


Abstract
In this study, we investigated the regulation of different G protein-coupled receptor (GPCR)-stimulated signaling pathways by GPCR kinase 2 (GRK2). We used thyrotropin receptor, which is coupled to different G proteins, to investigate the regulation of Gαs- and Gαq-mediated signaling (assessed by cAMP and inositol phosphate production, respectively). In transfected cells, both pathways were desensitized by GRK2. However a kinase-dead GRK2 mutant (GRK2-K220R) only decreased inositol phosphate production, indicating that GRK2 could regulate Gαq signaling through a phosphorylation-independent mechanism. Similar results were obtained with serotonin receptor 5-hydroxytryptamine2C, which is coupled to Gαq. This effect was mimicked by the N-terminal domain of GRK2 (GRK2-Nter), but not by the C-terminal domain. In cells transfected with Gαq, direct activation of Gαq signaling (by AlF4 −) was desensitized by GRK2-Nter, indicating an effect at the Gα-level. For comparison, in parallel samples we studied a protein regulator of G protein signaling RGS4 and we found a similar regulatory profile. We therefore hypothesized that the GRK2-Nter could directly interact with the Gαq subunit to regulate its signaling, as demonstrated for several RGS proteins. This hypothesis is further supported by the presence, within the GRK2-Nter, of an RGS homology domain. In direct binding experiments, we found that GRK2-Nter interacts with Gαq (only when activated) but not with Gαs and Gαo. We conclude that GRK2, besides desensitizing the GPCR by phosphorylation, is able to selectively bind to Gαq and to regulate its signaling.
G protein-coupled receptors (GPCR) mediate the signal transduction of a wide array of molecules ranging from neurotransmitters, hormones, chemokines, lipids to light and odorants. Upon binding of the agonist to its GPCR, the heterotrimeric G protein dissociates into the Gα- and Gβγ-subunits (Hamm, 1998). The dissociated subunits can either activate or inhibit a number of effector enzymes such as adenylyl cyclase, phospholipase C, ion channels, tyrosine kinases, and many others (Hamm, 1998).
Signal transduction by G protein-coupled receptors is strictly regulated by multiple mechanisms acting at different levels of signal propagation. After agonist stimulation, the receptor is desensitized by G protein-coupled receptor kinase (GRK) phosphorylation and subsequent binding of arrestin proteins (homologous desensitization) (Iacovelli et al., 1999b). The activated α-subunit of the G protein (Gα) can in turn be inhibited by regulators of G protein signaling (RGS) proteins (Berman and Gilman, 1998; De Vries and Farquhar, 1999; Hepler, 1999). These RGS proteins work by interacting with Gα and by increasing the intrinsic GTPase activity of Gα, acting as GTPase-activating proteins.
GRK family consists of six cloned members, named GRK1 to 6 (Iacovelli et al., 1999b). Three of these kinases were previously known as rhodopsin kinase (GRK1), βARK1 (GRK2), and βARK2 (GRK3), whereas GRK4, 5, and 6 were more recently cloned. Most of the studies on GPCR regulation by GRK have used GRK2 as a prototype kinase. It was shown by several laboratories that a variety of GPCRs can be regulated by GRK2 probably through phosphorylation, although a phosphorylation-independent mechanism has been reported for desensitization of parathyroid hormone receptor (Dicker et al., 1999). The general structure of GRK2 consists of a central catalytic domain (Phe191-Gly443), a C-terminal domain (Gly443-Leu689) involved in membrane targeting, and an N-terminal domain (Met1-Phe191) whose functional role is poorly defined. GRK2 interacts directly with the dissociated Gβγ and this interaction is important for membrane translocation and activation of the kinase. The pleckstrin homology domain that is present within the GRK2 C terminus is important for the binding to Gβγ. An RGS homology domain has been identified within the N terminus of GRK2 (Siderovski et al., 1996).
The existence of multiple mechanisms that regulate GPCR signaling may have relevant functional consequences. For example, GRK, which acts at the receptor level, should desensitize all the signaling pathways activated by one receptor, whereas the RGS proteins only desensitize the signaling activated by a given Gα-protein. However, this paradigm has not been tested experimentally.
In this study, we investigated the regulation of different GPCR-stimulated signaling pathways by GRK2. We found that GRK2, besides desensitizing the receptor by phosphorylation, is able to selectively regulate Gαq. GRK2 N-terminal domain directly interacts with the activated Gαq and regulates its signaling in a phosphorylation-independent manner. We conclude that GRK2 regulates different GPCR-mediated responses by multiple mechanisms that in part depend on the coupling to different G proteins.
Materials and Methods
Vectors.
To generate a fusion protein between glutathioneS-transferase (GST) and the N-terminal domain of GRK2 (GST-Nter), we used a polymerase chain reaction product of the GRK2 N-terminal region (Ala2-Thr187, GRK2-Nter) using as forward and reverse primer 5′-CGGGATCCGCGGACCTGGAGGC-3′ and 5′-TCAGGTCAGGTGGATGTTGAGC-3′, respectively (the native sequences are underlined). The polymerase chain reaction product was restriction digested with BamHI and ligated into pGEX-4T1 digested with BamHI and SmaI. Subsequently, the GRK2-Nter was subcloned in the eukaryotic expression vector pCDNA3HisC (Invitrogen, Carlsbad, CA) with the BamHI andNotI restriction enzymes to be used for transfection. The following plasmids were generous gifts: GRK2-K220R and GRK2 C-terminal domain (Gly495-Leu689, GRK2-Cter) from C. Scorer (Glaxo Wellcome, Stevenage, UK); GST-RGS4 in pGEX-4T2 from R. Neubig (University of Michigan, Ann Arbor); RGS4 in pCMV vector from J. Hepler (Emory University, Atlanta, GA); eukaryotic expression vectors bearing Gαs cDNAs, and baculoviruses encoding the Gαq subunit, the β1, and the His6-γ2 subunits of G protein from A. Gilman (University of Texas, Dallas); constitutively active Gαq-Q209L from N. Dhanasekaran (Temple University, Philadelphia, PA) (Dhanasekaran et al., 1994); thyrotropin receptor (TSHr) cDNA from L. D. Kohn (National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD); and 5-hydroxytryptamine (HT)2C receptor cDNA from A. Saltzman (Rhone-Poulenc Rorer Central Research, King of Prussia, PA).
Cell Culture, Transfection, and Second Messenger Measurements.
Human embryonic kidney (HEK)293 and COS7 cells were cultured in Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY) supplemented with 10% fetal calf serum and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin). We used COS7 cells for initial studies with TSHr, whereas subsequent experiments (5-HT2C receptor signaling) were performed on HEK293 cells. This was done for practical reasons because these two cellular models were already in use in our laboratory for the transfection of these receptors. One day before transfection, the cells were subcultured into the appropriate petri dishes at a density of 50,000 cells/cm2. HEK293 cells were transfected with the calcium phosphate precipitation method, and COS7 cells were transfected by DEAE-dextran procedure (Iacovelli et al., 1996). In cotransfection experiments performed in COS7 cells (TSHr regulation), 3 μg of TSHr plasmid was used along with 4 μg of GRK2, 2.5 μg of GRK2-K220R, 8 μg of GRK2-Nter, 4 μg of GRK2-Cter, 4 μg of RGS4, or 8 μg of empty vector, each 1 × 106COS7 cells. When experiments were performed in HEK293 cells (5-HT2C receptor regulation) half amounts of the cDNAs indicated above were used. The day after the transfection, cells were seeded into 24-well plates (180,000 cells/well) for inositol phosphate (IP) assay and into 48-well plates (80,000 cell/well) for cAMP assay. Seventy-two hours post-transfection, the cells were harvested and IP and cAMP accumulation were measured as previously described (Iacovelli et al., 1999a). Briefly, cells plated for IP assay were loaded overnight withd-myo-[3H]inositol (5 μCi/ml) in M199 serum-free medium. Cells were washed twice with prewarmed Hanks' balanced salt solution (HBSS), incubated for 15 min at 37°C in HBSS buffer containing 10 mM HEPES and 10 mM LiCl, pH 7.3, and then stimulated with the indicated agonists for 30 min. Total [3H]IP was extracted with percloric acid, neutralized, and analyzed by ion exchange chromatography on Dowex column. Experiments were performed in duplicate. For cAMP assay, cells were washed twice with prewarmed HBSS and added with HBSS containing 0.4% BSA, 10 mM HEPES, and 0.5 mM 3-isobutyl-1-methylxanthine. Incubations with indicated agonists were continued at 37°C for 30 min and the reaction was stopped by aspirating the incubation medium and adding ice-cold ethanol. The intracellular cAMP content was measured with a commercial radioimmunoassay. Experiments were performed in duplicate.
Preparation of Bacterial Recombinant Proteins.
Recombinant GST-RGS4 and GST-Nter fusion proteins were expressed inEscherichia coli BL21 by induction for 3 h with 1 mM isopropyl β-d-thiogalactoside. The fusion proteins were purified essentially according to Frangioni and Neel (1993). Briefly, the bacteria were lysed by sonication in STE buffer [10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM dithiothreitol (DTT), 1 mM EDTA] containing 100 μg/ml lysozyme, 1.5%N-laurylsarkosine, and protease inhibitor cocktails. After clarification by a centrifugation step, the lysate was incubated with glutathione agarose beads for 1 h at 4°C, washed eight times with ice-cold PBS, and placed in storage buffer I (75 mM HEPES, 150 mM NaCl, 5 mM DTT, 10% glycerol) until used.
Purification of Gαq Protein.
The purification of Gαq was performed according to Kozasa and Gilman (1995). Briefly, Sf9 cells were infected with baculoviruses encoding the Gαq subunit, the β1 subunit, and the His6-γ2 subunit (1 plaque-forming unit per cell for each virus). Seventy-two hours later, the cells were harvested in ice-cold lysis buffer and loaded onto a Ni-NTA column (Qiagen, Chatsworth, CA). The Gαq was eluted by an AlF4 −-containing buffer. The Gαq was inactivated by exchanging the elution buffer with storage buffer II [20 mM HEPES, pH 8.0, 100 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 3 mM DDT, 0.7% 3-[(3-cholamidopropyl)dimethylammino]propanesulfonate, 0.5 μM GDP] and used for protein-protein interaction experiments with GST-Nter or GST-RGS4.
Binding of G Proteins to RGS4 and GRK2 N-Terminal.
Cytosolic proteins (150 μg) from HEK293 cells (Iacovelli et al., 1996) transfected with the indicated Gα-subunits were mixed with 40 μl of slurry containing GST-RGS4, or GST-Nter fusion proteins bound to glutathione agarose beads in a final volume of 400 μl of binding buffer (20 mM Tris-HCl, pH 7.5, 1 mM DTT, 100 mM NaCl, 0.1% Lubrol, 10 μM GDP, 3 mM MgCl2), in the presence or absence of 47 mM MgCl2, 30 μM AlCl3, and 20 mM NaF. After 1 h at 4°C, the beads were washed three times with 1 ml of ice-cold binding buffer and the resins containing the eventual bound proteins were analyzed by immunoblotting (Iacovelli et al., 1996) after SDS-polyacrylamide gel electrophoresis, with anti-Gαq, anti Gα-common, or anti-Gαs serum (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-GST (Pharmacia Biotechnology, Arlington Heights, IL) antibodies. One fraction of starting material (30–40 μg, ∼25% of the total cytosolic proteins used for binding) also was included in the gel (indicated as S in Fig.5). All these experiments were performed with cytosol from HEK293 cells transfected with different Gα, which represents a convenient source of cytosolic Gα for binding experiments. Untransfected cells were not tested because the majority of Gα is membrane-associated.

Selective binding of GRK2-Nter to activated Gαq. A, schematic representation of GRK2 with the relevant functional domains (numbers indicate the amino acid position). B, top: recombinant purified GST-Nter fusion protein was incubated with untransfected HEK293 membranes in the absence (−) or presence (+) of 30 μM AlCl3 and 20 mM NaF to activate the Gα. Incubation (1 h at 4°C) was stopped by centrifugation (300,000g) and the GST-Nter bound to membranes was revealed by immunoblot with anti-GST antibody. Middle: recombinant purified GST conjugated to glutathione agarose beads was incubated (1 h at 4°C) with cytosolic proteins (150 μg) from Gαq-transfected HEK293 cells in the absence (−) or presence (+) of 30 μM AlCl3 and 20 mM NaF. After three extensive washings, Gαq bound to the column was revealed by immunoblot with anti-Gαq antibody. Starting material (S) (30–40 μg of cytosolic preparation, about one-fourth of total cytosol used for binding) also is included in the immunoblot. Bottom: recombinant purified GST-Nter conjugated to glutathione agarose beads was incubated (1 h at 4°C) with purified Gαq (100 ng) in the absence (−) or presence (+) of 30 μM AlCl3 and 20 mM NaF. After extensive washing, Gαq bound to the column was revealed by immunoblot with anti-Gαq antibody. C, recombinant purified GST-Nter or GST-RGS4 conjugated to glutathione agarose beads was incubated with cytosolic proteins from HEK293 cells transfected with the indicated Gα. Specific antibodies were used for different Gα. All procedures are as described in B. The experiments shown were repeated at least three times with similar results.
Results and Discussion
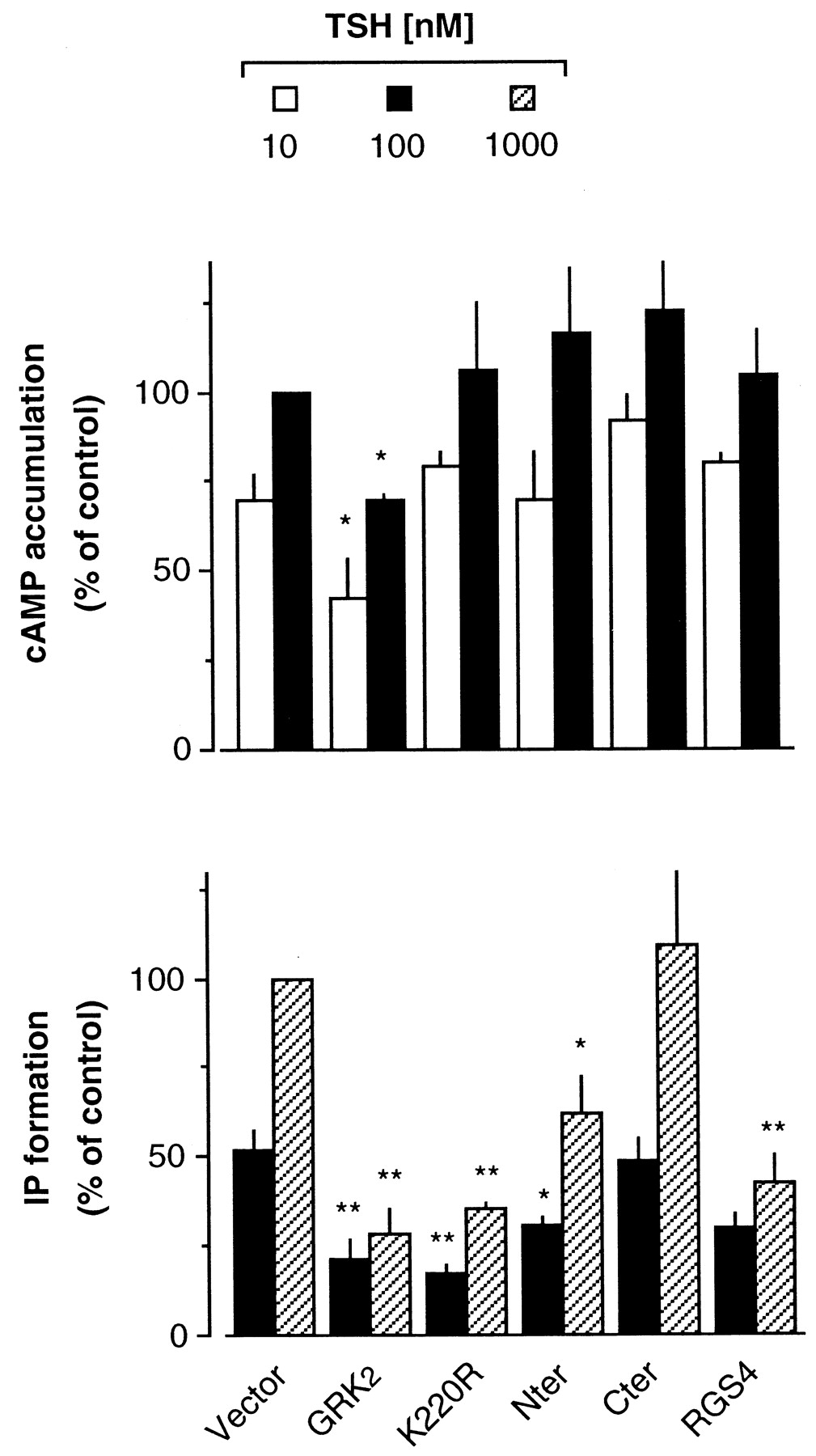
To investigate the regulatory role of GRK2 on different GPCR-stimulated second messengers, we used TSHr, which is able to couple to different G proteins and to activate different signaling pathways (Tornquist and Ahlstrom, 1993). We analyzed the regulation of Gαs- and Gαq-mediated signaling (assessed by measuring cAMP accumulation and IP production, respectively) when activated by TSH in COS7 cells transfected with TSHr. As previously reported (Tornquist and Ahlstrom, 1993) the doses of TSH that stimulate IP formation are about one order of magnitude higher than those that activate cAMP accumulation (Fig. 1). Cotransfection of GRK2 significantly decreased the TSH-stimulated responses (Fig. 1), consistent with desensitization of TSHr by GRK2, as previously reported by our group (Iacovelli et al., 1996, 1999a). The effect of GRK2 on Gαs- and Gαq-mediated responses (cAMP accumulation and IP production, respectively) was somehow different, and the TSH-stimulated cAMP response was desensitized to a lower extent than the receptor-stimulated IP production (Fig. 1). This may indicate that TSHr coupling to Gαs is more efficient than the coupling to Gαq, thus resulting in a lower sensitivity of Gαs to desensitizing mechanisms. Alternatively, one can hypothesize that there are additional mechanisms, possibly phosphorylation-independent as suggested in a recent report (Dicker et al., 1999), by which GRK2 could desensitize the TSHr-Gαq-stimulated response. To test the latter possibility, we used a kinase-dead GRK2 mutant (GRK2-K220R) in which the catalytic activity was disrupted (Kong et al., 1994). Overexpression of GRK2-K220R did not affect TSH-stimulated cAMP response, indicating that the levels of endogenous GRK2 are not sufficient to desensitize overexpressed TSHr, similar to what observed with other GPCRs (Ito et al., 1999). In contrast, TSH-stimulated IP production was substantially blunted by GRK2-K220R (Fig. 1), consistent with the idea that a phosphorylation-independent mechanism is selectively involved in the regulation of TSHr-Gαq-mediated signaling. To determine which is the domain of GRK2 involved in this effect, GRK2-Nter and GRK2-Cter were coexpressed with TSHr and the TSH-stimulated cAMP and IP responses were measured (Fig. 2). TSH-stimulated cAMP accumulation was not affected by the expression of these domains, further indicating that a phosphorylation-dependent mechanism is involved in the desensitization of this pathway. Gαq-mediated response was not affected by GRK2-Cter, but it was substantially blunted by GRK2-Nter, indicating that this is the domain involved in the regulation of TSH-Gαq-mediated response. For comparison, we studied the effect of RGS4 (coexpressed in parallel samples) on the different pathways stimulated by TSHr. RGS4 is known to bind to Gαq (but not to Gαs) and to desensitize the Gαq-stimulated intracellular response (Berman and Gilman, 1998; De Vries and Farquhar, 1999; Hepler, 1999). In our experimental model, RGS4 significantly reduced TSH-stimulated IP production, whereas it was ineffective on cAMP response stimulated by TSH (Fig. 2). This is in agreement with what previously reported for other receptors (Hepler et al., 1997).

Regulation of TSHr-mediated signaling by GRK2. COS7 cells were transiently cotransfected with TSHr plus vector (control, ●) or GRK2 (○) or GRK2-K220R (▪) cDNAs, and TSH-stimulated (30 min at 37°C) cAMP accumulation (top) or IP formation (bottom) were measured. Data (means ± S.E. of three experiments) are expressed as percentage of maximal stimulation (TSH, 100 nM for cAMP and 1000 nM for IP) in control cells. See Fig. 2 for statistical analysis.

Regulation of different TSHr-mediated signaling pathways. TSH-stimulated cAMP accumulation and IP formation in COS7 cells cotransfected with TSHr plus vector (control), GRK2, GRK2-Nter, GRK2-K220R, GRK2-Cter, or RGS4. TSH (30 min at 37°C) was used at 10 and 100 nM for cAMP and at 100 and 1000 nM for IP stimulation. Data (means ± S.E. of three or four experiments) are expressed as percentage of maximal stimulation (TSH, 100 nM for cAMP and 1000 nM for IP) in control cells. Statistically significant differences versus control are indicated (*P < .05 and **P < .01; Dunnett's test).
To see whether the effect of GRK2-Nter on GPCR-stimulated-Gαq signaling can be observed with other receptors, we used 5-HT2C, which is primarily coupled to Gαq (Berg et al., 1994) (Fig. 3). We transfected HEK293 cells to transiently express 5-HT2Creceptor and we measured the effect of GRK2-Nter coexpression on 5-HT-stimulated IP production. Untransfected HEK293 did not respond to 5-HT, whereas in transfected cells 5-HT (10 μM) stimulated IP production by ∼5-fold. Cotransfection of GRK2-Nter significantly reduced the 5-HT-induced IP accumulation, further indicating that the interaction of this domain of GRK2 with Gαq results in a negative regulation of Gαq-mediated signaling. Consistently, the coexpression of the kinase-dead mutant GRK2-K220R also inhibited 5-HT-induced IP production, further indicating that GRK2 can desensitize 5-HT2C-mediated signaling by a phosphorylation-independent mechanism. The coexpression of GRK2-Cter did not affect the 5-HT-stimulated IP production, confirming that this domain is not involved in Gq signaling regulation. Finally, 5-HT2C receptor-mediated signaling was desensitized by overexpression of wild-type GRK2. As expected, the overexpression of RGS4 significantly reduced 5-HT-stimulated IP accumulation. Overexpression of different proteins was confirmed by Western blot analysis (data not shown).

Regulation of 5-HT2C receptor-mediated signaling. 5-HT2C receptor-stimulated IP formation in HEK293 cells transfected with 5-HT2C receptor and cotransfected with the vector (control) or with GRK2, GRK2-Nter, GRK2-K220R, GRK2-Cter, and RGS4. Data (means ± S.E. of three or four experiments) are expressed as percentage of maximal-stimulation by 5-HT (10 μM for 30 min) in control cells. Statistically significant differences versus control are indicated (**P < .01; Dunnett's test).
These results indicate that the GRK2-Nter is able to desensitize GPCR-stimulated Gαq signaling. To assess whether GRK2-Nter regulates directly Gα, rather than acting at the level of the receptor, we overexpressed Gαq and activated its signaling by AlF4 − (Fig. 4). Coexpression of GRK2-Nter decreased Gαq-dependent IP formation stimulated by AlF4 −, indicating that GRK2-Nter directly inhibits Gαq signaling. In a parallel set of samples, we additionally cotransfected 5-HT2C receptor and included 10 μM 5-HT to activate the receptor. The presence of the receptor and 5-HT did not further increase IP formation, confirming that Gαq was already maximally stimulated by AlF4 −(IP formation was 21,700 and 22,800 cpm/well in cells with Gαq alone or Gαq plus 5-HT2C, respectively). However, the presence of the receptor enhanced the ability of GRK2-Nter to decrease Gαq signaling, indicating that the receptor contributes to the ability of GRK2-Nter to interact with Gαq. Cotransfection of RGS4 affected Gαq signaling in a similar manner, consistent with previous findings showing that the direct interaction of RGS4 with the receptor contributes to the ability of RGS4 to regulate G protein signaling (Zeng et al., 1998).

Regulation of Gαq signaling by GRK2-Nter and RGS4. HEK293 cells were transfected with Gαq (▪) plus the indicated amounts of GRK2-Nter (top) or RGS4 (bottom). In a parallel set of samples, cells were additionally cotransfected with 5-HT2Creceptor (■). In all the conditions, 30 μM AlCl3 and 10 mM NaF (30 min at 37°C) were included to activate Gαq. In cells expressing 5-HT2C receptor (■), 10 μM 5-HT was added to make the receptor in the active (agonist bound) conformation. Because Gαq was maximally activated by AlF4 −, the addition of 5-HT did not result in any further increase in IP formation. Data are expressed as percentage of maximal stimulation of cells transfected with Gαq plus 5-HT2C. The experiment shown is representative of two similar results.
The ability of GRK2-Nter to selectively regulate Gαq-mediated response is similar to the effect of RGS4 on signaling. We therefore hypothesized that this domain of GRK2 could directly interact with the Gαq subunit to regulate its signaling, as previously shown for several RGS proteins (Tesmer et al., 1997). This hypothesis is further supported by the presence, within the N-terminal domain of GRK2, of an RGS homology domain (Siderovski et al., 1996) (Fig.5A). To assess the possible interaction between GRK2-Nter and the activated Gα, we measured the binding of GST-Nter to plasma membranes obtained from untransfected HEK293 cells. The binding of GRK2-Nter to plasma membranes was substantially increased in the presence of AlF4 − used to activate endogenous Gα (Fig. 5B). This indicates a possible interaction of GRK2-Nter with the activated Gα. To directly assess this possibility, we prepared a GST-Nter fusion protein, which was used in binding experiments. The cytosolic proteins from cells transiently transfected with different Gα-subtypes were incubated with agarose-conjugated GST-Nter. Unbound proteins were removed by extensive washing and the Gα bound to GST-Nter was revealed by immunoblot. When the incubation was done in the presence of AlF4 − to activate the Gα, a substantial fraction of Gαq (estimated ∼25% of starting material) was bound to the GST-Nter (Fig. 5C). When incubated in buffer without AlF4 − (i.e., Gα is in the inactive state), only a negligible interaction between Gαq and GRK2-Nter was detected. The ability to bind to GRK2-Nter, when in the activated state, appeared to be specific for Gαq because Gαs and Gαo failed to bind to GST-Nter even when activated by AlF4 − (Fig.5C). The interaction between GRK2-Nter and Gαq is probably due to direct interaction between these proteins because it also was observed when using purified Gαq (Fig. 5B). In parallel experiments we measured the binding of GST-RGS4 fusion protein to different Gα (Fig.5C). According to previous reports (Tesmer et al., 1997), in the presence of AlF4 − we observed a significant binding of RGS4 to Gαo and Gαq, but not to Gαs (Heximer et al., 1997). We also assessed the ability of GST-Nter to bind to a Gαq-Q209L mutant that is constitutively active because this mutation abolishes its intrinsic GTPase activity (Dhanasekaran et al., 1994). This mutant is thought to be in the GTP-bound form (Berman et al., 1996). Expression of Gαq-Q209L in HEK293 cells increased IP levels by ∼6-fold (7,000 and 44,000 cpm for vector and Gαq-Q209L-transfected cells, respectively) in the absence of AlF4 −, confirming that this mutant is constitutively active. Similar level of expression of wild-type Gαq did not affect IP levels (7000 and 8000 cpm for vector and Gαq-transfected cells, respectively). A substantial binding of Gαq-Q209L to GST-Nter was observed even in the absence of AlF4 − and was not enhanced by the activating solution (Fig. 5C). In parallel experiments, we found that Gαq-Q209L failed to bind to GST-RGS4 both in the presence and in the absence of AlF4 −, consistent with the evidence that RGS4 did not bind to the constitutively activated Gαi (Berman et al., 1996). As negative control we show that Gαq does not bind to GST (Fig. 5B).
The interaction of GRK2-Nter with Gαq is probably due to the RGS box that is present in this region of GRK2. Although GRK2-Nter and RGS4 interact with Gαq in a similar manner, there are substantial differences. For example, GRK2-Nter was more selective than RGS4 in interacting with different Gα because it did not bind to Gαo, which is a member of the Gαi subfamily. Additionally, we showed that GRK2-Nter is able to bind to Gαq both in the AlF4 −- and in the GTP-bound form (i.e., Gαq-Q209L mutant), whereas RGS4 only interacted with the AlF4 −-activated form of Gαo and Gαq. The similarity between the GRK2 RGS homology domain and several RGS proteins ranged between 15 and 25% (data not shown). Some amino acids involved in the interaction between RGS4 and Gαi, including N128 that probably participates to the GTPase-activating protein activity (Tesmer et al., 1997), are not conserved in GRK2-Nter. This limited level of similarity probably accounts for the differences between GRK2-Nter and RGS4 in interacting with different Gα.
In this study, we have shown that GPCR-signaling cascade is regulated by multiple mechanisms and that GRK2 can exert different regulatory roles through different functional domains. The ability of GRK2 to phosphorylate the receptor in an agonist-dependent manner desensitizes the receptor, and as a consequence, all the receptor-activated intracellular signaling pathways. The ability of GRK2-Nter to interact selectively with Gαq decreases the Gαq-mediated signaling. When one receptor, such as TSHr, can activate different G proteins, only the Gαq-mediated pathway is inhibited by GRK2-Nter.
These findings provide a possible explanation from the results recently reported by another group (Dicker et al., 1999). They found that parathyroid hormone receptor-stimulation of IP production was inhibited by GRK2 in a phosphorylation-independent manner because the GRK2-K220R was also able to desensitize this signaling. The GRK2-Cter construct was ineffective. We suggest that the phosphorylation-independent regulation of this receptor is probably due to the ability of GRK2-Nter to selectively inhibit Gαq-mediated signaling, as extensively documented by this study.
GRK2 is a multidomain kinase that regulates GPCR signaling interacting with different proteins of the signaling cascade: although the catalytic domain phosphorylates the receptor (important for homologous desensitization), the pleckstrin homology domain interacts with dissociated Gβγ (important for kinase targeting to membranes), and the N terminus, probably through the RGS box, interacts with the activated Gαq (important for Gαq signaling regulation). Although many subtypes of GPCR and Gβγ interact with GRK2, we show herein that the interaction of GRK2-Nter with Gαq appears to be selective. Therefore, GRK2 regulates GPCR signaling in a complex manner and multiple interacting mechanisms are probably important for the fine-tuning of this machinery in living cells.
Acknowledgments
We thank J. R. Hepler and H. G. Dohlman for helpfull discussion and Giuliana Martarelli and Lucia Simigliani for expert assistance in the preparation of the figures.
Footnotes
- Received October 21, 1999.
- Accepted January 3, 2000.
-
Send reprint requests to: Antonio De Blasi, Consorzio Mario Negri Sud, via Nazionale 66030 S. Maria Imbaro, Italy. E-mail:deblasi{at}cmns.mnegri.it
-
↵1 Recipient of a fellowship granted by Progetto Speciale Ricerca Scientifica e Applicata nel Mezzogiorno PS35–93/IND.
-
This study was supported by Telethon-Italy (Grant 1238), the Associazione Italiana per la Ricerca sul Cancro, Consiglio Nazionale delle Richerche Target Project on Biotechnology, and EC Biomed 2 program-PL 963566.
Abbreviations
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- RGS
- regulator of G protein signaling
- GST
- glutathione S-transferase
- GST-Nter
- GST fusion protein with N-terminal domain of GRK2
- GRK2-Nter
- N-terminal domain of GRK2
- GRK2-Cter
- C-terminal domain of GRK2
- TSHr
- thyrotropin receptor
- 5-HT
- 5-hydroxytryptamine
- HEK
- human embryonic kidney
- IP
- inositol phosphate
- HBSS
- Hanks' balanced salt solution
- DTT
- dithiothreitol
- GRK2-K220R
- kinase-dead GRK2 mutant
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}