


Abstract
The effects of treatment with squalestatin 1, a potent inhibitor of squalene synthase, the first committed enzyme of sterol biosynthesis, were examined on cytochrome P450 expression in primary cultured rat hepatocytes and rat liver. Incubation of cultured hepatocytes with squalestatin 1 caused marked accumulations (maximal elevations that were ∼25–100% of phenobarbital-elicited increases) of CYP2B mRNA and immunoreactive protein but not of CYP1A, CYP3A, or CYP4A. Squalestatin 1 treatment increased CYP2B and 3-hydroxy-3-methylglutaryl coenzyme A reductase mRNA content in hepatocyte cultures with comparable potencies (ED50 = 5.0 and 18 nm, respectively), and significantly induced CYP2B (mRNA, immunoreactive protein, and pentoxyresorufin O-dealkylase activity) in the livers of treated rats, producing maximal increases at a dose of 25 mg/kg/day that were ∼32–87% of phenobarbital-induced increases. Squalestatin 1 treatment induced both CYP2B1 and CYP2B2 and activated reporter gene expression in cultured hepatocytes transiently transfected with a plasmid containing ∼2.4 kb of CYP2B1 gene 5′-flanking region or containing a previously described phenobarbital-responsive region. Coincubation of cultured hepatocytes with 25-hydroxycholesterol suppressed squalestatin 1-mediated CYP2B and 3-hydroxy-3-methylglutaryl coenzyme A mRNA induction with approximately the same potency. Treatment of cultures with SQ-34919, a structurally distinct squalene synthase inhibitor, produced the same selective CYP2B mRNA induction as did squalestatin 1. These results suggest that inhibition of hepatic sterol synthesis activates processes that culminate in increased CYP2B gene transcription.
The enzymes of the P450 superfamily catalyze the oxidative metabolism of a diverse array of substrates, both endogenous (e.g., steroid hormones, sterols, fatty acids, and prostaglandins) and xenobiotic (e.g., drugs and environmental contaminants, including procarcinogens). Each P450 is characterized not only by its substrate specificity but also by its particular pattern of tissue-, gender-, and development-specific regulation, as well as by its ability to undergo induction, an adaptive response whereby the presence of a chemical “inducer” evokes an increase in the amount of one or more P450 enzymes, which often, but not always, metabolize the inducer. For example, polycyclic aromatic hydrocarbons (e.g., 3-methylcholanthrene) and polyhalogenated hydrocarbons (e.g., 2,3,7,8-tetrachlorodibenzo-p-dioxin and planar polychlorinated biphenyls) induce “aromatic hydrocarbon-inducible” P450s (i.e., CYP1A1, CYP1A2, and CYP1B1), whereas PB and “PB-like” chemicals (e.g., organochlorine pesticides, nonplanar polychlorinated biphenyls, and certain imidazole antimycotic drugs) induce “PB-inducible” P450s (primarily CYP2B1 and CYP2B2 in rat) (Gonzalez, 1989). Synthetic steroids (e.g., pregnenolone 16α-carbonitrile and dexamethasone) and peroxisome proliferators (e.g., clofibrate) induce primarily CYP3A and CYP4A enzymes, respectively (Gonzalez, 1989).
Knowledge of how diverse foreign chemicals induce P450s, especially the initial steps whereby the chemicals interact with cellular “receptors,” is at various stages of understanding. Much progress has been made in understanding aryl hydrocarbon receptor-mediated induction of CYP1A1 (Whitlock et al., 1996). Also, a receptor, termed the peroxisome proliferation associated receptor α, is now known to mediate peroxisome proliferator-inducible CYP4A gene expression (Green and Wahli, 1994). For steroid-inducible CYP3A induction, although the evidence has not supported a role for the classic glucocorticoid receptor (Schuetz and Guzelian, 1984; Quattrochiet al., 1995; Huss et al., 1996), a recently described orphan nuclear receptor, termed the pregnane X receptor, appears to be a likely mediator of the response (Kliewer et al., 1998). However, no receptor that mediates PB-inducible CYP2B induction has been identified. Indeed, even standard pharmacological evidence for a receptor-mediated mechanism is lacking, such as a clear set of structure-activity relationships for the inducing agents or enantioselectivity (Nims et al., 1994).
One proposed mechanism for PB-mediated P450 induction is that PB and PB-like inducers interfere with the metabolism of an endogenous regulator of P450 expression, possibly a steroid (Waxman and Azaroff, 1992). In this light, several pieces of evidence suggest that there is a relationship between cholesterol metabolism and PB-inducible P450 expression. For example, Plewka and Kaminski (1996) recently reported that rats fed a high cholesterol diet developed suppressed levels of hepatic basal and PB-inducible P450 content, aniline hydroxylase activity, and 4-aminopyrine N-demethylase activity, relative to rats fed a standard diet. Reduced hepatic levels of several P450-dependent monooxygenase activities were also observed in spontaneously hyperlipidemic rats that had ∼3- 4-fold higher serum cholesterol levels than did wild-type rats (Watanabe et al., 1996). In addition, PB treatment of rats was reported to result in increased plasma cholesterol levels (Thomas, 1984) and increased hepatic expression of several genes encoding cholesterol-metabolizing enzymes (Andersson et al., 1994; Sudjanasugiaman et al., 1994).
To test whether inhibition of cholesterol biosynthesis could result in altered P450 expression, we examined the effects of several drugs that competitively inhibit HMG-CoA reductase, the rate-limiting enzyme of cholesterol biosynthesis, in which HMG-CoA is converted to mevalonate. We reported that certain HMG-CoA reductase inhibitors, such as lovastatin and fluvastatin, represent a unique class of P450 inducers that primarily induce CYP2B and CYP4A expression, both in primary cultured rat hepatocytes and (in the case of fluvastatin, at least) in rat liver in vivo (Kocarek et al., 1993; Kocarek and Reddy, 1996).
By inhibiting mevalonate formation, HMG-CoA reductase inhibitors prevent the synthesis not only of sterols but also of other biologically active isoprenoids. Therefore, in the present study, we tested the possibility that altered P450 expression derives specifically from inhibited sterol biosynthesis by examining the effects of treatment with squalestatin 1 (also known as zaragozic acid A), a potent inhibitor of squalene synthase, the first committed enzyme of sterol biosynthesis (Baxter et al., 1992), on P450 expression in primary cultured rat hepatocytes and in rat liver. We report that squalestatin 1 is the most potent inducer of rat hepatic CYP2B expression yet described, inducing CYP2B mRNA in primary cultured rat hepatocytes with an ED50 value of ∼5 nm.
Experimental Procedures
Materials.
Squalestatin 1 was a gift from Glaxo Wellcome Research and Development (Hertfordshire, UK), and SQ-34919 was a gift from Bristol-Myers Squibb Pharmaceutical Research Institute (Princeton, NJ). Fluvastatin was a gift from Novartis Pharmaceuticals (Summit, NJ). Matrigel and Matrisperse were purchased from Collaborative Research Products (Bedford, MA). Vitrogen (95–98% type I collagen with the remainder type III collagen) was purchased from The Collagen Corporation (Palo Alto, CA). Opti-MEM culture medium and Lipofectin reagent were purchased from GIBCO BRL (Grand Island, NY). The oligonucleotide probes directed against CYP2B1 or CYP2B2 mRNA previously described by Omiecinski (1986) were purchased from National Biosciences (Plymouth, MN). Terminal deoxynucleotidyl transferase was purchased from Promega (Madison, WI). [α-32P]ATP (3000 and 6000 Ci/mmol) was purchased from DuPont NEN (Boston, MA). 25-Hydroxycholesterol was purchased from Steraloids (Wilton, NH). Pentoxyresorufin, resorufin, NADPH, and mevalonic acid lactone were purchased from Sigma Chemical (St. Louis, MO). Solvents for lipid extraction and chromatography (Burdick and Jackson) and thin layer chromatography plates (Baker silica gel, 250 μm) were purchased from VWR Scientific Products (Chicago, IL). [2-14C]Acetate (58 mCi/mmol) was purchased from Amersham Life Science (Arlington Heights, IL). Plasmids containing cDNA inserts to CYP1A1 (p210), CYP2B1 (pSR-p450), CYP3A1 (pDex12), CYP4A1 (p46), and 7S RNA (pA6) were gifts from Dr. John Fagan (Maharishi International University, Fairfield, IO), Dr. Milton Adesnik (New York University, NY), Dr. Philip Guzelian (University of Colorado, Denver, CO), Dr. Frank Gonzalez (National Cancer Institute, Bethesda, MD), and Dr. Allan Balmain (Beatson Institute, Glasgow, Scotland), respectively. A cDNA probe to rat HMG-CoA reductase was prepared as described previously (Kocarek and Reddy, 1996). Polyclonal antibodies to CYP1A1 and CYP2B1 were purchased from Xenotech LLC (Kansas City, KS). A polyclonal antibody to CYP3A1 was a gift from Dr. Janis Hulla (University of North Dakota, Grand Forks, ND). A polyclonal antibody to CYP4A1 was purchased from GenTest (Woburn, MA). Other supplies and reagents were obtained from the sources previously described (Kocarek and Reddy, 1996, 1998).
Primary hepatocyte culture.
Adult male Sprague-Dawley rats, weighing 200–250 g, were purchased from Harlan Sprague-Dawley (Indianapolis, IN) and maintained in an AAALAC-approved animal facility with free access to chow and water for ∼1 week before use. Hepatocytes were isolated from the livers of the rats, weighing 220–280 g at the time of isolation, by collagenase perfusion, and plated onto 60-mm tissue culture dishes that were precoated with 1.5 mg of Matrigel, as described previously (Kocarek and Reddy, 1996). Cultured hepatocytes were incubated in a humidified chamber maintained at 37° and 5% CO2 with serum-free Williams’ Medium E supplemented with 0.25 units/ml insulin, 10−7 m triamcinolone acetonide, 100 units/ml penicillin, 100 μg/ml streptomycin, and 15 mmHEPES, unless otherwise indicated. Culture medium was renewed every 24 hr. Drug incubations were performed beginning 48 hr after plating the hepatocytes, as described in the individual figure legends. Drugs were added to the culture medium as concentrated stock solutions in water (squalestatin 1, SQ-34919, PB, fluvastatin, and mevalonate), dimethylsulfoxide (β-naphthoflavone, dexamethasone, and ciprofibrate), or ethanol (25-hydroxycholesterol). The final concentration of organic solvent in the culture medium was 0.1%.
In vivo treatment of rats.
Adult male Sprague-Dawley rats (260–300 g) were treated with saline (1, 3, or 5 daily doses intraperitoneal), β-naphthoflavone (80 mg/kg in 2% Tween-20 × 3 days intraperitoneal), PB (80 mg/kg in water × 3 days intraperitoneal), dexamethasone (150 mg/kg in 2% Tween-20 × 3 days intraperitoneal), ciprofibrate (5 mg/kg in 2% Tween-20 × 3 days intraperitoneal), or squalestatin 1 (1, 5, 10, 25, or 50 mg/kg in water × 3 days or 25 mg/kg × 1, 3, or 5 days intraperitoneal). At 24 hr after the last treatment, rats were injected with pentobarbital (120 mg/kg intraperitoneal), and livers were perfused briefly with ice cold phosphate-buffered saline, dissected, and frozen in liquid nitrogen for subsequent preparation of RNA and microsomes.
Northern and slot blot analyses.
Total RNA was prepared from samples of frozen rat liver or from three pooled dishes of cultured hepatocytes and analyzed by Northern blot hybridization or was prepared individually from triplicate dishes of hepatocytes and analyzed by slot blot hybridization, as described previously (Kocarek and Reddy, 1996,1998). For oligonucleotide hybridizations, poly(A)+ RNA was prepared from the pooled total RNA isolated from 16 dishes of hepatocytes, using a commercially available kit (Oligotex mRNA kit, Qiagen, Santa Clarita, CA). Poly(A)+ RNA samples (2 μg) were resolved and transferred to nylon membranes. Oligonucleotides were radiolabeled by tailing with [α-32P]ATP (6000 Ci/mmol), as described by Collins and Hunsaker (1985). Blots were prehybridized overnight at 50° in 6× SSPE (0.9 m NaCl, 60 mm NaH2PO4, 6 mm EDTA, pH 7.4), 5× Denhardt’s solution (0.1% Ficoll, 0.1% polyvinylpyrrolodone, 0.1% bovine serum albumin), 1% SDS, 200 μg/ml sonicated and denatured salmon sperm DNA, and 200 μg/ml polyadenylic acid (Collins and Hunsaker, 1985). Blots were hybridized overnight at 50° in the above solution containing 20 × 106 cpm/ml radiolabeled oligonucleotide. Blots then were washed twice for 1 hr at 50° in 5× SSC (0.75 mNaCl, 50 mmNaH2PO4, 5 mmEDTA, pH 7.4)/0.5% SDS, and once for 1 hr at 50° in 5× SSC/0.1% SDS. After autoradiography, filters were incubated briefly at 90° in 1% SDS to remove hybridized probes. Blots containing total RNA then were rehybridized with 7S cDNA to control for RNA loading and transfer, as described previously (Kocarek and Reddy, 1996). Band intensities on slot blots were quantified using a scanning laser densitometer (Molecular Dynamics, Sunnyvale, CA) equipped with ImageQuant software (version 3.3). To ensure that band intensities were quantified within the linear capacity of the film, several serial dilutions of a standard RNA sample were loaded onto each slot blot. For each blot, multiple film exposures were prepared, and only those experimental samples on a given film that fell within a linear range of standard dilutions were quantified.
Western blot analysis.
Microsomes were isolated from samples of frozen rat liver or pooled dishes of primary cultured hepatocytes (five or six dishes per treatment group), as described previously (Kocarek and Reddy, 1996). Protein concentrations were determined by the bicinchonic acid assay (Smith et al., 1985), using bovine serum albumin as standard. Samples of microsomal proteins (10 or 20 μg for microsomes prepared from rat liver or cultured rat hepatocytes, respectively) were resolved by SDS-polyacrylamide gel electrophoresis (10% acrylamide), using either a mini or a standard Protean II vertical electrophoresis apparatus (BioRad, Hercules, CA) and transferred electrophoretically onto nitrocellulose membranes. Blots were incubated with polyclonal antibodies to CYP1A1, CYP2B1, CYP3A1, or CYP4A1 and then with peroxidase-conjugated anti-rabbit or anti-goat IgG, as appropriate, and immunoreactive bands were visualized by enhanced chemiluminescence, according to the manufacturer’s instructions (Amersham Life Science), as described previously (Kocarek and Reddy, 1996). Band intensities were quantified by scanning laser densitometry as described for Northern blot analysis.
PROD activity.
The PROD assay was performed using the continuous fluorometric method and conditions described by Lubetet al. (1985) and 10–60 μg of rat liver microsomes, depending on the activity of the sample.
Lipid biosynthesis.
The 48-hr-old rat hepatocyte cultures were incubated for 24 hr with medium alone or containing 10−7 m squalestatin 1 (three 60-mm dishes per treatment group). At 2 hr before harvest at 72 hr, [14C]acetate (10 μCi, 57.5 μm) was added to the culture medium of each dish. After incubation for 2 hr, cells were washed twice with cold phosphate-buffered saline, scraped into 1 ml of Matrisperse, and allowed to stand on ice for ∼30 min (with periodic gentle inversion of the tubes), until the Matrigel had dissolved. After centrifugation and aspiration of supernatants, cell pellets were dissolved in 500 μl of 0.5 n NaOH, and 200 μl of the cell lysates, 300 μl of H2O, and 10 μl of lipid standards (mixture of 10 mg/ml cholesterol, lanosterol, and squalene in ethanol) were transferred to 16 × 125-mm siliconized glass screw-cap tubes. Protein concentrations were measured in aliquots of the remaining lysates (Smith et al., 1985). Lipids were saponified by adding 1.5 ml of 15% KOH/95% ethanol to the glass tubes and heating for 1 hr at 80°. After allowing the samples to cool to room temperature, nonsaponifiable lipids were extracted four times, each with 4 ml petroleum ether. The combined ether phases were transferred to 13 × 100-mm siliconized glass tubes, evaporated to <100 μl, and spotted onto SilicaGel 250-μm thin layer chromatography plates, which were developed with hexane/diethyl ether (1:1). Lipids were visualized with iodine vapor, and spots corresponding to cholesterol, lanosterol, and squalene were scraped and transferred to 20-ml plastic scintillation vials. After dissolving the silica scrapings in 0.5 ml of hydrofluoric acid, 10 ml of scintillation cocktail (ScintiSafe, Fisher Scientific, Itasca, IL) was added to each vial, and radioactivity was determined by liquid scintillation counting.
Transfection of primary cultured rat hepatocytes.
A 2451-bp fragment of the CYP2B1 gene, spanning bp −2413 to +23 relative to the transcription start site, was prepared by polymerase chain reaction amplification, using primers corresponding to bases 1465–1483 and 3900–3881 of the CYP2B1 sequence (GenBank accession number U30327), rat genomic DNA as template, and the proofreading Pfupolymerase (Stratagene Cloning Systems, La Jolla, CA). The PCR fragment was ligated into the BglII site of the pGL3-Basic luciferase (firefly) reporter plasmid (Promega), and a clone containing the CYP2B1 insert in the forward orientation was sequenced. The sequence of this cloned fragment was identical to that published by Sommer et al. (1996) at all except five positions. From this cloned CYP2B1 PCR fragment, we isolated the 163-bp Sau3AI fragment (bp −2299 to −2137) corresponding to the PB-responsive region (i.e., the PBRE) of CYP2B2 reported by Trottier et al. (1995) and ligated this fragment (forward orientation) into the BglII site of the pGL3-Promoter (SV40 promoter) luciferase reporter plasmid. The sequence of this PBRE region is highly conserved between CYP2B1 and CYP2B2 (2 mismatches of 163 bp). Primary cultured rat hepatocytes were transiently transfected using a modification of the method described byBurger et al. (1992). Hepatocytes were plated in standard Williams’ Medium E onto Vitrogen-coated 12-well plates (5 × 105 hepatocytes/well). At ∼21 hr after plating, culture medium was replaced with 0.6 ml of Opti-MEM containing a premixed complex of 5.5 μg of Lipofectin reagent and 0.8 μg of pGL3-Basic, CYP2B1(−2413 to +23)pGL3-Basic, pGL3-Promoter, or CYP2B1(−2299 to −2137)pGL3-Promoter, in combination with 0.08 μg of the pRL-TK reporter plasmid (Promega), which expresses theRenilla luciferase under the control of the herpes simplex virus thymidine kinase promoter, to allow for normalization among samples due to differences in transfection efficiency. Transfection incubations were continued for 5 hr, after which culture medium was replaced with standard Williams’ Medium E for 2 hr. Culture medium was then aspirated, and hepatocytes were overlaid with 0.8 mg of Matrigel. After incubation of the cultures at 37° for 30 min to allow for Matrigel gelation, standard culture medium (1 ml) was added to each well, and cultures were incubated overnight. At 48 hr after plating, fresh medium, either alone or containing 10−4 m PB or 10−7 msqualestatin 1, was added to each well. After 24 hr, hepatocytes were harvested for measurement of luciferase activity (firefly andRenilla) using the Dual Luciferase Reporter Assay System, according to the manufacturer’s instructions (Promega), and a Dynex model MLX luminometer.
Data analysis.
Numerical data were analyzed using either the unpaired Student’s t test or one-way analysis of variance followed by the Dunnett’s test. ED50 and ID50 values and 95% CIs were estimated by fitting dose-response data to a sigmoidal function, using Prism (version 2) software (GraphPAD, San Diego, CA).
Results
To extend our previous findings on the effects of HMG-CoA reductase inhibitor treatments on rat hepatic P450 expression (Kocareket al., 1993; Kocarek and Reddy, 1996), we examined the effect of incubating primary cultured rat hepatocytes with squalestatin 1 (see Fig. 1 for chemical structure), an inhibitor of squalene synthase, which catalyzes the first committed step in sterol biosynthesis. Inhibition of squalene synthase prevents the synthesis of sterols, but (unlike HMG-CoA reductase inhibition) not biologically active nonsterol isoprenoids (e.g., isopentenyl adenine, ubiquinone, dolichol, farnesol, and the farnesyl and geranylgeranyl groups used in post-translational modification reactions). Squalestatin 1, one of a family of fungal isolates, is a competitive inhibitor of squalene synthase with respect to farnesyl pyrophosphate and was originally reported to inhibit rat liver microsomal squalene synthase activity with an IC50 value of ∼12 nm (Baxter et al., 1992), although a later study demonstrated that the inhibitory mechanism of squalestatin 1 proceeded through competitive inhibition that was followed by mechanism-based irreversible inactivation and estimated the IC50value to be 0.75 nm (Lindsey and Harwood, 1995).

Chemical structures of squalene synthase inhibitors squalestatin 1 (zaragozic acid A) and SQ-34919.
Primary cultured rat hepatocytes were treated with a range of squalestatin 1 doses beginning 48 hr after plating and were harvested 24 hr later for measurement of biological end points. Preliminary experiments revealed that squalestatin 1 doses up to 3 × 10−5 m were not grossly toxic to the hepatocytes, as judged by phase-contrast microscopic examination, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium activity measurements, and recovery of total RNA. Doses of squalestatin 1 ranging from 3 × 10−9 to 10−7 m produced essentially a full-range dose-response relationship for induction of the mRNA encoding HMG-CoA reductase (Fig. 2), a gene known to be regulated by cellular sterols, suggesting that these squalestatin 1 doses produced a graded inhibition of squalene synthase, and of cholesterol biosynthesis, in the cultured hepatocytes. This supposition was supported by the finding that incubation of primary cultured rat hepatocytes with a squalestatin 1 dose of 10−8 or 10−7 m produced the expected decreases in cellular biosynthesis of squalene, lanosterol, and cholesterol (Table1). Thus, treatment with 10−8 or 10−7 m squalestatin 1 decreased the cellular content of [14C]squalene by 36% or 99% relative to control, respectively. These same two doses also decreased lanosterol and cholesterol biosynthesis by 72% and 87% and by 19% and 91%, respectively, in agreement with an earlier report that squalestatin 1 treatment inhibited cholesterol biosynthesis in freshly isolated rat hepatocytes with an IC50 value of 39 nm (Baxter et al., 1992).

Effects of squalestatin 1 treatment on P450 and HMG-CoA reductase mRNA levels in primary cultured rat hepatocytes.Left, 48-hr-old primary cultured rat hepatocytes were treated for 24 hr with medium alone (UT); medium containing one of the following positive control (PC) inducers: 10−5 m β-naphthoflavone (CYP1A1), 10−4 m PB (CYP2B), 10−5 m dexamethasone (CYP3A), 10−4 mciprofibrate (CYP4A), or 3 × 10−5 mfluvastatin (HMG-CoA reductase mRNA); or medium containing squalestatin 1 at concentrations ranging from 3 × 10−8 m to 10−7 m (shown as logs). Hepatocytes were harvested from pooled dishes (three dishes per treatment group) for preparation of total RNA, and CYP1A1, CYP2B, CYP3A, CYP4A, and HMG-CoA reductase (HMG-Red) mRNA levels were analyzed by Northern blot hybridization. Also shown is an autoradiograph of a blot that had been rehybridized with a cDNA to 7S RNA.Right, 48-hr-old primary cultured rat hepatocytes were treated for 24 hr with medium alone (UT) or medium containing 10−4 m PB or 10−7 m squalestatin 1 (Squal1). Hepatocytes were harvested from pooled dishes (16 dishes per treatment group) for preparation of poly(A)+ RNA, and CYP2B1 and CYP2B2 mRNA levels were analyzed individually by Northern blot hybridization, using gene-specific oligonucleotide probes.
Effects of squalestatin 1 treatment on sterol biosynthesis in primary cultured rat hepatocytes
For Northern blot analyses of squalestatin 1 treatment effects on P450 mRNA levels, we initially used cDNA probes that cross-hybridize with multiple P450 mRNAs belonging to the same subfamily to permit rapid evaluation of whether squalestatin 1 treatment affected expression of any P450s belonging to the major inducible classes. These analyses revealed that squalestatin 1 treatment produced a clear dose-dependent increase in the cellular content of CYP2B mRNA over the 3 × 10−9 to 10−7 m dose range (Northern blot results obtained using cDNA probes that hybridize to multiple related P450 mRNAs and Western blot results obtained under conditions in which individual immunoreactive proteins were not identified are described generically using the P450 subfamily designations) but had no effect on CYP1A1 or CYP3A mRNA levels and had little effect (estimated ≤2-fold increase) on CYP4A mRNA levels (Fig. 2, left). Squalestatin 1 treatment induced both CYP2B1 and CYP2B2 mRNA, as assessed by hybridization of poly(A)+ RNA samples with oligonucleotide probes that discriminate between these closely related forms (Omiecinski, 1986) (Fig. 2, right). The dose-dependency of the squalestatin 1-mediated CYP2B mRNA induction was characterized further by fitting the dose-response data obtained from slot blot analysis to a sigmoidal function, which yielded an ED50 value of ∼5.0 nm (95% CI, 2.5–10 nm) (Fig.3). This value was comparable to (although ∼3.6-fold lower than) the ED50 value that was calculated for induction of HMG-CoA reductase mRNA (18 nm; 95% CI, 12–27 nm) (Fig. 3). The effect of squalestatin 1 on CYP2B mRNA content was greater (∼2.2-fold at the 10−7 m dose) in hepatocytes cultured in the presence of glucocorticoid (10−7 m triamcinolone acetonide) (Fig. 3), as has been documented for PB-mediated CYP2B induction (Sidhu and Omiecinski, 1995), but different from what has been recently reported for fluvastatin-mediated CYP2B mRNA induction (Kocarek and Reddy, 1998). Potentiation of CYP2B induction by glucocorticoid treatment supports a role for the previously identified glucocorticoid-responsive region in the CYP2B2 gene (Jaiswal et al., 1990). Although in this experiment, the maximal level of mRNA induction observed after squalestatin 1 treatment approximated that occurring after treatment with a maximally effective dose of PB (10−4 m) (Kocarek et al., 1990), in many experiments (e.g., Figs. 2,5, and 10) squalestatin 1-induced mRNA levels were ∼25–50% of those induced by PB.
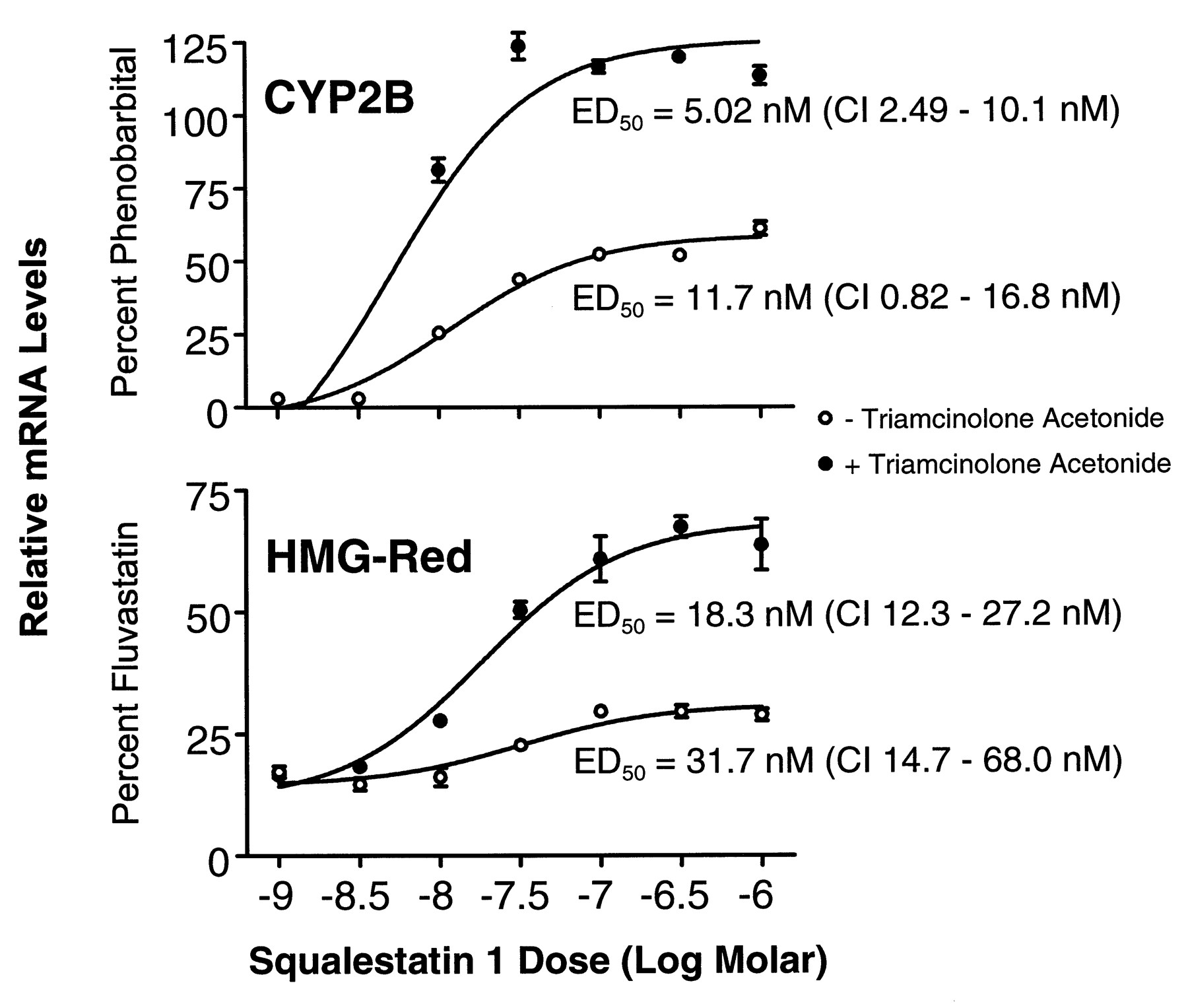
Dose-dependent effects of squalestatin 1 treatment on CYP2B and HMG-CoA reductase mRNA levels in primary cultured rat hepatocytes. Freshly isolated rat hepatocytes were incubated for 48 hr in serum-free Williams’ Medium E that was supplemented with 10−7 m triamcinolone acetonide for either the initial 24-hr period only (○) or throughout the entire experiment (•). The 48-hr-old cultures were treated for 24 hr with medium containing 10−4 m PB, 3 × 10−5 m fluvastatin, or squalestatin 1 at concentrations ranging from 10−9 to 10−6 m (shown as logs). Hepatocytes were harvested from individual dishes (three dishes per treatment group) for isolation of total RNA, and CYP2B and HMG-CoA reductase (HMG-Red) mRNA levels were analyzed by slot blot hybridization. After stripping, blots were rehybridized with 7S probe. Autoradiographic band intensities were quantified by scanning laser densitometry, and intensities of the P450 and HMG-CoA reductase bands relative to the intensities of the corresponding 7S bands were calculated. The normalized CYP2B and HMG-Red data were expressed as percentages of the values obtained for cells treated with PB or fluvastatin, respectively, fit to a sigmoidal function, and ED50 values with 95% CIs were calculated. All values are presented as the mean ± standard deviation of three dishes of hepatocytes. Error bars that are not visible are contained within the boundaries of the data points.

Time-dependent effects of squalestatin 1 treatment on P450 and HMG-CoA reductase mRNA levels in primary cultured rat hepatocytes. Left, 48-hr-old primary cultured rat hepatocytes were incubated for 6, 12, 24, 48, or 72 hr with medium alone (○) or medium containing 10−4 m PB (▴) or 10−7 m squalestatin 1 (•). Hepatocytes were either harvested from individual dishes (three dishes per treatment group) for preparation of total RNA and analysis of CYP2B mRNA levels by slot blot hybridization (as described in the legend to Fig. 3) or were harvested from pooled dishes (five dishes per treatment group) for preparation of microsomes and analysis of CYP2B immunoreactive protein levels by Western blot. All values are presented as percentages of the values measured in hepatocyte cultures treated for 24 hr with 10−4 m PB. mRNA values are presented as the mean ± standard deviation of three dishes of hepatocytes. Error bars that are not visible are contained within the boundaries of the data points.Right, hepatocytes were incubated with medium alone or containing squalestatin 1, as described above, and harvested from individual dishes (three dishes per treatment group) for preparation of total RNA and analysis of CYP1A1 (■), CYP3A (▾), CYP4A (⋄), and HMG-CoA reductase (*) mRNA levels by slot blot hybridization. At each time point, the squalestatin 1-inducible mRNA level is presented as the mean percentage of the mRNA level estimated in the corresponding untreated hepatocyte cultures.
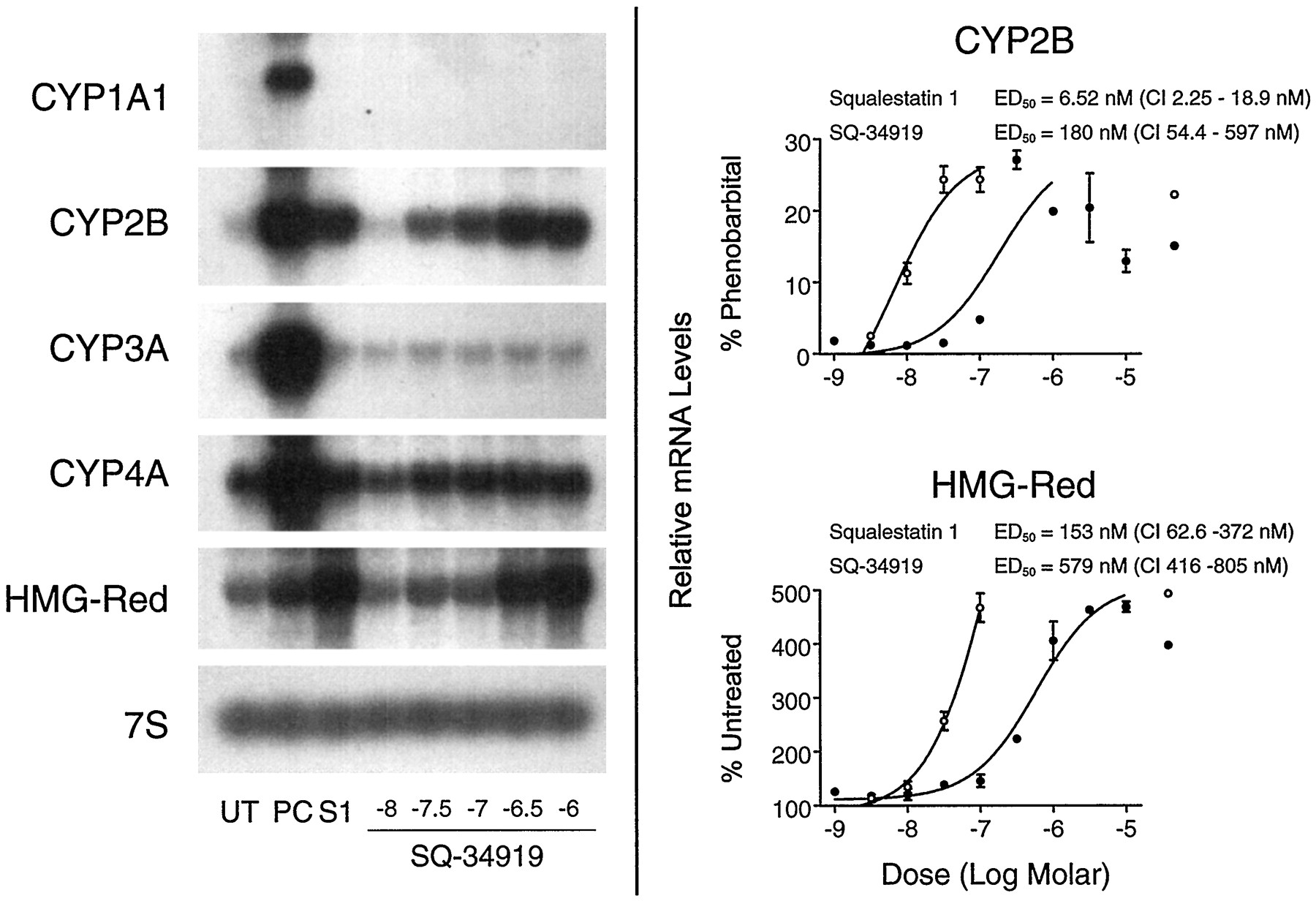
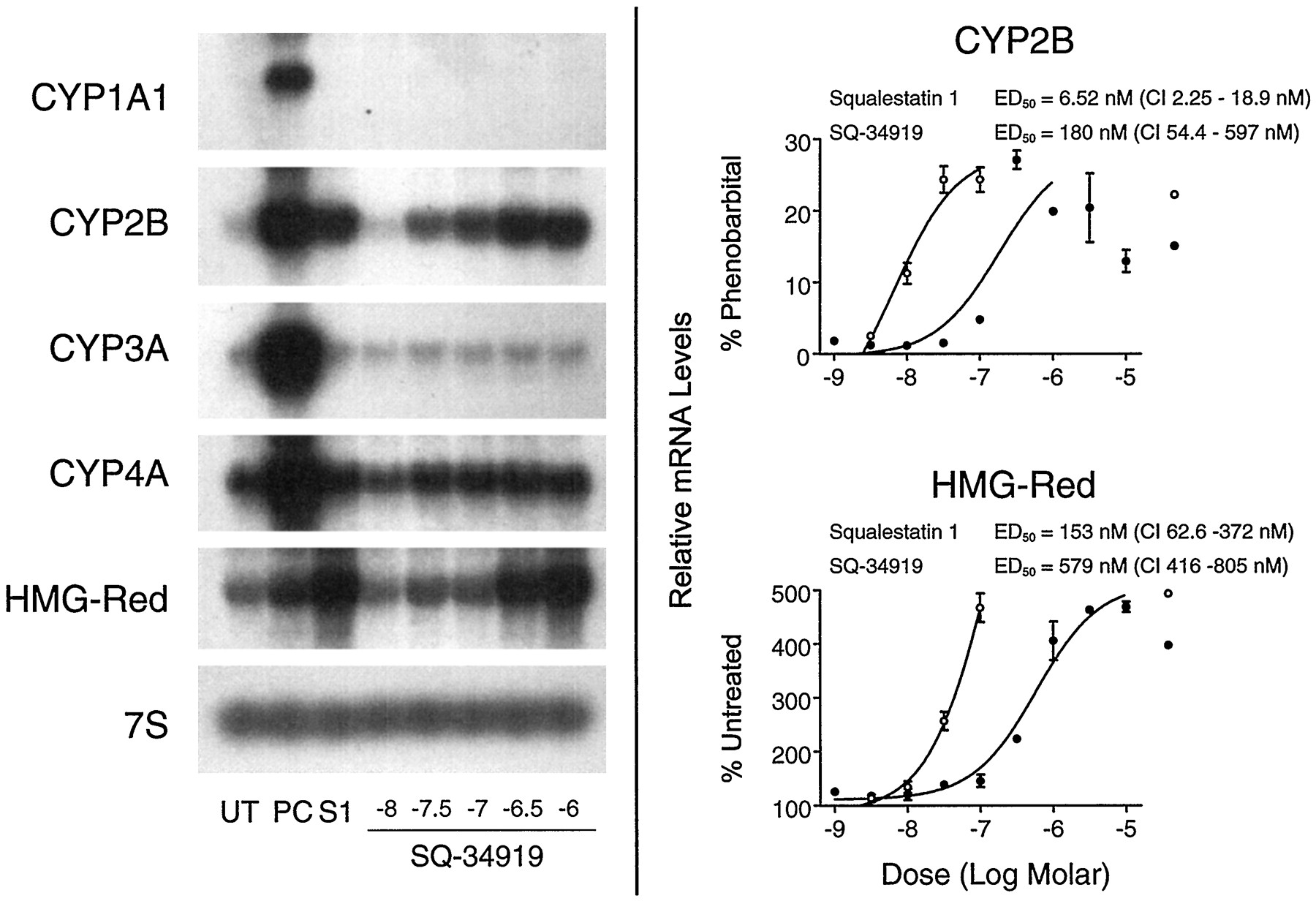
Effects of SQ-34919 treatment on P450 and HMG-CoA reductase mRNA levels in primary cultured rat hepatocytes.Left, 48-hr-old primary cultured rat hepatocytes were treated for 24 hr with medium alone (UT); medium containing one of the following positive control (PC) inducers: 10−5 m β-naphthoflavone (CYP1A1), 10−4 m PB (CYP2B), 10−5 m dexamethasone (CYP3A), 10−4 mciprofibrate (CYP4A), or 3 × 10−5 mfluvastatin (HMG-CoA reductase mRNA); or medium containing 10−7 m squalestatin 1 (S1) or SQ-34919 at concentrations ranging from 10−8 m to 10−6 m (shown as logs). Hepatocytes were harvested from pooled dishes (three dishes per treatment group) for preparation of total RNA, and CYP1A1, CYP2B, CYP3A, CYP4A, and HMG-CoA reductase (HMG-Red) mRNA levels were analyzed by Northern blot hybridization. Also shown is an autoradiograph of a blot that had been rehybridized with a cDNA to 7S RNA. Right, 48-hr-old primary cultured rat hepatocytes were treated for 24 hr with medium alone or containing 10−4 m PB, 3 × 10−9 to 10−7 m squalestatin 1 (○), or 10−9 to 10−5 m SQ-34919 (•). Hepatocytes were harvested from individual dishes (three dishes per treatment group) for preparation of total RNA, and CYP2B and HMG-CoA reductase mRNA levels were analyzed by slot blot hybridization, as described in the legend to Fig. 3. Normalized data for CYP2B or HMG-CoA reductase were expressed as percentages of the values obtained for PB-treated or untreated cultures, respectively, fit to a sigmoidal function, and ED50 values with 95% CIs were calculated. All values are presented as the mean ± standard deviation for three dishes of hepatocytes. Error bars that are not visible are contained within the boundaries of the data points.
Examination of squalestatin 1 treatment effects on P450 immunoreactive protein levels yielded essentially the same results that were seen at the mRNA level, with large increases in CYP2B, but no other P450, being apparent (Fig. 4). Time course analysis demonstrated that although PB-mediated increases in CYP2B mRNA levels were clearly apparent after 6-hr treatment and maximal after ∼12-hr treatment, squalestatin 1-induced CYP2B mRNA levels were first detectable after 12 hr and maximal after ∼24-hr treatment (Fig.5, left). Similarly, squalestatin 1-mediated increases in CYP2B immunoreactive protein occurred more slowly than did those induced by PB (Fig. 5,left). Squalestatin 1-mediated increases in HMG-CoA mRNA levels were maximal after 24-hr treatment. However, even after incubation times as long as 72 hr, no induction of CYP1A1, CYP3A, or CYP4A mRNA exceeding ∼2-fold was observed (Fig. 5, right).

Effects of squalestatin 1 treatment on P450 immunoreactive protein levels in primary cultured rat hepatocytes. The 48-hr-old primary cultured rat hepatocytes were treated as described in the legend to Fig. 2. Hepatocytes were harvested from pooled dishes (five dishes per treatment group) for preparation of microsomes, and CYP1A, CYP2B, CYP3A, and CYP4A immunoreactive protein levels were analyzed by Western blot hybridization. PC, positive control inducer treatment for each P450, as described in the legend to Fig. 2.
We performed three experiments examining the effects of in vivo treatments with squalestatin 1 on rat liver P450 expression. In the first two experiments, rats (three per treatment group) were treated for 3 days (intraperitoneal) with doses of squalestatin 1 ranging from 1 to 50 mg/kg/day (Fig. 6,top). Increased levels of CYP2B immunoreactive protein were detected at the lowest dose tested (1 mg/kg/day), whereas increased CYP2B mRNA levels were apparent at the 5 mg/kg/day dose (Fig. 6,top). Induced levels of CYP2B mRNA and immunoreactive protein were maximal at the 25 mg/kg/day dose (Fig. 6, top). In the third experiment, rats were treated for 1, 3, or 5 days with 25 mg/kg/day squalestatin 1 (Figs. 6, bottom, and 7). The highest level of CYP2B expression occurred after 5 days of treatment, resulting in CYP2B mRNA levels, CYP2B immunoreactive protein levels, and PROD activities that were ∼32%, ∼87%, and ∼33% of those observed in PB-treated rats. In vivo treatment with squalestatin 1 also produced small, but definite, increases in CYP4A expression (Figs. 6, bottom, and 7) but produced no remarkable increase in the hepatic content of CYP1A1 or CYP3A mRNA or immunoreactive protein.

Effects of squalestatin 1 treatment on rat liver P450 expression. Male Sprague-Dawley rats (three rats per treatment group) were treated for 3 days intraperitoneally with one of the following positive control (PC) inducers: β-naphthoflavone (CYP1A), PB (CYP2B), dexamethasone (CYP3A), ciprofibrate (CYP4A), or fluvastatin (HMG-CoA reductase), as described in Experimental Procedures, or with 0.9% saline or squalestatin 1 as described below. Top, rats were treated for 3 days intraperitoneally with 0.9% saline or with squalestatin 1 at doses of 1, 5, or 10 mg/kg/day or 10, 25, or 50 mg/kg/day. At 24 hr after the last treatment, rats were killed, and livers were dissected for preparation of total RNA and analysis of CYP2B and HMG-CoA reductase mRNA levels by Northern blot hybridization (also shown is an autoradiograph of a Northern blot that had been rehybridized with a cDNA to 7S RNA) and for preparation of microsomes and analysis of CYP2B immunoreactive protein levels by Western blot. Northern and Western blot analyses were performed using RNA or microsome samples that were pooled within each treatment group. Bottom, rats were treated for 1, 3, or 5 days intraperitoneally with saline or 25 mg/kg/day squalestatin 1. At 24 hr after the last treatment, rats were killed, and livers were dissected for preparation of total RNA and analysis of CYP1A1, CYP2B, CYP3A, CYP4A, and HMG-CoA reductase mRNA levels by Northern blot hybridization or for preparation of microsomes and analysis of CYP2B immunoreactive protein levels (by Western blot) and PROD activities. Left, all CYP2B data (i.e., mRNA, immunoreactive protein and PROD activity). Right, mRNA data for CYP1A1, CYP3A, CYP4A, and HMG-CoA reductase. All mRNA data are normalized to 7S RNA. All values are presented as mean ± standard deviation percentages for three livers of the values measured in rats treated with the appropriate positive control inducer, and data were analyzed by unpaired Student’s t tests. Significantly different from the corresponding saline control, ∗,p < 0.05, ∗∗, p < 0.01, ∗∗∗, p < 0.001.

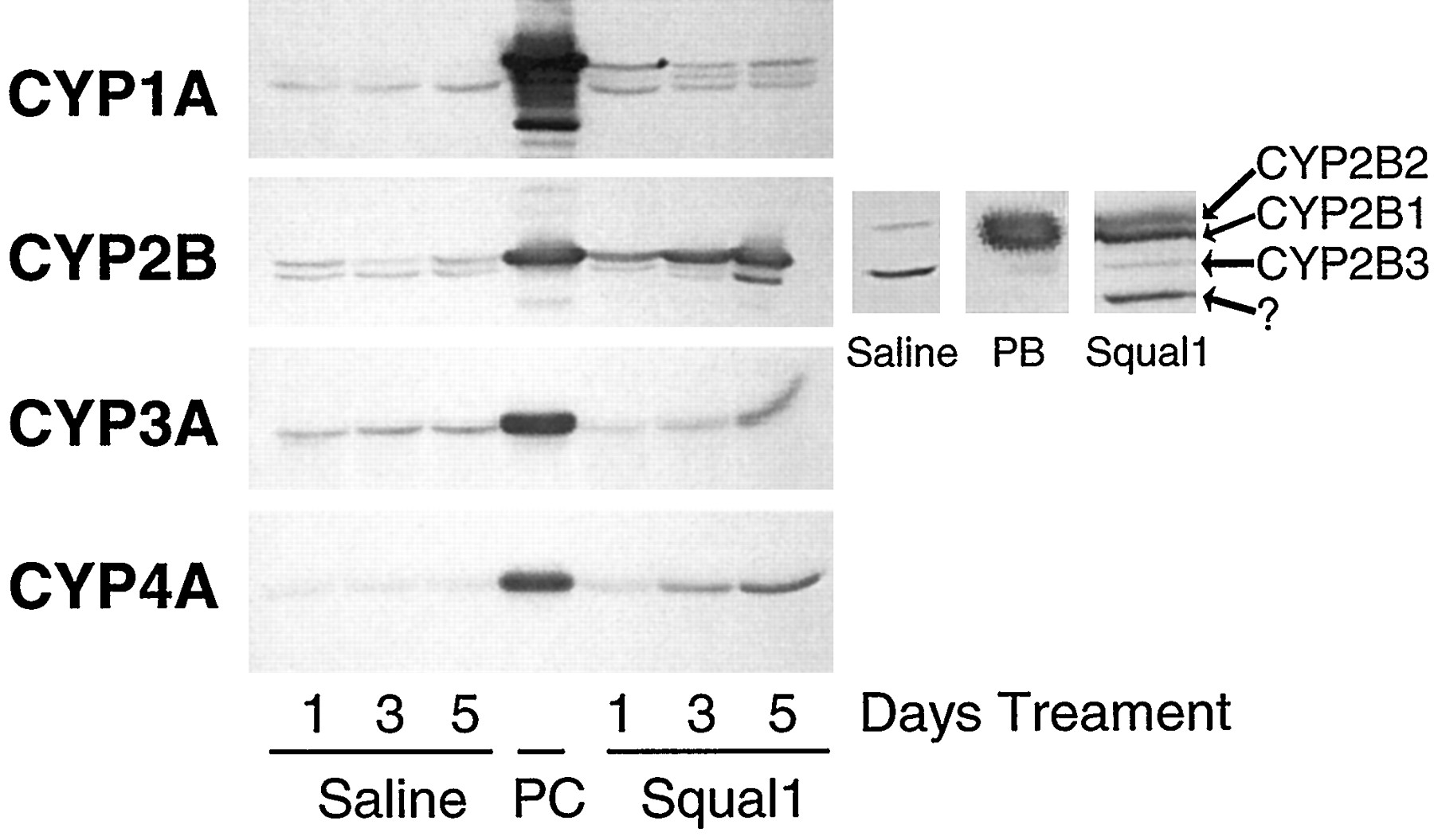
Time-dependent effects of squalestatin 1 treatment on rat liver P450 immunoreactive protein levels. Male Sprague-Dawley rats were treated as described in the legend to Fig. 6. At 24 hr after the last treatment, rats were killed, and livers were dissected for preparation of microsomes. Pooled samples of microsomes (10 μg/lane) were resolved on SDS-polyacrylamide gel electrophoresis (10% acrylamide) minigels, and levels of CYP1A, CYP2B, CYP3A, and CYP4A were measured by Western blot hybridization. Alternatively, microsome samples were resolved on a standard gel to permit detection of individual CYP2B proteins. Positions of CYP2B1, CYP2B2, CYP2B3, and an unidentified cross-reactive protein (?) are indicated (arrows). Squal1, squalestatin 1;PC, positive control inducer of each P450, as described in the legend to Fig. 6.
Squalestatin 1 treatment caused the accumulation of both CYP2B1 and CYP2B2 protein in liver microsomes (Fig.7, right), consistent with the finding (presented in Fig. 2) that both CYP2B1 and CYP2B2 mRNA were elevated in squalestatin 1-treated hepatocyte cultures. Treatment of rats or primary cultured rat hepatocytes with squalestatin 1, but not PB, also resulted in the detection on Western blots probed with the anti-CYP2B1 antibody of an unidentified protein that migrated more rapidly than CYP2B1 or CYP2B2 or than a band corresponding to the expected size of CYP2B3 (Jean et al., 1994) (Figs. 4 and 7). Evidence suggesting that this unidentified band does not represent a degradation product of CYP2B proteins formed during microsome preparation includes the failure to observe this band in microsomes prepared from untreated or PB-treated rat liver samples or cultured hepatocytes and the continued observance of this band when microsomes from squalestatin 1-treated samples were prepared in the presence of a protease inhibitor cocktail (data not shown).
Cotreatment of cultured hepatocytes with PB and squalestatin 1 revealed that at a submaximally effective dose of PB (10−5 m), the effects of squalestatin 1 and PB on CYP2B mRNA content were essentially additive, whereas at a maximally effective dose of PB (10−4 m), cotreatment with squalestatin 1 did not further increase the accumulation of CYP2B mRNA (data not shown), suggesting that the mechanisms whereby PB and squalestatin 1 induce CYP2B mRNA expression may share common features, possibly involving transcriptional activation. To test this possibility further, we examined the abilities of PB and squalestatin 1 to activate reporter gene expression in primary cultured rat hepatocytes transiently transfected with either of two CYP2B1-reporter constructs, the first containing ∼2.4 kb of the 5′-flanking region of the CYP2B1 gene driving luciferase expression through the endogenous CYP2B1 promoter (pGL3-Basic plasmid) and the second containing the 163-bpSau3A1 fragment (located −2299 to −2137 bp upstream of the CYP2B1 transcription start site) previously shown to be PB-responsive (Trottier et al., 1995), driving luciferase expression through the SV40 promoter (pGL3-Promoter plasmid). Neither PB nor squalestatin 1 treatment induced luciferase expression in hepatocytes transfected with the pGL3-Basic or pGL3-promoter plasmid lacking CYP2B1 sequence (Fig. 8). PB treatment induced luciferase expression ∼20- and ∼4.4-fold in hepatocytes transfected with vector containing the 2.4-kb fragment or the 163-bp PBRE fragment, which is in close agreement with the previous findings of Trottieret al. (1995). Squalestatin 1 treatment produced increases of ∼10- and ∼2.9-fold in hepatocytes transfected with the two vectors (Fig. 8), consistent with the lower magnitude of mRNA induction typically observed after squalestatin 1 treatment, compared with PB treatment, of hepatocyte cultures. These results demonstrate that squalestatin 1 treatment is able to activate CYP2B1 gene transcription through elements contained within the CYP2B1 flanking region and suggest that both PB and squalestatin 1 activate CYP2B1 gene transcription through elements contained within the PBRE.

Effects of PB or squalestatin 1 treatment on luciferase activity in primary cultured rat hepatocytes transiently transfected with CYP2B1-reporter constructs. Freshly isolated rat hepatocytes were plated in standard Williams’ Medium E onto Vitrogen-coated 12-well plates and transiently transfected with one of four reporter plasmids: pGL3-Basic, CYP2B1(−2413 to +23)pGL3-Basic, pGL3-Promoter or CYP2B1(−2299 to −2137)pGL3-Promoter, as described in Experimental Procedures. After transfection, hepatocyte cultures were overlaid with Matrigel and treated for 24 hr with medium alone or containing 10−4 m PB or 10−7 m squalestatin 1. Hepatocytes were harvested from individual wells (three wells per treatment group) for measurement of luciferase activity (firefly and Renilla), using the Dual Luciferase Reporter Assay System. Data are expressed as the mean ratios of firefly to Renilla luciferase activity ± standard deviation for three wells of hepatocytes and were analyzed by one-way analysis of variance followed by the Dunnett’s test. Significantly different from untreated group transfected with the same plasmid, ∗, p < 0.05, ∗∗,p < 0.01. These data are representative of those obtained in three independent experiments (i.e., experiments performed using hepatocytes isolated from three different rats).
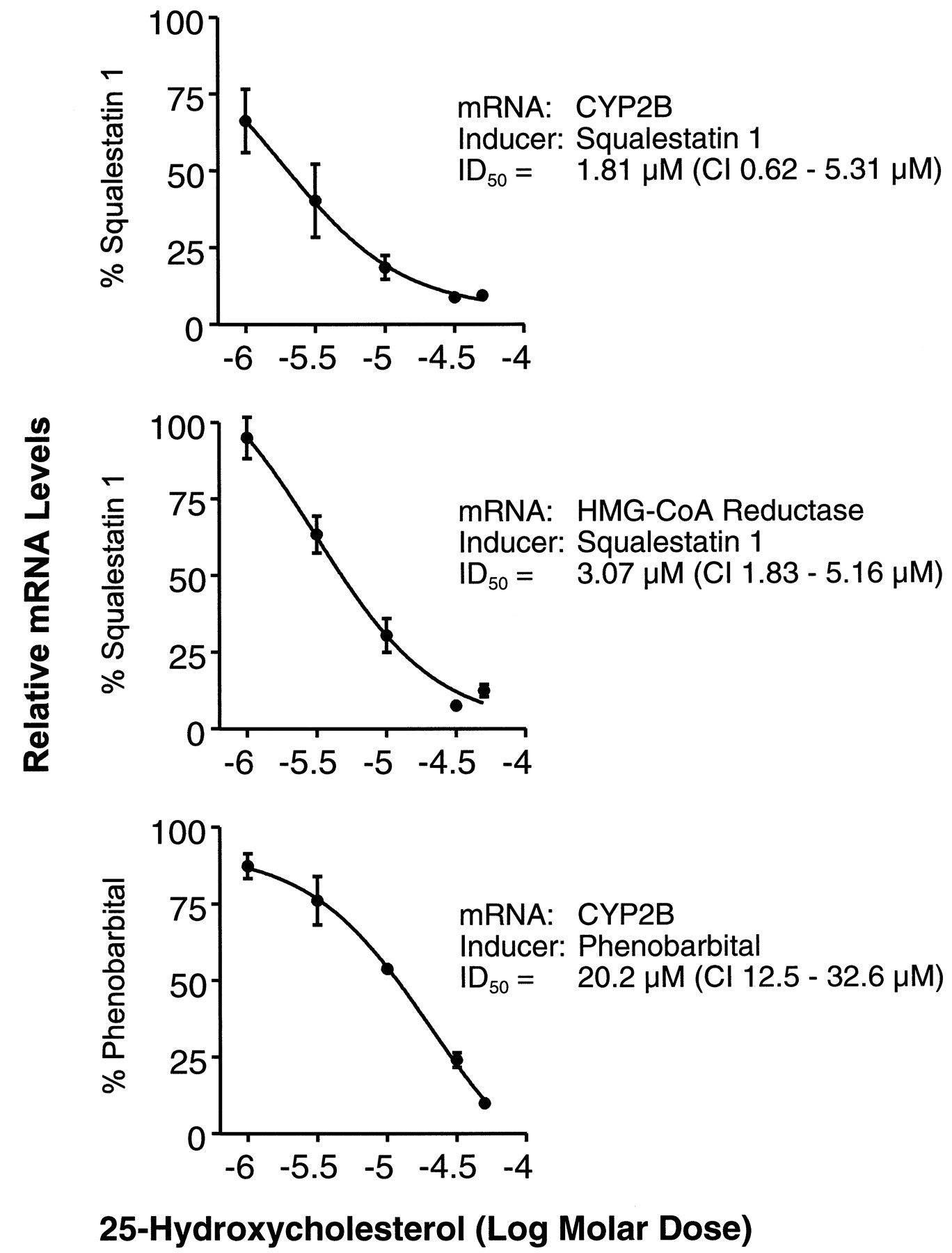
Demonstration that squalestatin 1-inducible CYP2B expression results from squalene synthase blockade and sterol synthesis inhibition requires that the induction process be reversed by cotreatment with a metabolite formed downstream (preferably immediately downstream) of the site of blockade, in this case, squalene or lanosterol. However, it also is essential that the exogenously added metabolite be able to distribute into the cellular pools, formed endogenously, that actively regulate gene expression. Unfortunately, the extreme lipophilicity of squalene and lanosterol severely limits the amounts that can be incubated with cell cultures and likely causes these metabolites to distribute into membrane compartments, where they are unable to incorporate into the regulatory sterol pools. However, cotreatment of squalestatin 1-treated cultured rat hepatocytes with 25-hydroxycholesterol, a model oxysterol that is widely used to demonstrate the sterol-dependency of gene expression, dose-dependently reversed the squalestatin 1-mediated increase in CYP2B mRNA levels (Fig. 9). The potency of 25-hydroxycholesterol for suppression of squalestatin 1-inducible CYP2B expression (ID50 ∼ 1.8 μm) was approximately the same as the potency of the sterol for suppressing squalestatin 1-mediated HMG-CoA reductase mRNA induction (ID50 ∼ 3.1 μm) and was ∼10-fold greater than its potency for suppressing PB-inducible CYP2B mRNA expression (ID50 ∼ 20 μm) (Fig. 9). By contrast, coincubation of cultured rat hepatocytes with squalestatin 1 and 10−4 or 10−3 m mevalonate, expected to cause accumulation of nonsterol isoprenoids, had no effect on the CYP2B mRNA induction dose-response relationship (data not shown). These findings are consistent with the interpretation that squalestatin 1-mediated CYP2B induction occurred as a consequence of squalene synthase inhibition and the associated depletion of cellular sterols rather than the accumulation of a nonsterol isoprenoid. To test further this possibility, we examined the effects of SQ-34919, a synthetic squalene synthase inhibitor representing a different class of chemical agent (i.e., lipophilic 1,1-bisphosphonate; see Fig. 1 for chemical structure) (Ciosek et al., 1993) on P450 mRNA levels in primary cultured rat hepatocytes. SQ-34919 treatment produced the same profile of effects on P450 mRNA expression as did squalestatin 1, potently increasing CYP2B mRNA levels (ED50 ∼ 180 nm; 95% CI, 54–597 nm) to the same magnitude as produced by squalestatin 1 (Fig.10). Although only four doses of squalestatin 1 were tested in this experiment, the ED50 value that was calculated for the induction of CYP2B mRNA was 6.5 nm (95% CI, 2.3–19 nm), which is in close agreement with the value calculated from the data presented in Fig. 3. Also, as calculated for the squalestatin 1 treatment effect data presented in Fig. 3, the potency of SQ-34919 for induction of CYP2B mRNA was ∼3.2-fold greater than was its potency for HMG-CoA reductase induction, suggesting that CYP2B expression is extremely sensitive to the effects of cholesterol biosynthesis inhibition.

Effects of 25-hydroxycholesterol treatment on squalestatin 1-inducible CYP2B and HMG-CoA reductase mRNA expression in primary cultured rat hepatocytes. The 48-hr-old primary cultured rat hepatocytes were treated for 24 hr with medium containing 10−4 m PB or 10−7 msqualestatin 1, each in the presence of ethanol or 25-hydroxycholesterol at media concentrations ranging from 10−6 to 5 × 10−5 m (shown as logs). Hepatocytes were harvested from individual dishes (three dishes per treatment group) for preparation of total RNA, and CYP2B and HMG-CoA reductase mRNA levels were analyzed by slot blot hybridization, as described in the legend to Fig. 3. For each panel, the mRNA and inducer are indicated. The normalized data were expressed as percentages of the values obtained for cells treated with PB or squalestatin 1 and ethanol and fit to a sigmoidal function, and ID50 values with 95% CIs were calculated. All values are presented as the mean ± standard deviation for three dishes of hepatocytes. Error bars that are not visible are contained within the boundaries of the data points. These results are representative of those obtained in two independent hepatocyte culture experiments.
Discussion
We report that squalestatin 1 is the most potent inducer of rat hepatic CYP2B expression yet described, inducing CYP2B mRNA in primary cultured rat hepatocytes with an ED50 value of ∼5 nm. This makes squalestatin 1 ∼2000-fold more potent than PB (ED50 ∼ 10 μm) and 20-fold more potent than the organochlorine trans-nonachlor or the imidazole antimycotic drug clotrimazole (ED50 ∼ 0.1–0.2 μm) (Kocareket al., 1990) as a CYP2B mRNA inducer in primary cultured rat hepatocytes. Another potent PB-like inducer is TCPOBOP, which was demonstrated to be ∼650 times as potent as PB as an inducer of aminopyrine N-demethylase activity in mouse liver (Polandet al., 1980). However, this agent seems to be a moderately potent PB-like inducer in rat, inducing CYP2B in rat liver and rat hepatocytes with a potency (EC50 based on serum TCPOBOP in rats; ED50 in primary cultured rat hepatocytes) of ∼1 μm (Nims et al., 1993). Therefore, squalestatin 1 seems to be ∼200 times more potent than TCPOBOP as an inducer of rat CYP2B expression.
Squalestatin 1 treatment induced CYP2B1 and CYP2B2 mRNA and immunoreactive protein, both in vivo and in primary cultured rat hepatocytes. Transfection analysis using CYP2B1-reporter constructs demonstrated that squalestatin 1 treatment activated CYP2B1 gene transcription and suggested that squalestatin 1 and PB may drive transcription using the same cis-acting elements. However, PB usually induced CYP2B mRNA levels and reporter gene activity to a higher level than did squalestatin treatment, suggesting that PB may activate the CYP2B transcription machinery more effectively than does squalestatin 1. It may prove informative to compare the effects of squalestatin 1 treatment on protein binding to PBRE sequences with those recently reported by Kim and Kemper (1997), who used in vivo DNase I footprinting to demonstrate that PB treatment altered the composition or structure of the protein complex binding to the PBRE in native chromatin. Unlike PB, squalestatin 1 treatment did not increase CYP3A expression, providing further support for the concept that CYP2B and CYP3A induction are dissociable processes (Schuetzet al., 1986; Burger et al., 1990; Kocareket al., 1990). However, the selectivity of squalestatin 1 treatment for CYP2B induction was not absolute, in that some elevation of CYP4A was apparent, particularly after in vivo treatment. The combination of CYP2B and CYP4A induction also occurred when hepatocytes were treated with fluvastatin and other HMG-CoA reductase inhibitors (Kocarek and Reddy, 1996), although the HMG-CoA reductase inhibitors produced more pronounced increases in CYP4A expression.
Several pieces of evidence support the conclusion that the squalestatin 1-mediated effects on CYP2B expression were the direct consequence of squalene synthase blockade and subsequent depletion of hepatocellular sterols. First, the ED50 value for squalestatin 1-inducible CYP2B mRNA expression was comparable to that for induction of the mRNA encoding HMG-CoA reductase, a gene known to be under sterol regulatory control. Second, the time course for squalestatin 1-mediated CYP2B induction exhibited a longer lag time than did PB-induced CYP2B expression, which is consistent with the interpretation that some time is required after squalestatin 1 treatment for endogenous suppressive sterols to become depleted. Third, the inductive effects of squalestatin 1 on CYP2B and HMG-CoA reductase mRNA expression were readily reversed when hepatocytes were coincubated with 25-hydroxycholesterol. Fourth, the same pattern of P450 induction produced by squalestatin 1 treatment was observed when primary cultured hepatocytes were treated with SQ-34919, a synthetic squalene synthase inhibitor differing in chemical structure from squalestatin 1 (Cioseket al., 1993). Because squalestatin 1 and SQ-34919 are both structural mimics of farnesyl pyrophosphate, the substrate for squalene synthase, these compounds do share some structural similarity (i.e., they both contain isoprene units), leaving open the possibility that the effects of these drugs on P450 expression result from a common ability to interact with a macromolecule other than squalene synthase. However, preliminary studies using structurally distinct inhibitors of squalene oxidase, which catalyzes the step in sterol biosynthesis after that catalyzed by squalene synthase, have yielded the same P450 induction profile as observed for squalestatin 1, namely, preferential induction of CYP2B mRNA expression (Kocarek TA, Reddy AB, unpublished observations). Taken together, these results suggest that induction of rat hepatic CYP2B expression can be achieved by inhibiting sterol biosynthesis. Notably, although incubation of primary cultured rat hepatocytes with 25-hydroxycholesterol suppressed PB-mediated CYP2B mRNA induction, higher doses of the sterol were required than were needed to suppress squalestatin 1-mediated induction, suggesting that PB-inducible CYP2B expression does not occur as a direct consequence of sterol biosynthesis blockade but rather that the positive effects of PB on CYP2B expression are merely opposed by the negative effects of sterols.
If CYP2B expression is under the control of regulatory sterols, the challenges will be to identify which specific cellular sterols mediate the effect and to determine how a sterol-sensitive signal is transmitted to the nucleus to modulate CYP2B gene expression. Substantial information is available on these issues as they relate to regulation of the genes encoding the cholesterol homeostatic proteins. Evidence supports the existence of at least two “classes” of regulatory oxysterols that are formed in normal cells. The first class could be called the oxycholesterols, which would include 25-hydroxycholesterol, 27-hydroxycholesterol, and 24(S),25-epoxycholesterol. The primary mode of action of these molecules on expression of cholesterol homeostasis genes is to suppress their rate of transcription (Brown and Goldstein, 1980). The second class of oxysterols could be called the oxylanosterols, which would include 24(S),25-epoxylanosterol and the lanosterol demethylation intermediates 32-oxolanosterol and 32-hydroxylanosterol. Oxylanosterol molecules have been shown to exert post-transcriptional effects on HMG-CoA reductase expression, acting either to increase the rate of protein degradation (Panini et al., 1992) or to decrease the rate of mRNA translation (Trzaskos et al., 1993).
Substantial information indicates that many sterol-mediated effects on gene expression are transduced through a set of transcription factors termed the SREBPs (Brown and Goldstein, 1997). In sterol-replete cells, the SREBPs exist as inactive precursors that are attached to the membranes of the endoplasmic reticulum and nuclear envelope (Brown and Goldstein, 1997). In sterol-depleted cells, a protease cascade becomes activated that culminates in release of the 68-kDa NH2-terminal peptides, which are transcription factors of the basic-helix-loop-helix-leucine zipper family. These transcription factors translocate into the nucleus and activate gene transcription through enhancer elements termed SREs (Brown and Goldstein, 1997). The SRE-1 element (5′-ATCACCCCAC-3′, containing a direct repeat of 5′-PyCAPy-3′), is found in the 5′-flanking region of several genes of cholesterol homeostasis, including the LDL receptor and HMG-CoA synthase. Sterol responsiveness to the farnesyl pyrophosphate synthase gene is conferred through a 10-bp sequence, which has been termed SRE-3 (CTCACACGAG) (Ericsson et al., 1996). The SRE-1 and SRE-3, as well as nonconsensus SRE-like sequences, have been shown to bind SREBP (Ericsson et al., 1996; Lopezet al., 1996). Although no consensus SRE-1 element is present in the published 5′-flanking region of the CYP2B1 gene, a sequence ∼1370 bp upstream of the transcription start site (5′-CACCCCCCACCCCAA-3′) seems to bear some resemblance to the SRE-1 element. Also, although the published CYP2B1 5′ gene sequence does not contain a perfect SRE-3, it contains a nearly identical sequence ∼1095 bp upstream of the transcription start site (5′-CTCACAGCAG-3′). However, neither of these “SRE-like” sequences are located within the 163-bp PBRE of the CYP2B1 gene, and their roles, if any, if regulation of CYP2B1 gene expression remain to be determined.
Substantial data suggest that sterol metabolism and P450 expression are interrelated processes, although some evidence suggests that the effects can be dissociated (Kocarek and Reddy, 1996, 1998). Because the “purpose” of the cholesterol homeostatic enzymes seems to differ substantially from that of the xenobiotic-metabolizing P450s, intuition suggests that regulation of these two enzyme systems would not be identical. If this is true, then by understanding how the sterol and P450 pathways coexist and interact, it should be possible to dissociate effects on cholesterol metabolism from those on P450 expression, which should facilitate the development of safer, and possibly more efficacious, anticholesterol drugs. The ultimate objective of these studies is to understand the physiological implications of a linkage among cholesterol biosynthesis, metabolism, and P450 gene expression. What are the processes whereby a cell recognizes, responds to, and metabolizes a foreign chemical? It is possible that the hepatocyte tackles this problem by commandeering the cellular machinery that is already in place to recognize and metabolize the endogenous lipophilic chemicals of the cell.
Acknowledgments
We thank Dr. Melissa Runge-Morris for her helpful comments during preparation of the manuscript.
Footnotes
- Received February 23, 1998.
- Accepted May 13, 1998.
-
Send reprint requests to: Thomas A. Kocarek, Ph.D., Institute of Chemical Toxicology, Wayne State University, 2727 Second Avenue, Room 4000, Detroit, MI 48201. E-mail:t.kocarek{at}wayne.edu
-
This work was supported by National Heart, Lung, and Blood Institute Grant HL50710 and National Institutes of Environmental Health Sciences Center Grant ES06639.
Abbreviations
- P450
- cytochrome P450
- CI
- confidence interval
- bp
- base pair(s)
- ED50
- dose producing 50% of maximal effect
- HMG-CoA
- 3-hydroxy-3-methylglutaryl coenzyme A
- ID50
- dose producing 50% of maximal inhibitory effect
- PB
- phenobarbital
- PBRE
- phenobarbital-responsive element
- PROD
- pentoxyresorufin O-dealkylase
- SDS
- sodium dodecyl sulfate
- SRE
- sterol regulatory element
- SREBP
- sterol regulatory element binding protein
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}