


Abstract
The aim of our study was to examine the effects of different purine nucleotides [GTP, ITP, and xanthosine 5′-triphosphate (XTP)] on receptor/G protein coupling. As a model system, we used a fusion protein of the β2-adrenergic receptor and the α subunit of the G protein Gs. GTP was more potent and efficient than ITP and XTP at inhibiting ternary complex formation and supporting adenylyl cyclase (AC) activation. We also studied the effects of several β2-adrenergic receptor ligands on nucleotide hydrolysis and on AC activity in the presence of GTP, ITP, and XTP. The efficacy of agonists at promoting GTP hydrolysis correlated well with the efficacy of agonists for stimulating AC in the presence of GTP. This was, however, not the case for ITP hydrolysis and AC activity in the presence of ITP. The efficacy of ligands at stimulating AC in the presence of XTP differed considerably from the efficacies of ligands in the presence of GTP and ITP, and there was no evidence for receptor-regulated XTP hydrolysis. Our findings support the concept of multiple ligand-specific receptor conformations and demonstrate the usefulness of purine nucleotides as tools to study conformational states of receptors.
The β2-adrenergic receptor (β2-AR) is a prototypical G protein-coupled receptor (GPCR) that interacts with the G protein Gs to activate adenylyl cyclase (AC; Gilman, 1987; Kobilka, 1992). GPCRs activate G proteins by promoting GDP release from and GTP binding to G protein α subunits (Iiri et al., 1998). GTP-liganded Gsα activates AC, and G protein deactivation is accomplished by GTP hydrolysis (Cassel and Selinger, 1976; Gilman, 1987). The extended ternary complex model assumes that GPCRs exist in an equilibrium between an inactive state (R) and an active state (R*) (Lefkowitz et al., 1993; Gether and Kobilka, 1998). According to this model, GPCRs can undergo R to R* isomerization in the absence of agonist, which gives rise to a receptor-dependent basal G protein and effector activity. Agonists stabilize the R* state and increase G protein activity above basal levels, whereas inverse agonists stabilize the R state and suppress basal G protein activity (see, e.g., Chidiac et al., 1994; Samama et al., 1994; Gether et al., 1995; Wenzel-Seifert et al., 1998a). The R* state is also stabilized by guanine nucleotide-free G protein α subunits (De Lean et al., 1980; Seifert et al., 1998a,b). The agonist-occupied receptor and nucleotide-free G protein α subunit form a ternary complex that is characterized by high agonist affinity. The ternary complex is disrupted by guanine nucleotide binding to the G protein (De Lean et al., 1980; Seifert et al., 1998a,b). An increasing number of experimental observations indicate that the extended ternary complex model cannot sufficiently explain the molecular mechanisms underlying GPCR activation. First, Chidiac et al. (1994) have shown that certain β2-AR agonists can either act as partial agonists or as inverse agonists depending on whether effector system activity is assessed in intact cells or in cell membranes. Second, the extended ternary complex model proposes that inverse agonists stabilize an inactive and G protein-uncoupled state of GPCRs (Lefkowitz et al., 1993; Gether and Kobilka, 1998). However, the results from various studies suggest that inverse agonists induce a specific conformation in the GPCR that actively inhibits G protein function (Bouaboula et al., 1997; Seifert et al., 1998b). Third, the extended ternary complex model cannot explain why defined mutations in the dopamine D2 receptor result in agonist-dependent changes in signaling (Wiens et al., 1998). Fourth, the observation that not only agonists but even antagonists can promote GPCR internalization (Roettger et al., 1997) and that some receptor ligands behave as antagonists with respect to G protein activation but as agonists with regard to ternary complex formation (Brown and Pasternak, 1998) cannot be reconciled with the extended ternary complex model. Finally, several reports showed that various synthetic and natural opioids interact differently with the μ-opioid receptor (Keith et al., 1996; Blake et al., 1997; Yu et al., 1997). Based on these and several other observations, it has been proposed that there are multiple, ligand-specific GPCR conformations (Kenakin, 1996; Tucek, 1997).
The aim of our study was to explore the usefulness of guanine, inosine, and xanthine nucleotides as experimental tools to explore ligand-specific GPCR conformations. Previous studies had shown that inosine and xanthine nucleotides can bind to various G proteins, although with lower affinity than guanine nucleotides (Northup et al., 1982; Kelleher et al., 1986; Florio and Sternweis, 1989; Klinker and Seifert, 1997). The idea to use nucleotides as tools for analyzing receptor conformations originated from previous studies showing that GTP, ITP, and xanthosine 5′-triphosphate (XTP) behave differently with respect to signaling mediated by different GPCRs that are coupled to the same G proteins and effector systems (Wolff and Cook, 1973;Bilezikian and Aurbach, 1974; Klinker and Seifert, 1997). In our study, we use purine nucleotides to examine ligand-specific differences in signaling mediated by a single GPCR. We examined the effects of different classes of ligands on β2-AR-modulated interactions between the G protein Gs and the purine nucleotides GDP, GTP, IDP, ITP, xanthosine 5′-diphosphate (XDP), and XTP. As an experimental system, we used a fusion protein of the β2-AR and the long-splice variant of Gsα (GsαL) expressed in Sf9 insect cells. Fusion of the two proteins to each other does not change the fundamental properties of either the β2-AR or Gsα and allows for sensitive analysis of GPCR/G protein coupling in terms of ternary complex formation, GTP hydrolysis, and AC regulation (Seifert et al., 1998a,b). The β2-AR coupled to GsαL, but not the β2-AR coupled to GsαS, possesses the hallmarks of constitutive activity (high basal GTPase activity and high efficacy of inverse agonists and partial agonists; Seifert et al., 1998a). The apparent constitutive activity of the β2-AR coupled to GsαL can be explained by the relatively low GDP affinity of GsαL compared with the short-splice variant of Gsα(GsαS). Specifically, GsαL is more often guanine nucleotide-free than GsαS and therefore is more often available to stabilize the R* state. Here, we report that the potency and efficacy of a series of β2-AR ligands at the β2-ARGsαL fusion protein is dependent on the purine nucleotide that binds to GsαL. Our results provide further evidence for ligand-specific receptor conformational states.
Experimental Procedures
Materials.
[γ-32P]GTP (6000 Ci/mmol), [γ-32P]ITP (4000 Ci/mmol), and [γ-32P]XTP (4000 Ci/mmol) were custom synthesized by DuPont-NEN (Boston, MA). ITP, IDP, XTP, and XDP were of the highest purity available and were purchased from Sigma Chemical Co. (St. Louis, MO). GTP, guanine 5′-O-(3-thiotriphosphate) (GTPγS), guanylyl imidodiphosphate (GppNHp), GDP, and ATP were of the highest purity available and were purchased from Boehringer Mannheim (Mannheim, Germany). Nucleotide stock solutions (10 mM) were stored at −20°C. Nucleotide dilutions were prepared fresh daily. Sources of other materials have been described elsewhere (Seifert et al., 1998a,b). The construction of the fusion protein of the β2-AR and GsαL is described in Seifert et al. (1998a,b).
Cell Culture and Membrane Preparation.
The β2-AR or β2-ARGsα fusion protein was expressed in Sf9 cells via recombinant baculovirus, as described (Seifert et al., 1998a,b). Sf9 membranes expressing β2-ARGsα were prepared according to Seifert et al. (1998a,b). The experiments described in this study were performed in the absence of mammalian βγ complex. The effect of mammalian β1γ2 complex on the function of β2-ARGsαwas described previously (Seifert et al., 1998b).
[3H]Dihydroalprenolol (DHA) Binding.
[3H]DHA binding studies were carried out as described (Seifert et al., 1998a,b). Tubes contained Sf9 membranes expressing β2-ARGsα at 5.0 to 7.5 pmol/mg of protein (15–30 μg of protein/tube), 1 nM [3H]DHA, 1 μM salbutamol (SAL) and various nucleotides at increasing concentrations. As reported before, theK d value for [3H]DHA at β2-ARGsα is 0.36 ± 0.03 nM (Seifert et al., 1998b). Nonspecific binding with 1 nM [3H]DHA, as assessed by the binding not competed for by 10 μM (−)-alprenolol, was less than 5% of total binding.
Steady-State Nucleoside 5′-Triphosphatase (NTPase) Activity.
Nucleoside 5′-triphosphate (NTP) hydrolysis was determined according toSeifert et al. (1998a,b). Unless stated otherwise, assay tubes contained Sf9 membranes expressing β2-AR at 6.1 pmol/mg of protein or β2-ARGsα at 7.0 to 7.5 pmol/mg of protein (10 μg of protein), 1.0 mM MgCl2, 0.1 mM EDTA, 0.1 mM ATP, 1 mM adenylyl imidodiphosphate, 5 mM creatine phosphate, 40 μg of creatine kinase, and 0.2% (w/v) BSA in 50 mM Tris/HCl, pH 7.4. Tubes additionally contained β2-AR ligands and unlabeled GTP, ITP, or XTP at various concentrations. Assay tubes (80 μl) were incubated for 3 min at 25°C before the addition of 20 μl of [γ-32P]GTP, [γ-32P]ITP, or [γ-32P]XTP (0.75–2.0 μCi/tube). Reactions were conducted for 20 min at 25°C. Reactions were terminated by the addition of 900 μl of a slurry consisting of 5% (w/v) activated charcoal and 50 mM NaH2PO4, pH 2.0. Charcoal absorbs nucleotides but not Pi. Charcoal-quenched reaction mixtures were centrifuged for 15 min at room temperature at 15,000g. Seven hundred microliters of the supernatant fluid of reaction mixtures was removed, and32Pi was determined by liquid scintillation counting. Enzyme activities were corrected for spontaneous degradation of [γ-32P]NTP. Spontaneous [γ-32P]NTP degradation was determined in tubes containing all of the above-described components plus a very high concentration of unlabeled NTP (1 mM) that, by competition with the trace concentrations of [γ-32P]NTP, prevents [γ-32P]NTP hydrolysis by enzymatic activities present in Sf9 membranes. Spontaneous [γ-32P]NTP degradation was <1% of the total amount of radioactivity added. Note that, for NTPase studies, β2-ARGsα was expressed at high levels to increase the sensitivity of the system (Seifert et al., 1998b).
AC Activity.
Cyclic AMP (cAMP) formation in Sf9 membranes was carried out as described (Seifert et al., 1998a,b). Tubes contained Sf9 membranes expressing β2-AR at 6.1 pmol/mg of protein or β2-ARGsαat 2.3 to 2.7 pmol/mg of protein (15–20 μg of protein/tube), 5 mM MgCl2, 0.4 mM EDTA, and 30 mM Tris/HCl, pH 7.4, and purine nucleotides and β2-AR ligands at various concentrations. Assay tubes (30 μl) were incubated for 3 min at 37°C before the addition of 20 μl of reaction mixture containing (final) 40 μM [α-32P]ATP (2.5–3.0 μCi/tube), 2.7 mM mono(cyclohexyl)ammonium phosphoenolpyruvate, 0.125 IU of pyruvate kinase, 1 IU of myokinase, and 0.1 mM cAMP. Reactions were conducted for 20 min. [32P]cAMP was separated from [α-32P]ATP as described (Seifert et al., 1998a,b). Note that, for AC studies, β2-ARGsα was expressed at considerably lower levels than for NTPase studies. This was done to avoid AC availability becoming limiting (Seifert et al., 1998b).
Miscellaneous.
Protein was determined with the Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA). Data were analyzed by nonlinear or linear regression with the Prism program (GraphPAD, San Diego, CA). In this article, we use the term efficacyto describe the phenomenon that different agonists and nucleotides may vary in their ability to produce a response, although they may occupy the same proportion of receptors and G proteins, respectively. The efficacies of ligands on AC in the presence of GTP versus GTPase and on AC in the presence of ITP versus ITPase and the effect of [erythro-dl-1-(7-methylindan-4-yloxy)-3-isopropylaminobutan-2-ol] (ICI) on AC in the presence of XTP were compared with the ttest.
Results
Regulation of High-Affinity Agonist Binding at β2-ARGsα by Guanine, Inosine, and Xanthine Nucleotides and ATP.
One of the earliest steps of the G protein activation/deactivation cycle is formation of a ternary complex consisting of agonist, GPCR, and guanine nucleotide-free Gα (De Lean et al., 1980; Seifert et al., 1998a,b). The ternary complex is characterized by high agonist affinity. On binding of a guanine nucleotide, be it GMP, GDP, GTP, or a GTPase-resistant GTP analog, the ternary complex is disrupted and agonist affinity decreases (De Lean et al., 1980; Seifert et al., 1998a,b). To determine whether inosine and xanthine nucleotides can also disrupt the ternary complex, we examined binding of a fixed concentration of the antagonist [3H]DHA in the presence of a subsaturating concentration of the full β2-AR agonist (−)- isoproterenol (ISO; Fig.1, A and B) and the strong partial agonist SAL in Sf9 membranes expressing β2-ARGsα. Nucleotides at increasing concentrations were added to the binding assays. Nucleotide binding to Gsα reduces the affinity of the β2-AR for agonist and thereby increases [3H]DHA binding.

Effects of GTP, ITP, XTP, and ATP on high-affinity agonist binding in Sf9 membranes expressing β2-ARGsα. Binding experiments were carried out as described in Experimental Procedures, with membranes expressing β2-ARGsα at 5.0 to 7.5 pmol/mg of protein. Reaction mixtures additionally contained 1 nM [3H]DHA, 1 μM SAL, and NTPs at the concentrations indicated on the abscissa. Data are means ± S.D. of three independent experiments performed in triplicate.
NTPs inhibited high-affinity binding of both (−)-ISO and SAL at β2-ARGsα in the order of potency GTP > ITP > XTP > ATP (ineffective). This rank order to potency is in agreement with the data obtained for nonfused Gsα (Northup et al., 1982). The lack of effect of ATP on high-affinity agonist binding indicates that nucleoside diphosphate kinase-catalyzed transphosphorylation of endogenous GDP to GTP by NTP cannot account for the effects of ITP and XTP. In a previous study (Seifert et al., 1998b), we showed that agonist binding in membranes expressing β2-AR alone is guanine nucleotide-insensitive, ruling out the possibility that the β2-AR coupling to endogenous insect Gsα-like G proteins is responsible for the observed NTP effects. In agreement with the concept that the guanine nucleotide-free G protein α subunits support ternary complex formation (De Lean et al., 1980; Seifert et al., 1998a,b), we found that nucleoside 5′-diphosphates (NDPs) also inhibited ternary complex formation (order of potency, GDP > IDP > XDP). Whereas the observed order of potency of nucleotides to inhibit high-affinity agonist binding was expected (Northup et al., 1982; Klinker and Seifert, 1997), differences in potency and efficacy of nucleotides between (−)-ISO and SAL were somewhat unexpected. Specifically, NTPs and GDP were more potent at disrupting the ternary complex with SAL than with (−)-ISO (compare Fig. 1A with Fig. 1C and Fig. 1B with Fig.1D). In addition, whereas ITP and GDP were less efficacious at inhibiting the high-affinity binding of SAL, these nucleotides were about similarly efficacious at inhibiting the high-affinity binding to (−)-ISO.
Regulation of Basal AC Activity by GTP, ITP, and XTP in Sf9 Membranes Expressing β2-AR and β2-ARGsα.
Membranes expressing β2-ARGsα at 2.3 to 2.7 pmol/mg of protein had ∼8-fold higher basal AC activity than membranes expressing β2-AR alone at a higher level (6.1 pmol/mg of protein; Fig. 2). In membranes expressing β2-ARGsα, GTP increased AC activity with an EC50 of 0.7 ± 0.1 μM. Compared with GTP, ITP was considerably less potent (EC50, 30 ± 5 μM) and effective at increasing basal AC activity in membranes expressing β2-ARGsα. XTP had virtually no stimulatory effect on basal AC activity in membranes expressing β2-ARGsα.
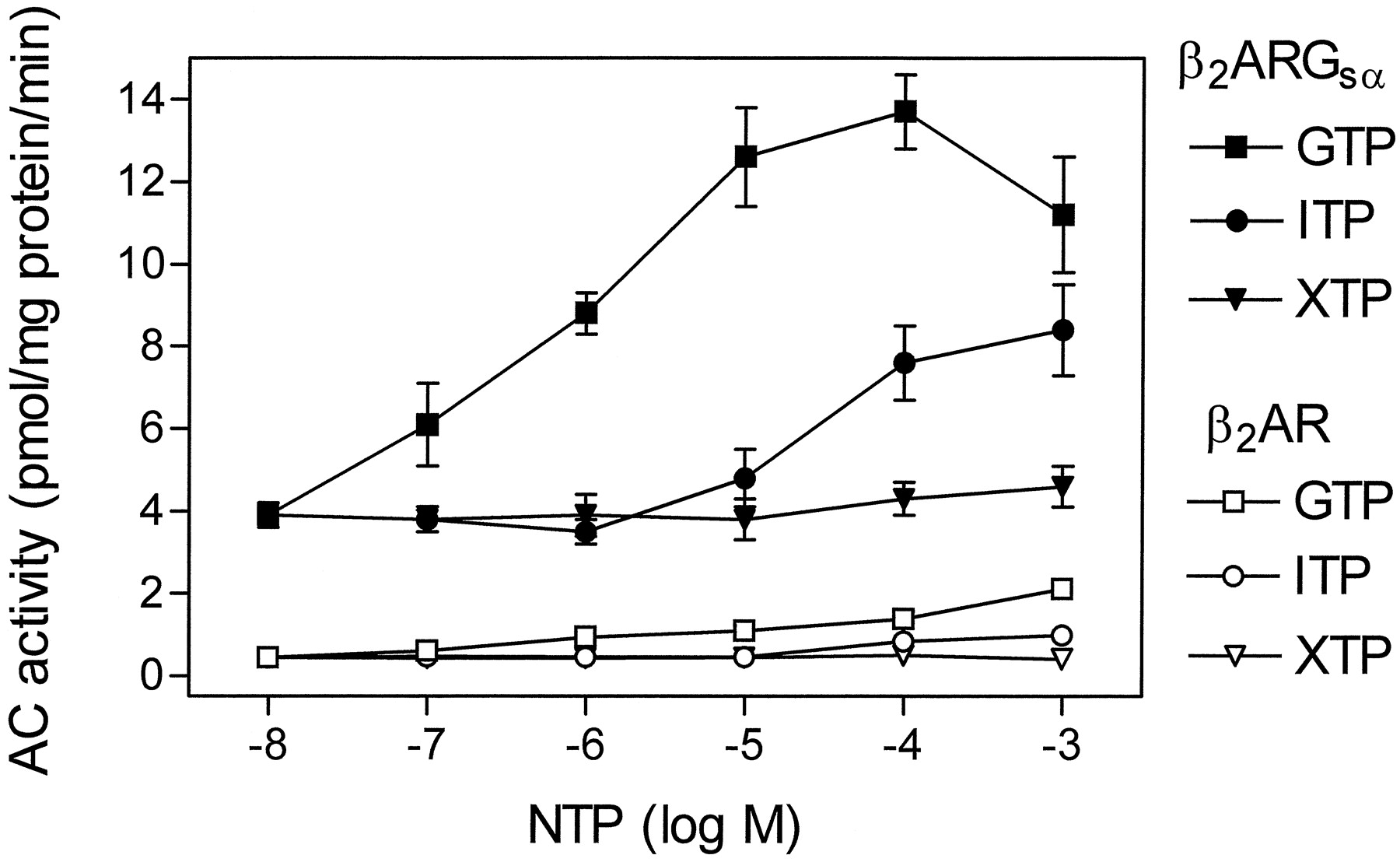
Effects of GTP, ITP, and XTP on basal AC activity in Sf9 membranes expressing β2-AR or β2-ARGsα. AC activity in membranes expressing β2-AR (6.1 pmol/mg of protein) or β2-ARGsα (2.3–2.7 pmol/mg of protein) was determined as described in Experimental Procedures. AC activity was determined in the presence of NTPs at the concentrations indicated on the abscissa. Data are means ± S.D. of three to six independent experiments performed in duplicate.
It has been shown that, in certain systems expressing fusion proteins, there can be cross talk between the fused GPCR and endogenous G proteins of the host cell (Burt et al., 1998). Could the stimulatory effects of GTP and ITP on basal AC activity in membranes expressing the β2-ARGsαL fusion protein be mediated by cross-activation of endogenous Gsα-like G proteins of Sf9 cells by the fused β2-AR? To address this question, we studied the effects of NTPs on AC activity in membranes expressing nonfused β2-AR. Note that, for these studies, we expressed the β2-AR at a level more than twice as high as β2-ARGsαL to increase the effects seen with the nonfused receptor. Despite the high expression level of β2-AR, the absolute stimulation of AC by GTP, ITP, and XTP was much less efficient in membranes expressing β2-AR than in membranes expressing β2-ARGsαL, and the maximal AC stimulation in membranes expressing nonfused β2-AR did not even approach basal AC activity in membranes expressing β2-ARGsαL in the absence of added nucleotides. We also had to rule out the possibility that the stimulatory effects of GTP and ITP in membranes expressing β2-ARGsαL had been caused by a nonspecific fusion-dependent perturbation of the β2-AR, resulting in high constitutive activity of the GPCR. If this were the case, we would observe a similar level of constitutive activity when the β2-AR is fused to either GsαS or GsαL. Therefore, we compared the effects of GTP (1 μM), ITP (10 μM), and XTP (100 μM) on AC activity in membranes expressing β2-ARGsαL (2.3–2.7 pmol/mg of protein) with the corresponding NTP effects in Sf9 membranes expressing β2-ARGsαS at a similar level (2.6 pmol/mg of protein). In membranes expressing β2-ARGsαS, NTPs did not have substantial effects on basal AC activity; i.e., the AC activities in the presence of the different nucleotides were in the same range (17–22 pmol · mg−1protein · min−1; data not shown). In contrast, with β2-ARGsαL, AC activities varied by 3-fold (4–12 pmol · mg−1protein · min−1; Fig. 2). In agreement with our previous study (Seifert et al., 1998a), the AC activities with β2-ARGsαS are considerably higher than the AC activities achieved with β2-ARGsαL. Collectively, these data indicate that the observed NTP effects on AC in membranes expressing β2-ARGsαL are attributable to the fused GsαL and not to activation of endogenous Gsα-like G proteins.
Regulation of AC Activity by (−)-ISO and ICI in Sf9 Membranes Expressing β2-ARGsα in Presence of GTP, ITP, and XTP.
The full β2-AR agonist (−)-ISO further increased AC activity in the presence of GTP, but the stimulatory effect of (−)-ISO did not exceed 50% (Fig.3A). The inverse agonist ICI suppressed GTP-dependent AC activity by ∼50%. In the presence of ITP, (−)-ISO increased AC activity by up to 100%, whereas ICI decreased basal AC activity by not more than 17% (Fig. 3B). The absolute agonist-stimulated AC activity with ITP was substantially lower than with GTP. In the presence of XTP (0.1–1 mM), (−)-ISO increased AC activity by up to 110%, but the absolute agonist-stimulated AC activity with XTP was lower than the corresponding AC activity with ITP (Fig. 3C). Whereas ICI behaved as an inverse agonist by suppressing AC activity in the presence of GTP and ITP, it behaved as a partial agonist in the presence of XTP. ICI increased AC activity by up to 20% in the presence of 100 μM XTP. In Fig. 3F, the stimulatory effect of ICI on AC in the presence of XTP is seen more clearly than in Fig. 3C, because Fig. 3F shows AC activities normalized to basal values.

Effects of (−)-ISO and ICI on AC activity in Sf9 membranes expressing β2-ARGsα in the presence of GTP, ITP, or XTP. AC activity in membranes expressing β2-ARGsα (2.3–2.7 pmol/mg of protein) was determined as described in Experimental Procedures. A–C, AC activity was determined in the presence of NTPs at the concentrations indicated on the abscissa without or with (−)-ISO (10 μM) or ICI (1 μM). D–F, AC activity was determined in the presence of purine nucleotides at the indicated concentrations and (−)-ISO or ICI at increasing concentrations. Data are means ± S.D. of four to six independent experiments performed in duplicate. The dotted lines in A–C are extrapolations of basal AC activities to illustrate the relative contributions of (−)-ISO and ICI at the ligand-regulated enzyme activities.
We also studied the concentration dependence of the effects of (−)-ISO and ICI on AC activity in the presence of GTP, ITP, and XTP at fixed concentrations. NTPs were used at concentrations that gave the highest relative agonist stimulation of AC. In the presence of GTP, (−)-ISO increased AC activity, with an EC50 of 18 ± 8 nM (Fig. 3D). Compared with GTP, the concentration-response curves for (−)-ISO were shifted to the right with ITP (EC50, 233 ± 34 nM; Fig.4E) and XTP (EC50, 416 ± 44 nM; Fig. 3F). The IC50 values of ICI to inhibit AC in the presence of GTP and ITP were similar (16 ± 8 and 22 ± 12 nM, respectively). The stimulatory effect of ICI on AC in the presence of XTP was half-maximal at 7 ± 4 nM and reached a maximum at 0.1 to 1.0 μM. At 0.1 and 1.0 μM, the stimulatory effect of ICI on AC in the presence of XTP was significant (p < .05).

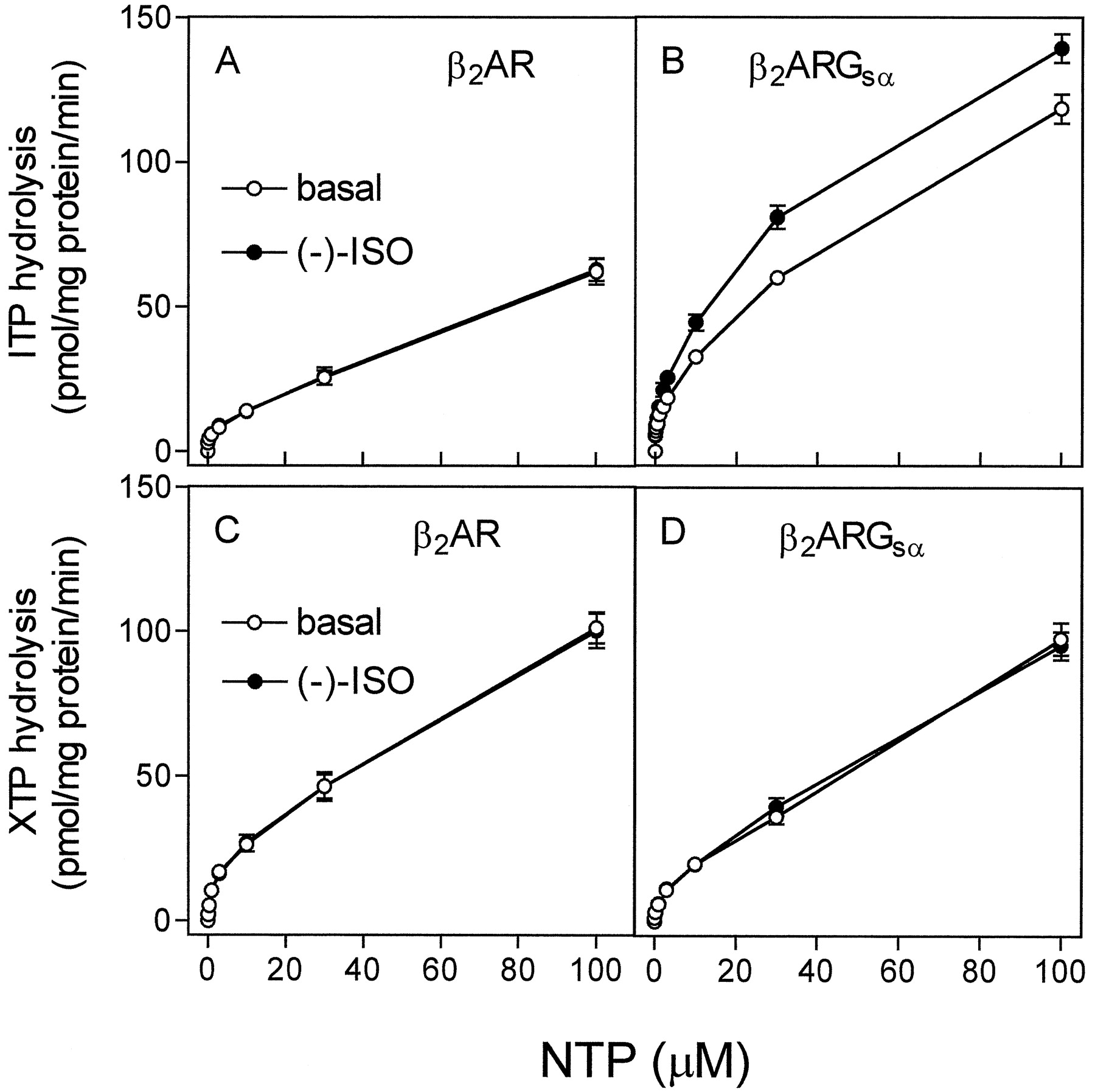
Kinetics of steady-state ITP and XTP hydrolysis in Sf9 membranes expressing β2-AR or β2-ARGsα. ITPase and XTPase activity in membranes expressing β2-ARGsα was determined as described in Experimental Procedures. Reaction mixtures contained 0.1 to 2.0 μM [γ-32P]ITP (1.0 μCi/tube; A and B) or 0.1 to 100 μM [γ-32P]XTP (2.0 μCi/tube; C and D). NTPase activities were determined in membranes expressing β2-AR (6.1 pmol/mg of protein; A and C) or β2-ARGsα (7.5 pmol/mg of protein). Reaction mixtures additionally contained solvent (basal) or (−)-ISO (10 μM). Data are means ± S.D. of two experiments performed in duplicate.
Analysis of GTPase, ITPase, and XTPase Activity in Membranes Expressing β2-ARGsα.
To obtain insight into the mechanism by which Gsα activation by ITP and XTP is terminated, we studied ITPase and XTPase activities in membranes expressing β2-ARGsα or β2-AR. The basal ITPase activity in membranes expressing β2-ARGsα was almost twice that of ITPase activity in membranes expressing β2-AR alone (Fig. 4, A and B). Whereas (−)-ISO had no stimulatory effect on ITP hydrolysis in membranes expressing β2-AR, (−)-ISO significantly increased ITP hydrolysis in membranes expressing β2-ARGsα. These data were the first indication that Gsα exhibits substantial ITPase activity.
Membanes expressing β2-AR and β2-ARGsα both exhibited significant basal XTPase activity. However, in contrast to the data obtained for ITPase, the XTPase activity in membranes expressing β2-ARGsα was not higher than the XTPase activity in membranes expressing β2-AR alone (Fig. 4, C and D). In addition, we did not detect a significant stimulatory effect of (−)-ISO on XTP hydrolysis in membranes expressing β2-AR or β2-ARGsα. It is unlikely that our failure to detect a stimulatory effect of (−)-ISO on XTP hydrolysis was because of an insensitive method, because we used high amounts of [γ-32P]XTP and membranes expressing β2-ARGsα at high levels (see Experimental Procedures). Varying the concentration of (−)-ISO from 1 nM to 1 mM, with another agonist (SAL at 10 nM to 1 mM) and changing the concentration of MgCl2 between 0.1 and 10 mM did not unmask β2-AR-ligand effects on XTP hydrolysis. The lack of ligand regulation of XTPase activity was also reported for Gi protein-coupled chemoattractant receptors in HL-60 membranes (Klinker and Seifert, 1997).
The XTPase experiments (Fig. 4, C and D) together with the agonist competition and AC studies (Figs. 1 and 3C) suggest that XTP binds to Gsα but is not hydrolyzed. To substantiate this hypothesis, we performed competition studies with [γ-32P]NTPs and various unlabeled NTPs. In a previous study, it had already been demonstrated that the GTPase-resistant GTP analog GTPγS efficiently inhibits β-AR-mediated GTP hydrolysis in turkey erythrocyte membranes (Cassel and Selinger, 1977a).
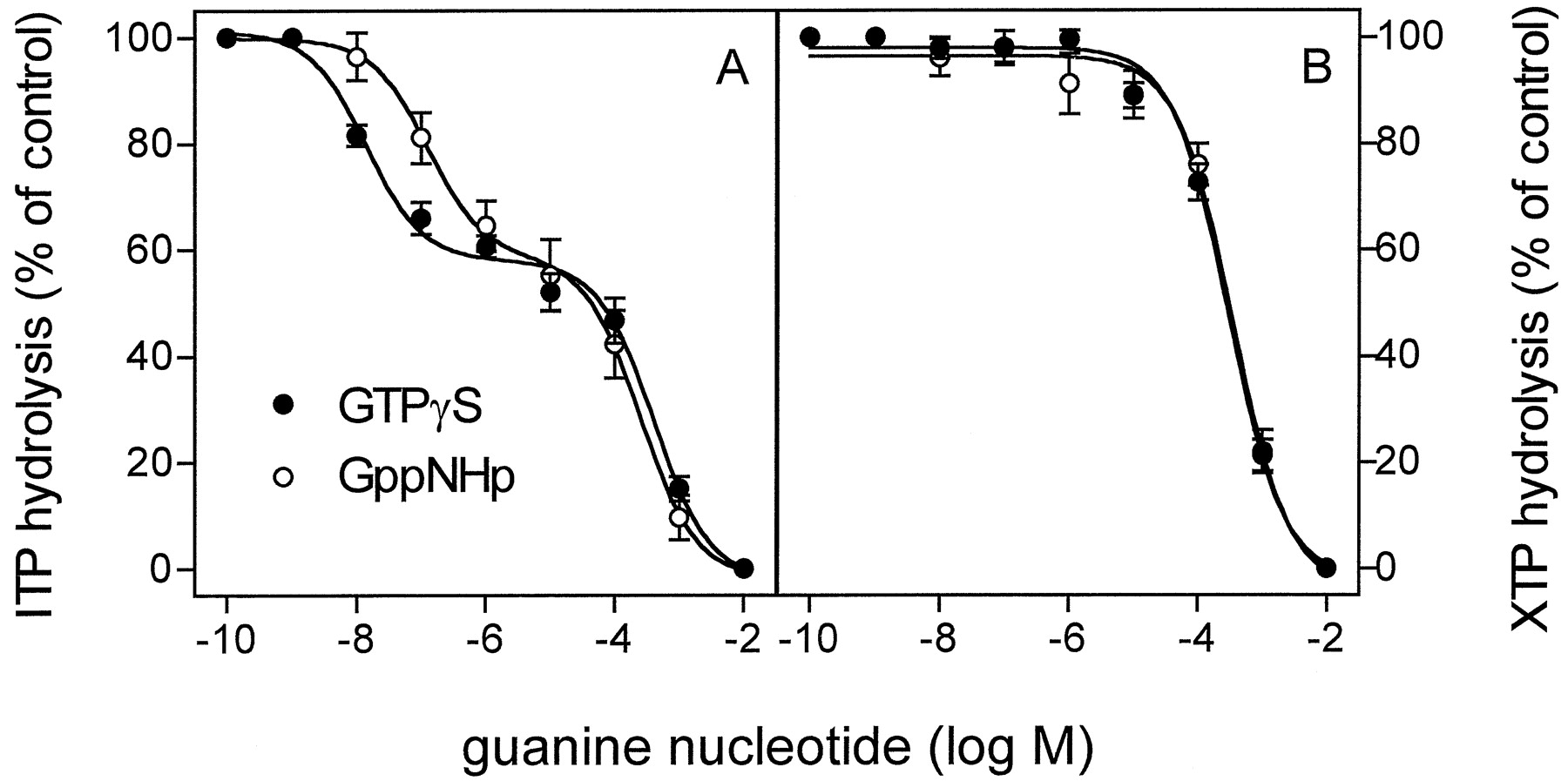
In a first set of experiments, we studied the effects of the nucleotidase-resistant GTP analogs GTPγS and GppNHp on ITP and XTP hydrolysis in the presence of (−)-ISO in membranes expressing β2-ARGsα. GTPγS and GppNHp bind to Gsα with high affinity, and GTPγS is ∼7-fold more potent in this regard than GppNHp (Northup et al., 1982). GTPγS and GppNHp inhibited ITP hydrolysis according to a biphasic function (Fig. 5A). About 40% of the inhibition of ITP hydrolysis by stable GTP analogs was attributable to a high-affinity interaction, whereas the remaining 60% was attributable to a low-affinity interaction. GTPγS inhibited the high-affinity component of ITP hydrolysis approximately nine times more potently than GppNHp, whereas, for the low-affinity component, no such difference in potency between GTPγS and GppNHp was observed (IC50, 402 ± 44 and 250 ± 33 μM, respectively). These findings show that GTPγS and GppNHp potently compete with ITP for binding to Gsα. The ITPase that is inhibited by GppHNp and GTPγS with low affinity presumably represents the activity of nonspecific nucleotidases of Sf9 cells.

Competition of ITP and XTP hydrolysis by GTPγS and GppNHp in Sf9 membranes expressing β2-ARGsα. ITPase and XTPase activity in membranes expressing β2-ARGsα (7.5 pmol/mg of protein) was determined as described in Experimental Procedures. Reaction mixtures contained 1 μM [γ-32P]ITP (1.0 μCi/tube; A) or 1 μM [γ-32P]XTP (1.0 μCi/tube; B). Reaction mixtures additionally contained (−)-ISO (10 μM) and GTPγS or GppNHp at the concentrations indicated on the abscissa. The activities observed in the presence of (−)-ISO were set 100% (control). Data are means ± S.D. of two experiments performed in duplicate.
In marked contrast to the biphasic competition of ITP hydrolysis by GTPγS and GppNHp, no high-affinity inhibition of XTPase by GTPγS and GppNHp was detected (Fig. 5B). The IC50values of GTPγS and GppNHp for XTP hydrolysis were 294 ± 23 and 351 ± 44 μM, respectively, and were similar to the IC50 values for the low-affinity inhibition of ITPase by GTPγS and GppNHp.
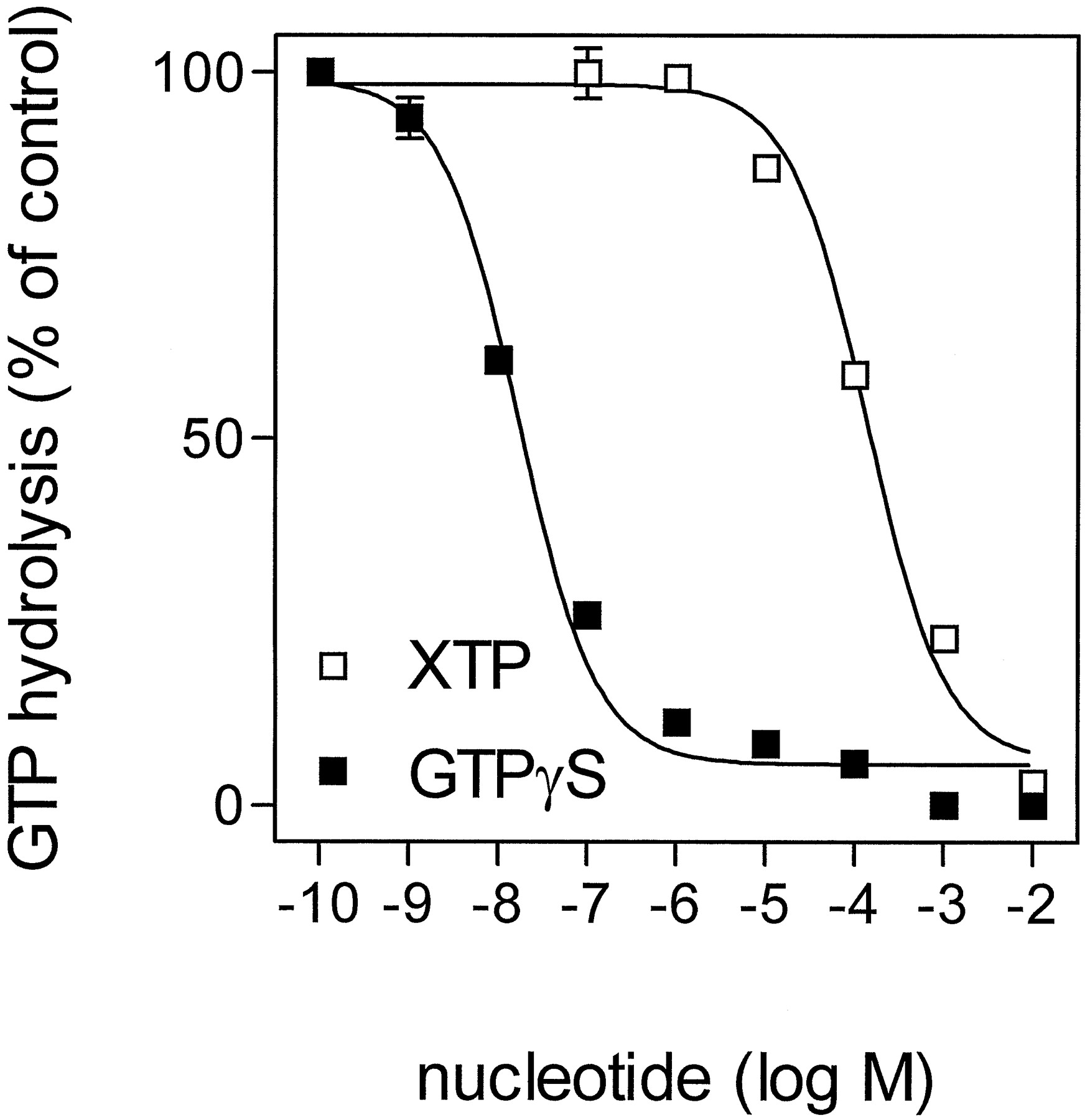
In a second set of experiments, we compared the effects of GTPγS and XTP on (−)-ISO-stimulated GTP hydrolysis (Fig.6). As expected from previous experiments (Cassel and Selinger, 1977a), GTPγS inhibited GTP hydrolysis (IC50, 17 ± 4 nM). If XTP binds to but is not hydrolyzed by Gsα, XTP is expected to block GTP hydrolysis, as does GTPγS. Indeed, XTP abolished (−)-ISO-stimulated GTP hydrolysis, although with a much higher IC50 value than GTPγS (IC50, 139 ± 22 μM). Taken together, the nucleotide competition data and the similar basal XTPase activities in membranes expressing β2-AR and β2-ARGsα indicate that basal XTP hydrolysis in Sf9 membranes is caused by endogenous nucleotidases and that Gsα does not hydrolyze XTP to a measurable extent.

Competition of GTP hydrolysis by GTPγS and XTP in Sf9 membranes expressing β2-ARGsα. GTPase activity in membranes expressing β2-ARGsα(7.5 pmol/mg of protein) was determined as described inExperimental Procedures. Reaction mixtures contained 100 nM [γ-32P]GTP (0.75 μCi/tube), 10 μM (−)-ISO, and GTPγS or XTP at the concentrations indicated on the abscissa. The activities observed in the presence of (−)-ISO were set 100% (control). Data are means ± S.D. of two experiments performed in duplicate.
Kinetics of Agonist-Stimulated GTP and ITP Hydrolysis in Membranes Expressing β2-ARGsα.
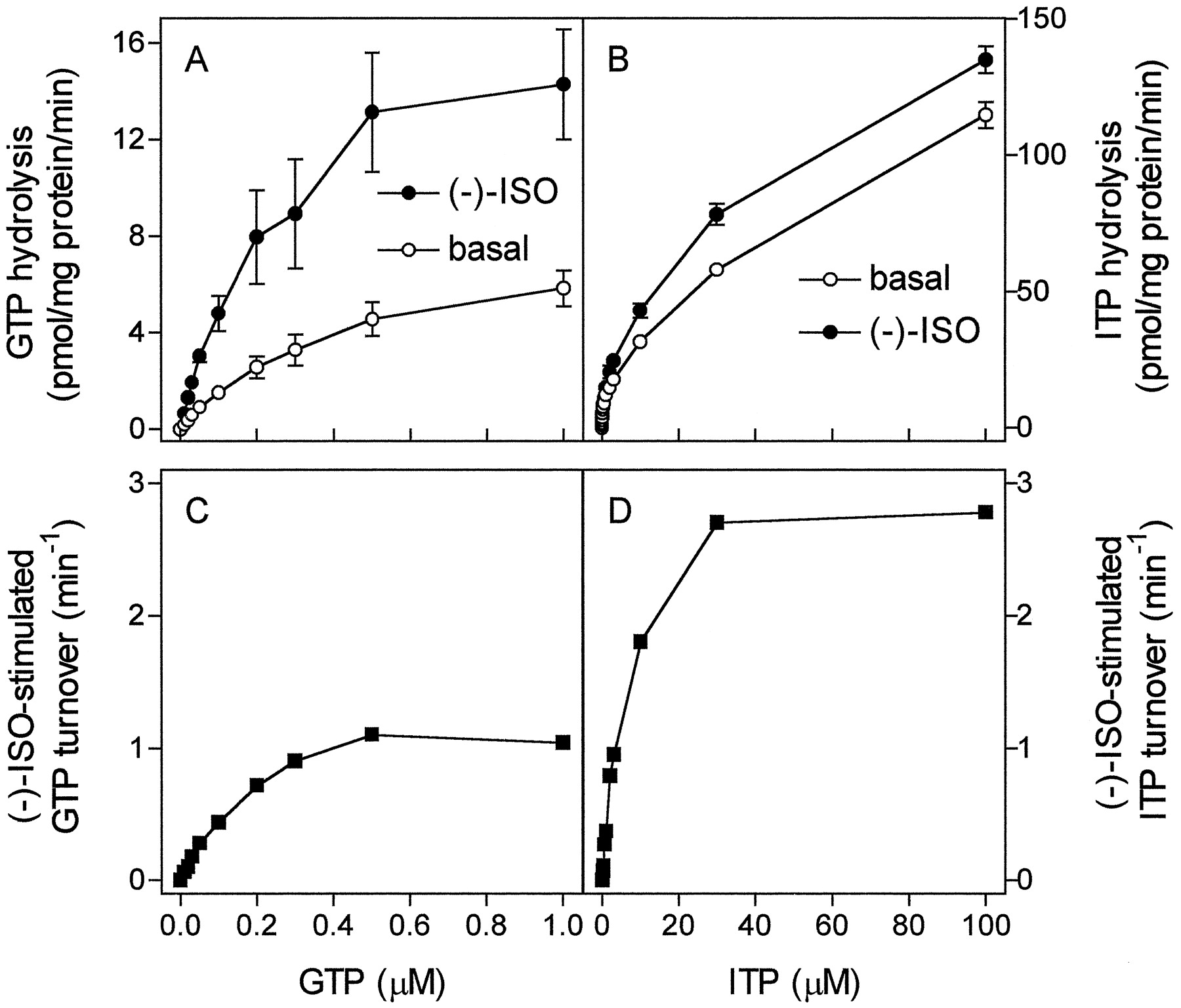
Because of the fixed 1:1 stoichiometry of GPCR to G protein in fusion proteins, the G protein concentration can be determined by receptor-antagonist saturation binding (Wise et al., 1997; Seifert et al., 1998a,b). These properties of fusion proteins allow calculation of agonist-stimulated NTP turnover of the fused G protein (Wise et al., 1997; Seifert et al., 1998b). With GTP at concentrations between 0.01 and 1.00 μM, (−)-ISO stimulated GTP hydrolysis up to 250% (Fig.7A). For each substrate concentration, the basal GTP hydrolysis rates were subtracted from the GTP hydrolysis rates observed in the presence of (−)-ISO and referred to the β2-ARGsα expression level. By doing so, a V max of (−)-ISO-stimulated GTP turnover of 1.37 ± 0.11 min−1 was obtained by nonlinear regression analysis (Fig. 7C). The K m value of the (−)-ISO-stimulated GTPase is 0.18 ± 0.04 μM. These kinetic properties of β2-ARGsαagree with data obtained for reconstituted purified β-AR and Gs (Brandt and Ross, 1986). Because the affinity of Gsα for ITP is lower than for GTP (Figs. 1and 2; Northup et al., 1982), ITP hydrolysis was studied with higher substrate concentrations than GTP hydrolysis. We readily detected stimulatory effects of (−)-ISO on ITP hydrolysis, with substrate concentrations from 0.1 to 100.0 μM (Fig. 7, B and D). TheV max of (−)-ISO-stimulated ITP hydrolysis was 3.06 ± 0.07 min−1, and theK m was 6.3 ± 0.5 μM.
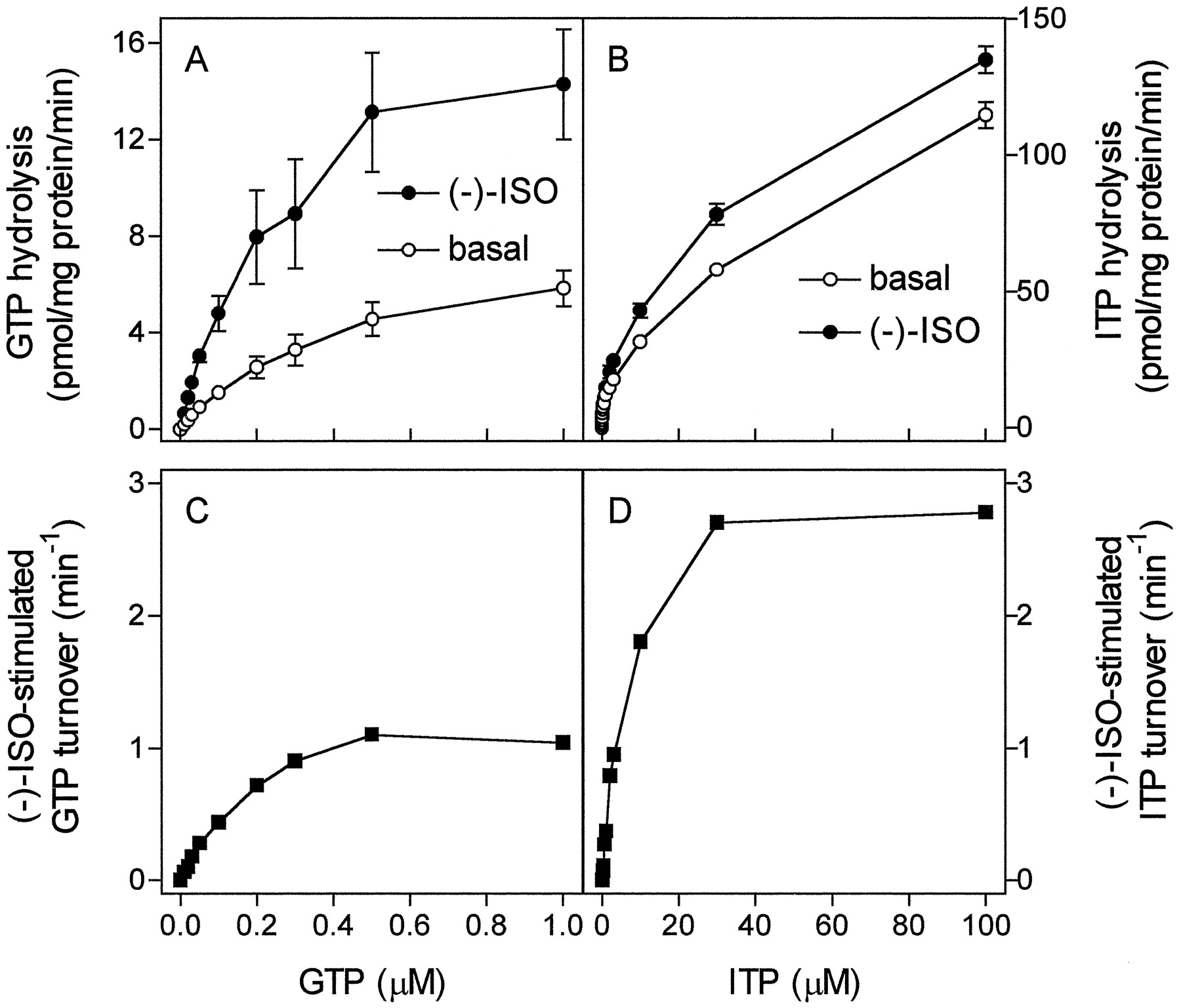
Kinetics of steady-state GTP and ITP hydrolysis in Sf9 membranes expressing β2-ARGsα . GTPase and ITPase activity in membranes expressing β2-ARGsαwas determined as described in Experimental Procedures. Reaction mixtures contained 0.01 to 1.00 μM [γ-32P]GTP (1.0 μCi/tube; A) or 0.1 to 100.0 μM [γ-32P]ITP (2.0 μCi/tube; B). Reaction mixtures additionally contained solvent (basal) or (−)-ISO (10 μM). Data are means ± S.D. of two experiments performed in duplicate. C, basal GTPase activity for each substrate concentration was subtracted from GTPase activity in the presence of (−)-ISO and normalized for β2-ARGsα expression level (7.5 pmol/mg of protein) to calculate molar GTP turnover of fused Gsα. D, basal ITPase activity for each substrate concentration was subtracted from ITPase activity in the presence of (−)-ISO and normalized for β2-ARGsα expression level (7.5 pmol/mg of protein) to calculate molar ITP turnover of fused Gsα.
Ligand Efficacies at β2-ARGsα in GTPase, ITPase, and AC Studies.
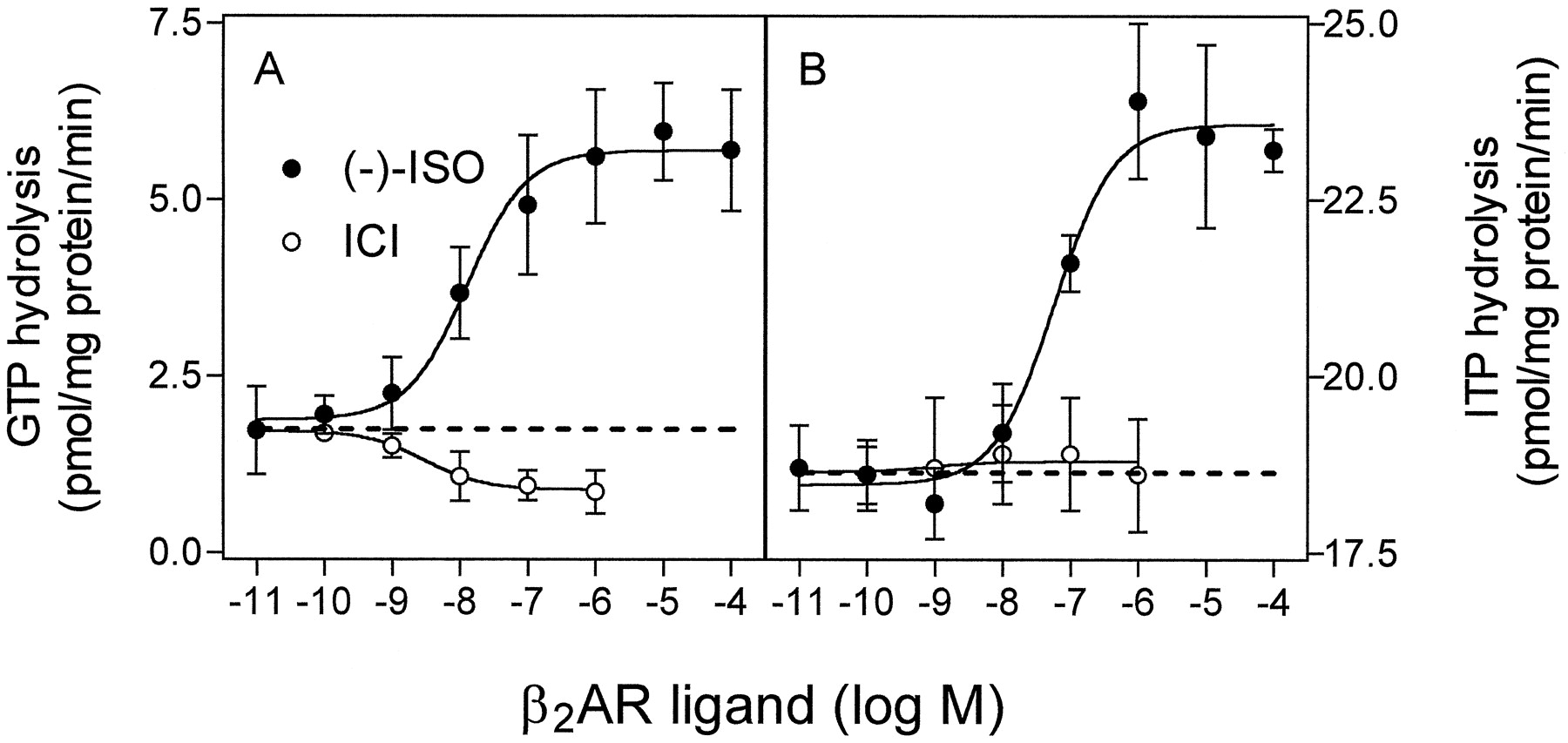
Fig.8 shows the concentration-response curves for the effects of (−)-ISO and ICI on GTP hydrolysis and ITP hydrolysis in membranes expressing β2-ARGsα (7.5 pmol/mg of protein). (−)-ISO stimulated GTPase, with an EC50 of 13 ± 3 nM, and ICI reduced GTP hydrolysis, with an IC50 of 3.0 ± 1.2 nM. (−)-ISO increased GTP hydrolysis by up to 230%, whereas ICI reduced GTP hydrolysis by up to 50%. In agreement with the reduced potency of (−)-ISO to stimulate AC in the presence of ITP (Fig. 3, D and E), the potency of (−)-ISO to activate ITPase was lower (EC50, 57 ± 10 nM) than the potency to stimulate GTPase (Fig. 8, A and B). In the presence of ITP at 3.0 μM, (−)-ISO increased ITP hydrolysis by ∼30% above basal (Fig. 8B). We could not detect an inhibitory effect of ICI on ITP hydrolysis, despite high assay sensitivity and presumably high basal ITPase activity of Gsα (see Fig. 4, A and B).

Concentration-response curves for the effects of (−)-ISO and ICI on GTPase and ITPase activity in Sf9 membranes expressing β2-ARGsα. GTPase and ITPase activity in membranes expressing β2-ARGsα(7.5 pmol/mg of protein) was determined as described inExperimental Procedures. Reaction mixtures contained 0.1 μM [γ-32P]GTP (0.75 μCi/tube; A) or 3.0 μM [γ-32P]ITP (1.5 μCi/tube; B) and (−)-ISO and ICI at the concentrations indicated on the abscissa. The dotted lines are extrapolations of basal GTPase and ITPase activities to illustrate the relative contributions of (−)-ISO and ICI at the ligand-regulated enzyme activities. Data are means ± S.D. of three or four independent experiments performed in duplicate. The data shown in A are identical with the data shown in Fig. 2B in Seifert et al. (1998a).
Table 1 summarizes the efficacies of a series of β2-AR ligands on GTPase and ITPase activity and AC activities measured in the presence of GTP, ITP, or XTP. A highly significant correlation was obtained when the efficacies of β2-AR agonists at activating GTPase and AC in the presence of GTP were plotted against each other (Fig. 8A). However, when the efficacies of β2-AR agonists at activating ITPase and AC in the presence of ITP were plotted against each other, the correlation was much less stringent than in the corresponding experiments with GTP (compare Fig.9, A and B). The efficacies of SAL, (−)-ephedrine (EPH), dichloroisoproterenol (DCI), and (−)-alprenolol at activating AC in the presence of ITP differed significantly from the respective efficacies of the ligands at activating ITP hydrolysis. The inverse agonists timolol and ICI were significantly more efficacious at reducing AC activity in the presence of GTP or ITP than at reducing hydrolysis of the respective NTP (see Table 1). Note that the efficacies of partial agonists on AC in the presence of XTP, most prominently the efficacies of dobutamine (DOB) and EPH, were very low.
Efficacies of β2-AR ligands at β2-ARGs α as assessed by AC Activity in presence of GTP, ITP, or XTP and by GTPase and ITPase activity

Correlation between the efficacies of agonists at activating steady-state GTPase or ITPase activity in Sf9 membranes expressing β2-ARGsα with the efficacies of ligands at activating AC in the presence of GTP and ITP, respectively. A, the efficacies of partial and full agonists at activating steady-state GTP hydrolysis in Sf9 membranes expressing β2-ARGsα (7.0–7.5 pmol/mg of protein) and AC in the presence of GTP in Sf9 membranes expressing β2-ARGsα (2.0–2.7 pmol/mg of protein) as shown in Table 1 were plotted against each other. Data were analyzed by linear regression analysis (r 2 = 0.990,p < .0001). The dotted line indicates the 95% confidence interval of the regression line. B, the efficacies of partial and full agonists at activating steady-state ITP hydrolysis in Sf9 membranes expressing β2-ARGsα (7.0–7.5 pmol/mg of protein) and AC in the presence of ITP in Sf9 membranes expressing β2-ARGsα (2.0–2.7 pmol/mg of protein) as shown in Table 1 were plotted against each other. Data were analyzed by linear regression analysis (r 2 = 0.644; p < .0092). The dotted line indicates the 95% confidence interval of the regression line. LAB, (±)-labetalol; PRO, (−)-propranolol; ALP, (−)-alprenolol.
Table 2 summarizes the EC50 values of selected β2-AR agonists on GTPase and ITPase activity and AC activities in the presence of GTP, ITP, or XTP. For comparison with agonist potencies, Table 2 also contains the high- and low-affinity K i values for the agonists studied. Moreover, we calculated potency ratios for the various parameters studied. For (−)-ISO, (+)-ISO, SAL, and DOB, the EC50 values for activation of AC in the presence of GTP were substantially lower than the EC50values for activation of AC in the presence of ITP. The same was true for the comparison of agonist potencies for activation of GTPase versus ITPase activation. With the exception of EPH, the EC50 values of agonists at activating AC in the presence of GTP were similar to the EC50 values of agonists at activating GTPase. Similarly, the EC50 values of agonists at activating AC in the presence of ITP were similar to the EC50 values of agonists at activating ITPase. When AC activation in the presence of XTP was considered, the EC50 values of (−)-ISO, (+)-ISO, and DOB were even higher than the EC50values for AC activation in the presence of ITP. When EPH was considered, we noted an unexpectedly low EC50value of the agonist at activating GTPase. However, the EC50 values of (−)-ISO, (+)-ISO, and EPH in certain functional assays were even higher than the low-affinityK i values. In contrast, the EC50 values for DCI in functional assays were lower than was expected from the K i values in the [3H]DHA competition-binding assay.
Potencies of selected β2-AR ligands at β2-ARGs α as assessed by AC activity in presence of GTP, ITP, or XTP and by GTPase and ITPase activity and agonist affinities
Discussion
Interaction of Guanine, Inosine, and Xanthine Nucleotides with Gsα.
Guanine, inosine, and xanthine nucleotides differentially form hydrogen bonds with a highly conserved aspartate residue in G protein α subunits (Noel et al., 1993; Fig.10). The reduced affinity of Gsα for IDP and ITP compared with GDP and GTP may be because the inosine ring forms only one hydrogen bond with Asp295 in Gsα (see Figs. 1, 2, and 10; Northup et al., 1982). Repulsion of the electronegative groups in the xanthine ring and Asp295 may explain why XDP and XTP have an even lower affinity for GSα than IDP and ITP (see Figs. 1, 2, and10). Factors that influence nucleotide affinity may also affect the efficacy of nucleotides. GTP, ITP, and XTP do not have the same maximal effect with respect to disruption of the ternary complex and AC activation (Figs. 1-3). Thus, there may be nucleotide-specific conformational changes in Gsα that influence interactions with the receptor and/or effector. Studies with fluorescent guanine nucleotides already provided evidence for the existence of nucleotide-specific G protein activation states (Remmers and Neubig, 1996).
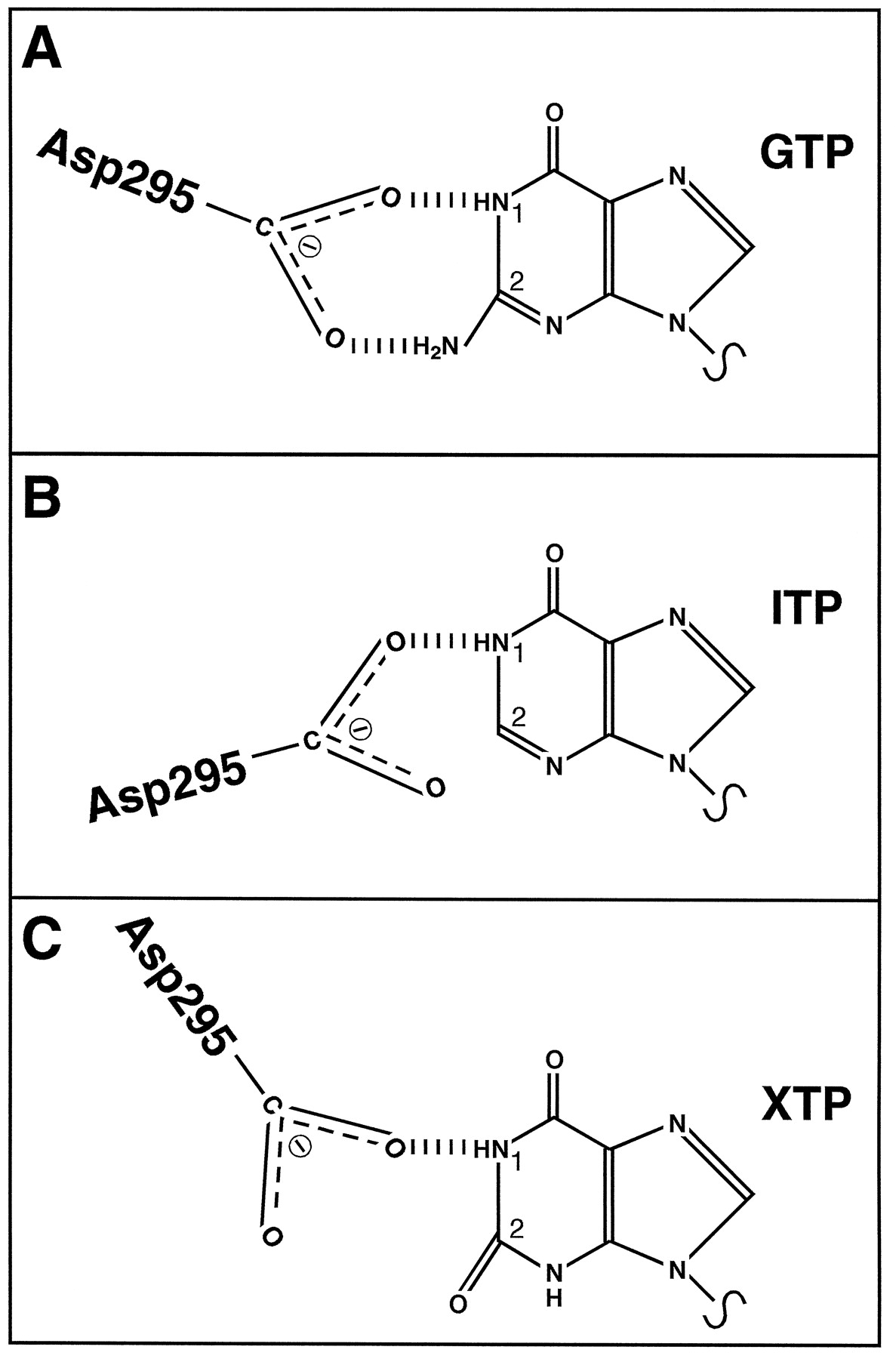
Model for the interaction of guanine, inosine, and xanthine nucleotides with Asp295 in Gsα. A, a highly conserved aspartate residue in G protein α subunits (Asp295 in GsαL) forms hydrogen bonds with the substituted nitrogen at position 1 of the guanine ring and the amino group in position 2. B, in inosine nucleotides, the amino group at position 2 is missing. Therefore, hydrogen bonding at this position cannot take place, and the orientation of IDP/ITP relative to Asp295 is presumably different from the orientation of GDP/GTP relative to Asp295. C, in xanthine nucleotides, position 2 of the purine ring is substituted with a keto group. Therefore, hydrogen bonding with Asp295 at position 2 of the purine ring cannot take place. The orientation of XDP/XTP relative to Asp295 may be different from the orientations of IDP/ITP relative to Asp295, because there is repulsion between the electronegative keto group of the xanthine ring and the carboxyl group of the aspartate residue. For the sake of simplicity, the sugar moiety and phosphate chain of NDPs/NTPs are not shown.
Differences in the kinetics of interaction of nucleotides with Gsα could also contribute to the different efficacies of nucleotides. NTP hydrolysis is the principal mechanism by which Gsα is deactivated (Cassel and Selinger, 1976; Gilman, 1987). The faster NTP hydrolysis proceeds, the shorter the time for which Gsα can stay in an active conformation (Wenzel-Seifert et al., 1998b). Thus, the lower efficacy of ITP compared with GTP at disrupting the ternary complex and activating AC could result from the higher ITP hydrolysis rate compared with GTP hydrolysis rate (see Figs. 1-3 and 7). Based on the dissociation rates of GDP and IDP from G proteins, it is also likely that the rate of ITP dissociation from Gsα is greater than the rate of GTP dissociation (Florio and Sternweis, 1989). Fast dissociation from Gsα could be another factor that contributes to the lower efficacy of IDP and ITP compared with GDP and GTP. For AC activation by GSα in the presence of XTP, nucleotide dissociation and not rapid hydrolysis appears to be the major mechanism by which Gsαis deactivated (Figs. 4, C and D, and 5B). Because of its low affinity for Gsα, XTP could be thought to dissociate from Gsα even before it can be cleaved. As a result of the rapid dissociation of XTP, Gsαstays in the active state only for short periods. NTP dissociation as a major mechanism of G protein deactivation is conceivable in view of the fact that even highly potent G protein ligands such as GTPγS or GppNHp can dissociate from G protein α subunits (Cassel and Selinger, 1977b; Hilf et al., 1992; Breivogel et al., 1998). We could not directly study dissociation of ITP and XTP because of the low affinity of these nucleotides for Gsα.
Another mechanism that could contribute to the observed differences in efficacy between GTP, ITP, and XTP is differential β2-AR regulation of NTP binding to Gsα. GTP binding to G protein α subunits does not passively follow GDP release but is actively catalyzed by GPCR (Iiri et al., 1998). The dual hydrogen bonding of the guanine ring at Asp295 could be envisaged to stabilize GTP binding to such an extent that even the agonist-free β2-AR can effectively increase GTP binding to Gsα (see Fig. 10A). This assumption is supported by the strong stimulatory effect of GTP on basal AC activity and the high inverse agonist efficacy of ICI and timolol (see Figs. 2 and 3; Table 1). Because of the weaker hydrogen bonding of the inosine ring to Gsα (see Fig. 10, A and B), binding of ITP to Gsα is less stable than binding of GTP so that substantial agonist occupancy of β2-AR is required to stabilize ITP binding. In accordance with this model are our findings that ITP is less efficient at increasing basal AC activity than GTP and that the inverse agonist efficacy in the presence of ITP is lower than in the presence of GTP (see Figs. 2, 3, and 8; Table 1). Additionally, agonist potency is lower in the ITPase assay and the AC assay with ITP than in the GTPase assay and the AC assay with GTP (see Figs. 4, D and E, and 9). Because of the repulsion of the electronegative groups, the energy barrier for XTP to bind to Gsα may be so high that the agonist-free β2-AR is virtually ineffective at promoting this XTP binding (see Fig. 10C). In support of this assumption, XTP only minimally increased basal AC activity, no inverse agonist effects were observed, and agonist potency was very low (see Figs. 2 and 3, C and E, and Table 1).
GTP, ITP, and XTP as Tools to Analyze Ligand-Specific GPCR Conformations.
If receptor-stimulated NTP hydrolysis is assumed to be a function of receptor-promoted nucleotide binding and the intrinsic nucleotidase activity of the G protein, then the efficacies and potencies of ligands at activating AC and NTP hydrolysis should be identical. In agreement with previously published data, we found that a strong correlation exists when AC activation in the presence of GTP and GTPase activation is considered (see Fig. 9A and Table 1; Pike and Lefkowitz, 1980). However, we made several observations that are not compatible with this model. First, there was a much weaker correlation between the efficacies of agonists at activating ITPase and activating AC in the presence of ITP (see Fig. 9B and Table 1). Moreover, despite the lack of agonist regulation of XTP hydrolysis, we observed efficient agonist regulation of AC activity in the presence of XTP (see Figs. 3F and 4D; Table 1). Furthermore, certain ligands exhibited unexpected pharmacological properties when we examined AC regulation in the presence of XTP. Most notably, ICI, although an inverse agonist in the presence of GTP and ITP, displayed weak partial agonistic activity in the presence of XTP (Fig. 1). As another example, DOB is a strong partial agonist with respect to AC activation in the presence of GTP and ITP but only a weak partial agonist in the presence of XTP, having an efficacy comparable to the efficacy of ICI. We also observed differences in the EC50 values of some agonists when comparing their stimulation of AC with their stimulation of GTPase or ITPase activity (Table 2). It is unlikely that these differences are the result of differences in the experimental conditions in the AC versus NTPase assay, because such differences should have affected EC50 values and efficacies of agonists in a systematic manner.
The divergent effects of agonists and inverse agonists on NTP hydrolysis and AC activity in the presence of various NTPs could be reconciled by proposing multiple ligand-specific GPCR conformations. Each ligand stabilizes a unique β2-AR conformation that allows the receptor to interact with Gsα in a ligand-specific manner, i.e., induces ligand-specific conformational changes in the G protein. As a result of this ligand-specific receptor/G protein coupling, all of the kinetic properties of NTP interaction with Gsα, i.e., NTP binding, dissociation, and hydrolysis, are differentially altered, ultimately leading to ligand-specific effector activation. Thus, in the presence of ITP, the efficacy of (−)-ISO is greater than that of SAL in stimulating AC, whereas SAL has a higher efficacy at stimulating ITP hydrolysis (Table 1). This might be explained by proposing that SAL and (−)-ISO are equally effective in promoting ITP binding, but SAL is more efficient than (−)-ISO in promoting ITP hydrolysis. As a result, Gsα activated by SAL-liganded β2-AR is more rapidly inactivated and thus less effective in stimulating AC. Perhaps most interesting is the behavior of ICI in the presence of XTP. Whereas the β2-AR conformation stabilized by ICI may be efficient at preventing GTP binding to Gsα, it promotes, at least to some extent, the binding of XTP (see Fig. 3, D and F). As another example, DOB may stabilize a specific β2-AR conformation that is much more efficient at enhancing GTP and ITP binding to Gsα than XTP binding (see Table 1).
The apparent discrepancies between agonist affinity as determined in [3H]DHA competition studies and agonist potencies in AC and NTPase studies also provide insight into the mechanisms underlying receptor/G protein coupling (Table 2). We expected that the EC50 values of agonists in AC assays would not be higher than the low-affinityK i values for agonist binding (which is actually assumed to represent the G protein-uncoupled state; De Lean et al., 1980; Lefkowitz et al., 1993). However, this was not the case; i.e., the EC50 of (−)-ISO and (+)-ISO in the AC assays with XTP were higher than their low-affinityK i values (Table 2). Similar discrepancies between agonist potency and agonist affinity have been observed before for several GPCRs, including the β2-AR (Gether et al., 1995; Albrecht et al., 1998; Wenzel-Seifert et al., 1998a). Most important, (−)-ISO binds to the purified β2-AR with a K ivalue of 1 μM, but (−)-ISO induces a conformational change in the receptor only with an EC50 of ∼30 μM (Gether et al., 1995). These findings indicate that the β2-AR (and other GPCRs) may exist in a state of ultralow agonist affinity that is difficult to detect in ligand-binding studies, either because this ultralow agonist-affinity state is in a rapid equilibrium with K l or the proportion of receptors in this state is small. Of interest in this context is the fact that, for the partial agonist DCI, EC50 values were always lower thanK i values in binding assays (Table 2). In this case, a fraction of the β2-AR that is too small to be detected in the binding assay may exhibit high affinity for DCI and mediate G protein coupling, regardless of whether GTP or ITP is the nucleotide present. Collectively, the dissociation of agonist affinities and agonist potencies provide further support for the existence of multiple GPCR conformations (Kenakin, 1996; Tucek, 1997) and indicate that ligand-binding studies cannot detect all existing and functionally relevant receptor states.
Conclusions.
Guanine, inosine, and xanthine nucleotides can be used as probes to detect ligand-specific G protein-coupling states of receptors. We observed that the efficacy and potency of a panel of β2-AR ligands are influenced by the nucleotide bound to Gsα and that purine nucleotides differentially disrupt the ternary complex stabilized by different ligands, supporting the concept of multiple active receptor conformations. Our results suggest that unique ligand-induced receptor states not only promote NDP release and NTP binding but may also influence NTP dissociation and hydrolysis. Moreover, the efficacy and potency of a ligand at regulating nucleotide binding may differ from its efficacy at promoting nucleotide hydrolysis or dissociation. This conclusion implies that G proteins retain a “memory” of the ligand-specific receptor conformation. The molecular basis for such G protein memory could be continuous physical interaction of a GPCR with the G protein during the entire G protein activation/deactivation cycle. The fact that ternary complex formation is at least partially preserved when Gsα binds ITP, XTP, GDP, IDP, or XDP clearly points to persistent receptor/G protein contact even after nucleotide binding. Although it is conceivable that such interaction of receptor and G protein during the entire cycle can happen in the conformationally constrained fusion protein system, such interaction may not be restricted to such systems. Specifically, guanine nucleotide-insensitive high-affinity-agonist binding has also been observed in nonfused systems (Szele and Pritchett, 1993; Wild et al., 1993; Seifert et al., 1998b), and guanine nucleotides do not prevent copurification of receptors and G proteins (Matesic et al., 1989). Moreover, cytoskeletal elements may restrict the mobility of receptors and G proteins in vivo, thereby forcing their close association(Neubig, 1994).
Acknowledgments
We thank Dr. M. Doughty (Department of Medicinal Chemistry, University of Kansas, Lawrence, KS) for stimulating discussions, the reviewers of the manuscript for constructive criticism, and M. Bakk for help with the cell culture.
Footnotes
- Received January 6, 1999.
- Accepted April 28, 1999.
-
Send reprint requests to: Brian Kobilka, M.D., Howard Hughes Medical Institute, B-157, Beckman Center, Stanford University Medical School, Stanford, CA 94305-5428. E-mail:kobilka{at}cmgm.stanford.edu
-
↵1 Current address: Department of Pharmacology and Toxicology, The University of Kansas, 5001 Malott Hall, Lawrence, KS 66046.
-
↵2 Current address: Department of Cellular Physiology, Institute of Medical Physiology 12.5, The Panum Institute, University of Copenhagen, Blegdamsvej 3, DK-2200 Copenhagen N, Denmark.
-
↵3 Current address: Higuchi Biosciences Center, The University of Kansas, 5003 Malott Hall, Lawrence, KS 66045.
-
R.S. and K.W.-S. were supported by a research fellowship of the Deutsche Forschungsgemeinschaft.
Abbreviations
- β2-AR
- β2-adrenergic receptor
- GPCR
- G protein-coupled receptor
- AC
- adenylyl cyclase
- XTP
- xanthosine 5′-triphosphate
- XDP
- xanthosine 5′-diphosphate
- GsαL
- long-splice variant of Gsα
- GsαS
- short-splice variant of Gsα
- GTPγS
- guanine 5′-O-(3-thiotriphosphate)
- GppNHp
- guanylyl imidodiphosphate
- DHA
- dihydroalprenolol
- SAL
- salbutamol
- NTP
- nucleoside 5′-triphosphate
- cAMP
- cyclic AMP
- ICI
- [erythro-dl-1-(7-methylindan-4-yloxy)-3-isopropylaminobutan-2-ol]
- ISO
- isoproterenol
- NDP
- nucleoside 5′-diphosphate
- EPH
- (−)-ephedrine
- DCI
- dichloroisoproterenol
- DOB
- dobutamine
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}