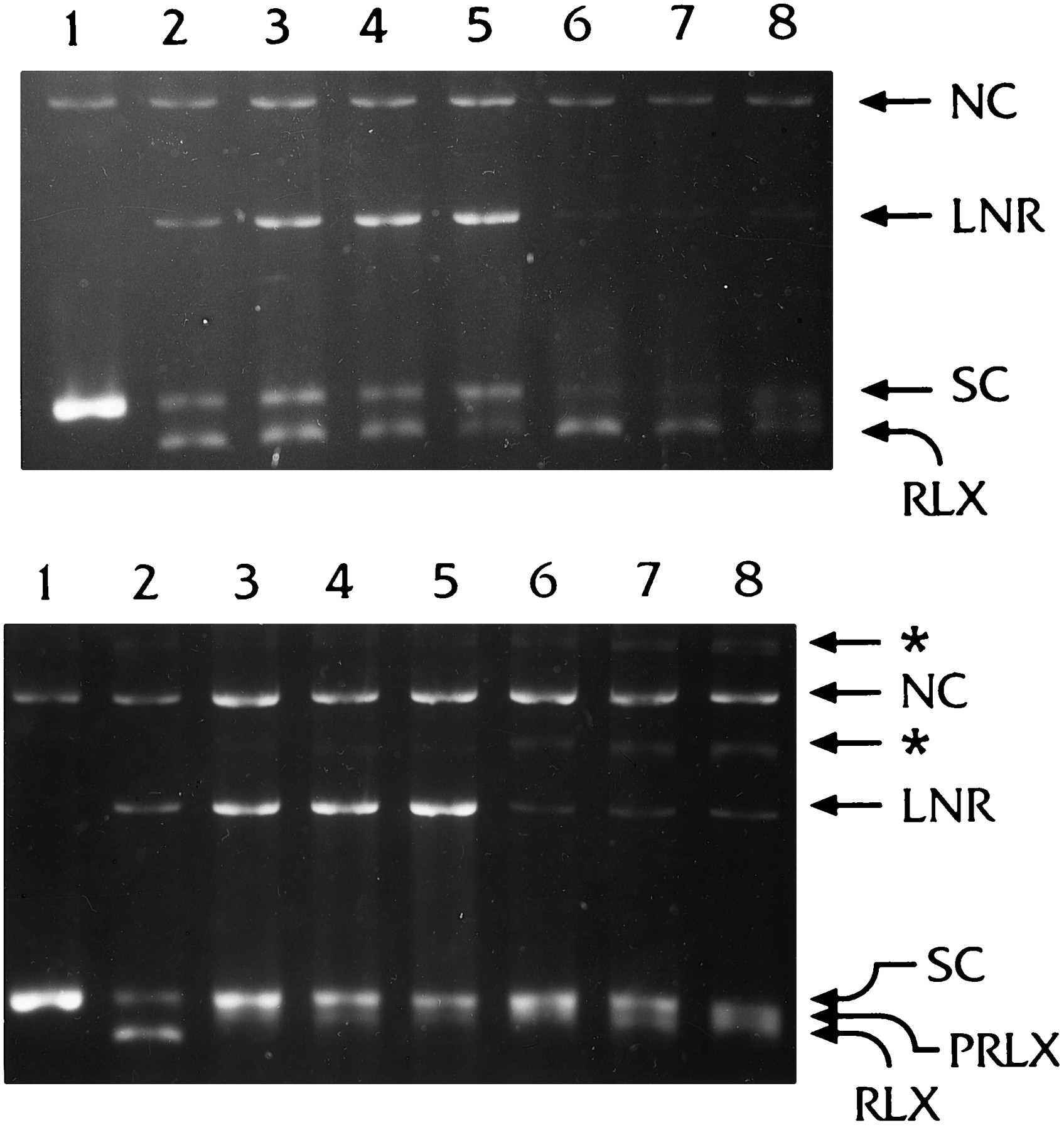
We next compared the effect of the addition order of reagents (topoisomerase, DNA, and inhibitors) on the rate of product formation (nicked and relaxed DNA forms). We explored all three possibilities: preincubation of drugs with the purified enzyme or with DNA before mixing to form the complex, and incubation of the already formed complex with the drugs. Typical gel photographs are shown in Fig.4, A and B, for VP-16 and VPQ, respectively. These gels also show the reversal of the trapped complexes induced by elevated temperature in the presence of EDTA (see below). Band intensities were measured and data are presented as percent difference from control (topoisomerase/DNA in the absence of drugs, Fig. 5). The levels of trapped topoisomerase/DNA complexes, which generated linear DNA after digestion with proteinase K, were highest when drugs were added directly to the already existing complex (stronger for VPQ), followed by the case when drugs were first preincubated with DNA. Changes in the levels of RLX and SC DNA forms for the individual drugs were less pronounced when drug addition order was changed. Incubation of VPQ with DNA alone, even at elevated temperatures, did not result in any detectable changes in the DNA migration pattern (not shown).

Effect of addition order of reagents on topoisomerase II processing of supercoiled pBR 322 DNA in the presence of VP-16 (A) and VPQ (B) and the effect of Mg2+ removal by EDTA at elevated temperature. Lane 1, 100 ng of DNA;lane 2, 6 units of topoisomerase II/100 ng of DNA (no drugs); lane 3, drugs were preincubated with the enzyme alone for 10 min at 0°; lane 4, drugs (25 μm) were preincubated with DNA for 10 min at 0°;lane 5, drugs were added to the mixture of topoisomerase/DNA and incubated for 10 min at 0°. After preincubation on ice the full component reactions were incubated for 6 min at 37°. Reactions in lanes 2–5 were terminated normally by addition of SDS. Reactions in lanes 6–8, where samples were treated identically to lanes 3–5, were additionally incubated for 5 min at 56° in the presence of EDTA/NaCl and terminated thereafter by addition of SDS.

Numerical presentation of the results obtained following different addition orders of reagents, as shown in Fig. 4,lanes 2–5. Results are presented as percent difference from control (topoisomerase/DNA without drugs). Bar groups:1, drugs preincubated with the enzyme; 2, drugs preincubated with DNA; and 3, drugs added to the existing complex. DNA forms as indicated. Averaged data from three independent experiments (mean ± standard deviation). ▧, RLX; ▩, SLC; ▤, LNR.
Reversibility of the trapped topoisomerase/DNA complexes and rates of DNA strand religation.
Enzyme-mediated DNA breaks resulting from trapping of the cleavable complex by VP-16 have been shown to be reversible [i.e., cleaved DNA strands can be religated upon addition of salt, EDTA, or by dilution or heating (Hsiang and Liu, 1989;Osheroff, 1989; Robinson and Osheroff, 1991)]. Experiments were performed to determine whether the cleavable complex trapped by VPQ could also be reversed under different conditions.
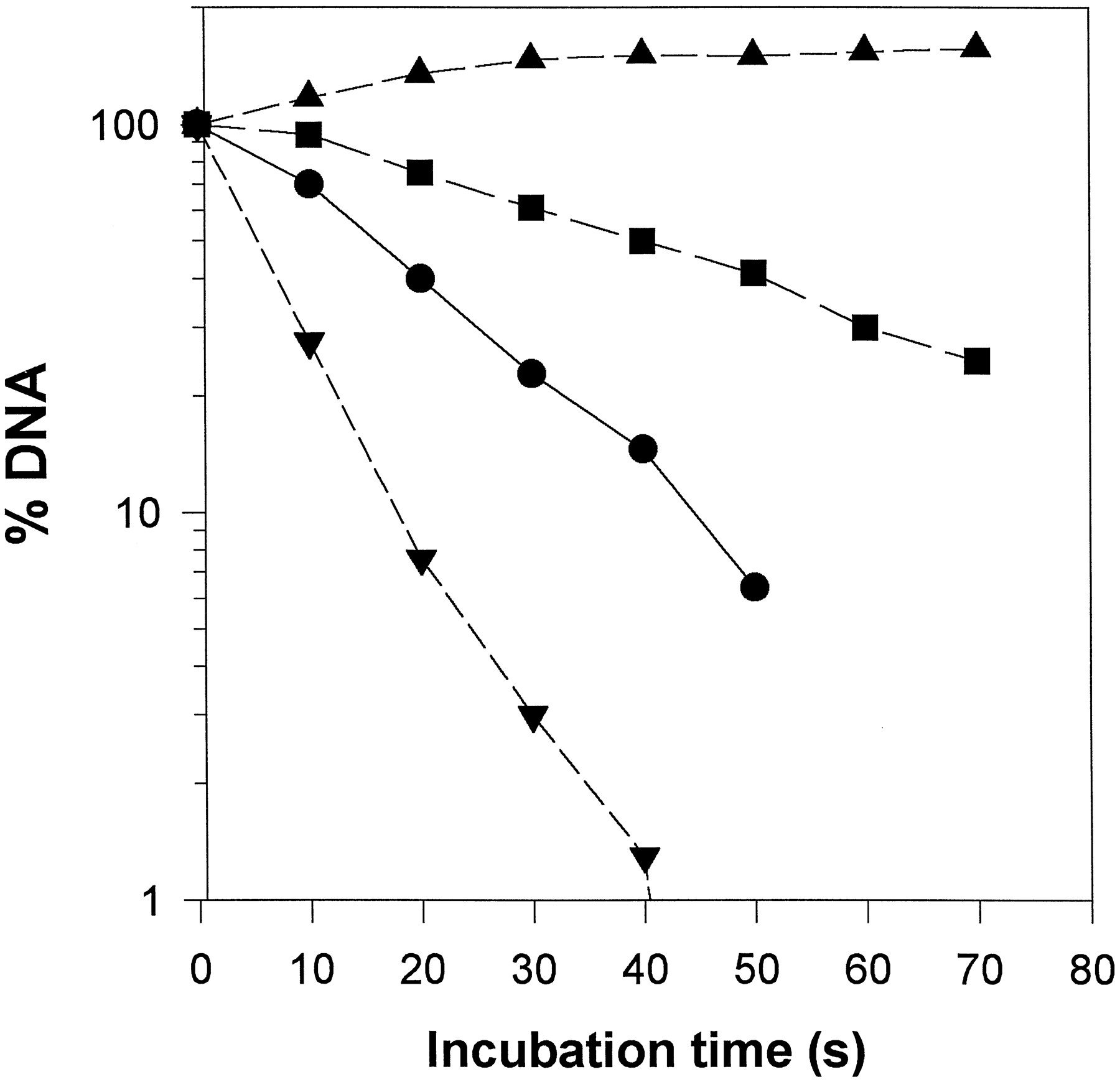
As shown in Fig. 4, if the reaction in the presence of either VP-16 or VPQ was stopped by EDTA and heating before addition of SDS, double-stranded DNA breaks were reversed (i.e., religation of one or both of the cleaved DNA strands occurred). Reversal of the double-stranded DNA breaks of the drugs trapped complex also took place when the reaction was stopped only by EDTA/NaCl at 37°, or when the reaction was performed in the absence of ATP, conditions that prevent enzyme turnover (Fig. 6). However, there were several important differences between VP-16 and VPQ, concerning both the effect of the two drugs on the product yields as well as the absence or presence of ATP. One of the major features during the reversal of the topoisomerase/DNA complex trapped by VPQ was the appearance of additional slow-moving bands when the reaction was performed in the presence of ATP (Fig. 4, *), but not when this cofactor was absent (Fig. 6). These new bands could not be eliminated by altering the digestion conditions with proteinase K. They probably represent partially digested DNA/(poly)peptide complexes containing VPQ(poly)peptide covalent adducts. Fig. 7compares the changes in product yields after the EDTA/NaCl reversal procedure of the cleavable complex trapped by either of the two drugs and in the presence or absence of ATP. Data are plotted as percent difference from control, where the controls are levels of each DNA form when the reaction was stopped by SDS, before the addition of EDTA/NaCl. Note that in the absence of ATP, no measurable levels of RLX DNA were formed in the presence of drugs (Fig. 6) (only minor quantities of PRLX were observed). In the absence of ATP, the nicked DNA products formed in the presence of VP-16 and VPQ (LNR and NC) change in a similar manner (i.e., LNR DNA decreases by 70–75%, and NC increases; the effects are more pronounced in the case of VPQ) (Fig. 7B). In all cases (topoisomerase, topoisomerase/VP-16, and topoisomerase/VPQ), reversal of the cleavable complex enhances the levels of SC DNA, as expected, indicating that under these conditions (no ATP), complexes were mainly trapped before the strand-passage event. In the presence of ATP, however, the effects of the two drugs were significantly different (Fig. 7A). When the complex was trapped by VPQ, the reversibility procedure resulted in a significant increase in SC DNA (i.e., inhibition of DNA strand passage) and a stronger increase in NC DNA (2–3 times higher than in the case of VP-16). This latter difference prompted us to perform experiments designed especially to monitor strand religation kinetics. Previous kinetic analyses revealed that topoisomerase II religates cleaved DNA by a sequential two-step mechanism (Robinson and Osheroff, 1991), and the apparent rate constant for first- and second-strand religation is reduced approximately 3-fold in the presence of VP-16 (Osheroff, 1989). In our experiments, DNA cleavage was performed in the presence of Ca2+ions, the complexes were trapped with EDTA, and religation was initiated by the addition of Mg2+ ions (Osheroff and Zechiedrich, 1987). Under these experimental conditions, religation of the first cleaved strand (disappearance of LNR DNA) in the absence of inhibitors (topoisomerase II only), or in the presence of VP-16 was fast and kinetically unresolved. A gradual disappearance of LNR DNA within about 50 sec after initiation of religation by Mg2+ was observed when the cleavable complex was trapped by VPQ (Fig. 8). This result indicates that VPQ decreases substantially the rate of first-strand religation compared with the enzyme alone or in the presence of VP-16. It has been shown previously that the religation rate of the second cleaved strand is about an order of magnitude lower than that of the first strand (Osheroff, 1989; Robinson and Osheroff, 1991). In this case, the kinetics were well resolved and we were able to monitor the time dependence of the changes of the levels of NC DNA (Fig. 8) in the absence or presence of drugs. The rate of religation of the second strand was about 2.8-fold lower in the presence of VP-16 compared with the enzyme alone. In contrast, when the complex was trapped by VPQ, instead of a time-dependent decrease in the levels of NC DNA, an initial (within the first 20–30 sec) increase in the amount of NC DNA was observed, followed by a plateau. This result is interpreted as evidence of a strong inhibition of the religation of the second DNA strand by VPQ.

Reaction products after cleavage of 100 ng of DNA in the presence of 6 units of topoisomerase II performed in a cleavage buffer containing Mg2+ and VP-16 or VPQ, but no ATP, and the effect of removal of Mg2+ ions by EDTA at 37° before protein denaturing by SDS. Lane 1, DNA; lanes 2–3, topoisomerase II/DNA; lanes 4–5, enzyme/DNA complexes trapped by 25 μm VP-16; lanes 6–7, enzyme/DNA complexes trapped by 25 μm VPQ. Drugs were added to already existing complexes and the reactions were incubated for 30 min at 37°. Lanes 2, 4, and 6, SDS was added before EDTA/NaCl; lanes 3, 5, and 7, EDTA/NaCl was added before SDS.
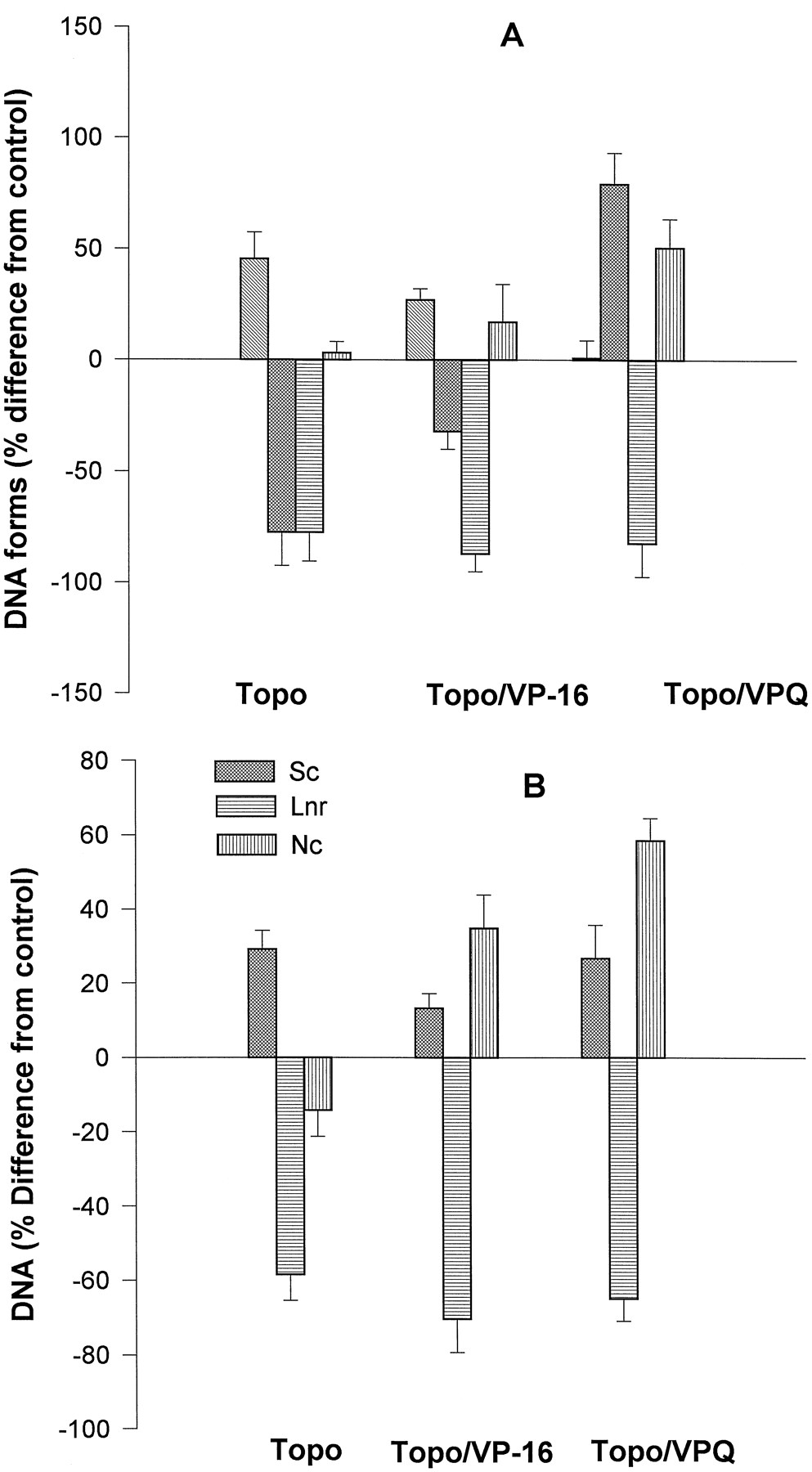
Comparison of the effects of the complexing of Mg2+ ions by EDTA on the levels of different DNA forms produced by topoisomerase II (6 U) during cleavage in the presence, or absence of drugs, and in the presence (A), or absence (B) of ATP. Reaction conditions as in Fig. 6. Data presented as % difference from controls (samples where the reaction was terminated by SDS before the addition of EDTA). Averaged data from triplicate experiments. ▧, RLX; ▩, SLC; ▤, LNR; ▥, NC.

Effects of VP-16 and VPQ on DNA strand religation reaction of topoisomerase II. The religation of the first strand (i.e., conversion of LNR DNA to NC) was kinetically resolved only in the case of the VPQ trapped complex (▾). Transformation of the second strand (i.e., conversion of NC DNA to negatively supercoiled form) was resolved for topoisomerase II without drugs (•); for topoisomerase II/DNA complex trapped by VP-16 (▪), and by VPQ (▴).
The role of GSH and GSSG.
It has previously been shown that GSH can form covalent adducts with VPQ, but not with VP-16 (Manset al., 1992). In the present work, we examined the effects of GSH and GSSG on the efficiency of the two drugs to trap the topoisomerase/DNA cleavable complex. Because glutathione and other reduced thiols are usually added to the enzyme assays to prevent oxidative inactivation of the topoisomerase, we performed the experiments so that the controls (topoisomerase/DNA without drugs) were in the presence or absence of thiols. Drugs (25 μm) were preincubated with 3 mm GSH or GSSG for 15 min before mixing with topoisomerase/DNA in cleavage buffer, containing ATP, followed by incubation for 6 min at 37°. As seen in Fig.9, the presence of GSH or GSSG does not induce changes in the trapping of the cleavable complex by VP-16 (i.e., LNR DNA formation); however, virtually no linear DNA is formed in the presence of VPQ and either GSH or GSSG. Although no further analysis of the interaction of VPQ with both thiol compounds was performed in this study, it can be suggested that VPQ, as a nucleophile, may attack the -SH groups of the reduced glutathione as well as the amino-terminal groups of both peptides (Kalyanaraman et al., 1987). In addition, it is clear that the VPQ-glutathione conjugates are not able to trap the topoisomerase/DNA cleavable complex.

Effects of thiols (GSH and GSSG) on the efficiency of VP-16 and VPQ inhibition of topoisomerase II/DNA complex. Data presented as percent difference from control (topoisomerase II/DNA without drugs, ± 3 mm thiols). DNA forms as indicated. Averaged data from triplicate experiments. ▧, RLX; ▩, SLC; ▤, LNR.
Discussion
The present study shows that the o-demethylated metabolic product of the anticancer drug etoposide, VP-16ortho-quinone (VPQ), is a potent inhibitor of the topoisomerase II/DNA cleavable complex. This is an important finding in view of structure-activity relationships and implies that substitution of one of the methoxy groups and the hydroxyl group on the VP-16 E-ring by keto groups does not adversely affect trapping of the topoisomerase/DNA cleavable complex. Several mechanisms of VPQ action against the topoisomerase/DNA cleavable complex are possible and may include noncovalent (Burden et al., 1996) or covalent interactions, or may involve slow transformations to a toxic semiquinone radical (Kalyanaraman et al., 1987). Theortho-quinone might also act as a metal ion chelator in Fenton-type oxidative reactions (Sakurai et al., 1991). The fact that the topoisomerase-mediated double-stranded DNA breaks were highly reversible indicates that, as for VP-16, theortho-quinone derivative inhibits DNA-strand rejoining mainly by forming a noncovalent complex with the topoisomerase homodimer/DNA duplex and does not inactivate the purified enzyme alone. However, our results point out several differences between the mode of action of the two drugs. VPQ is obviously a stronger inhibitor and traps the complex predominantly before the strand-passage event (in the presence of ATP). Previously, it has been shown that VP-16 inhibits the complex, both before and after strand passage (Robinson and Osheroff, 1991), which is consistent with our experimental data. Thus, in the reversibility experiments performed in the presence of ATP, trapping of the complex by VPQ resulted in an increased formation of SC and NC DNA compared with the corresponding controls and in contrast to VP-16, where reversal resulted in an increase in RLX and a decrease in SC DNA. These data indicate that, although VPQ may interfere with the topo II-DNA cleavage reaction, it predominantly interacts with the cleavable complex and affects the strand-passage event. The involvement of multi-step conformational rearrangements is a prerequisite for topoisomerase processing of DNA (Roca and Wang, 1994). Consequently, it is not surprising that VPQ may exhibit additional mode(s) of action against the topoisomerase/DNA complex in interactions that depend on the stage of the enzyme cycle. Apart from the residues that are important for ATP binding (Lys 103 and Lys 337), the ATPase carboxyl-terminal regions of DNA gyrase and topoisomerase II are unusually enriched with arginine residues (i.e., potential targets for nucleophilic attack) (Wigley et al., 1991; Berger et al., 1996). Therefore, possible interference of VPQ with ATP binding and/or modification of the DNA strand-passage channel of the complex might be expected. Different interactions are conceivable, including coordination with metal ions and/or blocking of essential amino acid side-chain groups by VPQ. The question of whether VPQ undergoes covalent chemical reactions with the complex cannot be answered unequivocally, but it is likely that covalent adducts are also formed, as evidenced by the appearance of new slow-migrating DNA bands observed only after removal of divalent ions by EDTA and in the presence of ATP before digestion with proteinase K. It has been shown that quinolone compounds cause asymmetric conformational changes in the gyrase/DNA complex (Orphanides and Maxwell, 1994). This may be also the case with VPQ and may explain the results obtained in the religation experiments, namely the strong (asymmetric) inhibition of the second strand rejoining step in the presence of this drug. Elucidation of the possible involvement of these additional modes of action will require further experimental work. It can be anticipated that when the interactions of VPQ with the topoisomerase/DNA complex are better understood, this drug may be a useful tool for mechanistic studies of topoisomerase II.
In this study, we have also shown that GSH and GSSG prevent the inhibition of the topoisomerase/DNA complex by VPQ but not by VP-16. In this case, as has been shown previously (Mans et al. 1992), the process is expected to involve formation of covalent conjugates between SH- and/or terminal NH2-groups of the peptides and VPQ. Interestingly, VPQ did not deactivate the topoisomerase itself, indicating that there are no accessible and sensitive amino acid groups in the protein in the absence of DNA. As hypothesized above, such groups might become exposed to drug attack after conformational rearrangements following the formation of the topoisomerase/DNA complex. The prevention of the topoisomerase/DNA complex inactivation by VPQ in the presence of GSH (or by other protein and nonprotein thiols) can be expected to play an important role in the cellular toxicity of this VP-16 metabolite (Yokomizo et al., 1995; Gantchev and Hunting, 1997a). In agreement with previously reported cytotoxicity data (Sinha et al., 1990), our experiments with cells in culture under similar conditions to those described in Gantchev and Hunting (1997a) showed that when drugs were incubated in RPMI 1640 medium, containing 1 mm GSH and 10% fetal bovine serum, VPQ was about 50% less toxic than its parent compound, VP-16 (results not shown). However, when the incubation was performed in phosphate buffered saline instead of medium, both drugs were equally toxic and promoted formation of DNA breaks at similar efficiencies. These results imply that VPQ itself probably cannot be administered as an anticancer drug, because it will exert its toxic potential primarily when formed directly in cells, where one of its effects will be to act as a powerful topoisomerase/DNA cleavable complex inhibitor. It can be also anticipated that the levels and distribution of intracellular VP-16 metabolizing enzymes (cytochrome P450 monooxygenases and peroxidases), as well as the antioxidant potential of a given cell type, will significantly alter the cytotoxic potency of VP-16.
Footnotes
- Received September 5, 1997.
- Accepted November 12, 1997.
-
Send reprint requests to: Dr. Tsvetan G. Gantchev, Department of Nuclear Medicine and Radiobiology, Faculty of Medicine, Université de Sherbrooke, Sherbrooke, QC, J1H 5N4, Canada. E-mail: gantchev{at}courrier.usherb.ca
-
This work was supported by the Medical Research Council of Canada.
Abbreviations
- VP-16
- etoposide
- VPQ
- ortho-quinone derivative of etoposide
- LNR
- linear DNA form
- NC
- nicked circular DNA form
- PRLX
- partially relaxed DNA form
- RLX
- relaxed DNA form
- SC
- supercoiled DNA form
- GSH
- reduced glutathione
- GSSG
- oxidized glutathione
- SDS
- sodium dodecyl sulfate
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}