


Abstract
Metabotropic glutamate (mGlu) 5 is a G-protein-coupled metabotropic glutamate receptor that plays an important role as a modulator of synaptic plasticity, ion channel activity, and excitotoxicity. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP) is a highly potent, noncompetitive, selective, and systemically active antagonist of mGlu5 receptors. It binds to a novel allosteric site that resides within the seven-transmembrane domain of mGlu5 receptors. Using site-directed mutagenesis, [3H]MPEP binding, a functional Ca2+ mobilization assay, and rhodopsin-based homology modeling, we identified eight residues (Pro-6543.36, Tyr-6583.40, Leu-7435.47, Thr-7806.44, Trp-7846.48, Phe-7876.51, Tyr-7916.55, and Ala-8097.47) that are crucial for MPEP-binding to rat mGlu5 receptors. Four mutations, Y6583.40V, W7846.48A, F7876.51A, and A8097.47V, caused complete loss of [3H]MPEP binding and also blocked the MPEP-mediated inhibition of quisqualate-induced intracellular Ca2+ mobilization. To visualize these experimental findings, we have constructed a homology model based on the X-ray crystal of bovine rhodopsin and have suggested a possible binding mode of MPEP. We propose that MPEP via its interactions with a network of the aromatic residues including Phe-6583.40 in transmembrane (TM) 3 helix and Trp-7986.48, Phe-7876.51, and Tyr-7916.55 in TM6 helix prevents the movement of TM6 helix relative to TM3 helix, a step that is required for receptor activation, and consequently stabilizes the inactive conformation of mGlu5 receptor. In the TM6 region, we observed a striking similarity between the critical residues involved in MPEP-binding site with those of previously identified as 1-ethyl-2-methyl-6-oxo-4-(1,2,4,5-tetrahydro-benzo[d]azepin-3-yl)-1,6-dihydropyrimidine-5-carbonitrile-binding pocket of mGlu1, pointing to a common mechanism of inhibition shared by both antagonists.
Metabotropic glutamate (mGlu) receptors belong to family 3 of G-protein-coupled receptors (GPCRs), which play central roles as modulators of both glutamatergic and other major neurotransmitter systems in the CNS. Other members of the family 3 GPCRs include the GABAB, Ca2+-sensing, vomero-nasal, pheromone, and putative taste receptors (Bockaert and Pin, 1999). To date, the eight cloned mGlu receptors are divided into three classes based on their sequence similarities, signal transduction, and agonist rank order of potency. Group I (mGlu1 and -5) receptors are coupled to the activation of phospholipase C, group II (mGlu2 and -3) and group III receptors (mGlu4, -6, -7, and -8) are negatively coupled to cAMP production (Conn and Pin, 1997; De Blasi et al., 2001). In contrast to family 1 GPCRs, in which the agonist-binding site is located within the seven-transmembrane domain (7TMD), the family 3 receptors (including the mGlu receptors) are characterized by two distinctly separated topological domains: an exceptionally long extracellular amino-terminal domain (500-600 amino acids) that contains a venus flytrap module for the agonist binding (Kunishima et al., 2000), and the 7TM helical segments plus intracellular carboxyl-terminal domain that is involved in receptor activation and G-protein coupling (Parmentier et al., 2002). The 7TMD region of group I mGlus has also been shown to form a binding pocket for the novel class of compounds acting as positive or negative allosteric modulators (Gasparini et al., 2002; Mutel, 2002).
Although our knowledge of the precise role played by different mGlu receptors in physiological and pathophysiological processes of the CNS is rather limited, the recent discovery of the allosteric modulators of group I mGlu receptors has provided invaluable tools to explore their potential as therapeutic targets. 7-(Hydroxyimino)cyclopropan[b]chromen-1acarboxylic acid ethyl ester was the first subtype-selective antagonist of the mGlu1 receptor (IC50 = 6.5 μM at hmGlu1b) to be described (Annoura et al., 1996). Litschig et al. (1999) elucidated the site of action of 7-(hydroxyimino)cyclopropan[b]chromen-1a-carboxylic acid ethyl ester, which binds within the 7TMD of mGlu1, in close contact with the residues Thr-815 and Ala-818 of TM7. Likewise, 2-methyl-6-(phenylethynyl)-pyridine (MPEP) was the first highly potent, noncompetitive, selective, and systemically active mGlu5 receptor antagonist (IC50 = 36 nM at hmGlu5a in the IP accumulation assay) to be described (Gasparini et al., 1999). The mutational analysis has also suggested that [3H]MMPEP makes close contact with the amino acid residues Ala-810 in TM7 and Pro-655 and Ser-658 in TM3 of the mGlu5 receptor (Pagano et al., 2000).
mGlu5 receptors have a widespread expression in the CNS, with the highest density in hippocampus, caudate/putamen, lateral septum, cortex, and olfactory bulb (Shigemoto et al., 1993; Romano et al., 1995). Given their high expression in the limbic system, mGlu5 receptors might be expected to have a beneficial therapeutic role in behavioral and emotional processes of brain function. Indeed, in many preclinical studies, MPEP has been shown to display anxiolytic and antidepressant activity in various rodent models of anxiety and depression test paradigms (Spooren et al., 2000; Tatarczynska et al., 2001; Pilc et al., 2002; Wieronska et al., 2002); to reverse the akinetic deficits in a rat model of Parkinson's disease (Breysse et al., 2002); to reverse the induced thermal hyperalgesia in three models of nerve injury (Hudson et al., 2002); and to reduce the conditioned rewarding effects of cocaine (Chiamulera et al., 2001; McGeehan and Olive, 2003). Therefore, mGlu5 antagonists might represent an exciting new therapeutic avenue for the treatment of psychiatric and neurological disorders, including anxiety and depression, addiction, Parkinson's disease, and inflammatory pain (Spooren et al., 2001).
We have determined previously the critical amino acids that are located in the allosteric antagonist-binding pocket of mGlu1 in a rhodopsin-based model of the mGlu1 7TMD complexed with EM-TBPC, a highly potent, subtype-selective, and noncompetitive antagonist of rat mGlu1. Moreover, our mutational studies have validated the application of the rhodopsin-based model to 7TMD of family 3 GPCRs (Malherbe et al., 2003). In the present study, we have extended these studies to probe the allosteric antagonist-binding site of mGlu5 using site-directed mutagenesis, [3H]MPEP binding, Ca2+ mobilization assay, and molecular modeling. Here, we report on the identification of additional amino acid residues in the TM3, -5, -6, and -7 regions of the mGlu5 receptor, which are important molecular determinants of the high-affinity binding site of MPEP. To visualize these experimental findings, we have constructed a homology model based on the X-ray crystal of bovine rhodopsin (Palczewski et al., 2000) and have suggested a possible binding mode of MPEP.
Materials and Methods
Materials
[3H]MPEP (specific activity, 36 Ci/mmol) and l-quisqualic acid were obtained from Tocris Cookson Ltd. (Bristol, UK). MPEP and l-glutamate (mono Sodium salt) were obtained from Sigma (Buchs, Switzerland).
Plasmids, Cell Culture, and Membrane Preparation
cDNA encoding the rmGlu5a receptors in pBlueScript II was obtained from Prof. S. Nakanishi (Kyoto, Japan). All point mutants were constructed using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The entire coding regions of all point-mutants were sequenced from both strands using an automated cycle sequencer (Applied Biosystems, Foster City, CA).
HEK-293 cells were transfected as described previously (Malherbe et al., 2001). Forty-eight hours after transfection, the cells were harvested and washed three times with ice-cold phosphate-buffered saline and frozen at -80°C. The pellet was suspended in ice-cold 50 mM Tris-HCl buffer containing 10 mM EDTA, pH 7.4, and homogenized with a Polytron homogenizer (Kinematica AG, Basel, Switzerland) for 10 s at 10 000 rpm. After centrifugation at 48,000g for 30 min at 4°C, the pellet was resuspended in ice-cold 50 mM Tris-HCl buffer containing 0.1 mM EDTA, pH 7.4, homogenized, and recentrifuged as above. This pellet was further resuspended in a smaller volume of ice-cold 50 mM Tris-HCl buffer containing 0.1 mM EDTA, pH 7.4. After homogenization for 10 s at 10,000 rpm, the protein content was measured using the bicinchoninic acid assay method (Pierce, Lausanne, Switzerland) with bovine serum albumin as the standard. The membrane homogenate was frozen at -80°C before use.
[3H]MPEP Binding
After thawing, the membrane homogenates were resuspended and homogenized in 15 mM Tris-HCl, 120 mM NaCl, 100 mM KCl, 25 mM CaCl2, and 25 mM MgCl2 binding buffer at pH 7.4 to a final assay concentration of 20 μg of protein/well. Saturation isotherms were determined by addition of 12 [3H]MPEP concentrations (0.04-100 nM) to these membranes (in a total volume of 200 μl) for 1 h at 4°C. At the end of the incubation, membranes were filtered onto unifilter (96-well white microplate with bonded GF/C filter preincubated 1 h in 0.1% PEI in wash buffer; PerkinElmer Life Sciences, Boston, MA) with a Filtermate 196 harvester (PerkinElmer Life Sciences) and washed three times with ice-cold 50 mM Tris-HCl, pH 7.4 buffer. Nonspecific binding was measured in the presence of 10 μM MPEP. The radioactivity on the filter was counted (3 min) on a TopCount microplate scintillation counter with quenching correction after addition of 45 μl of MicroScint 40 (Canberra Packard GmbH, Dreieich, Germany) and shaking for 20 min. Saturation experiments were analyzed by Prism 3.0 (GraphPad Software, San Diego, CA) using the rectangular hyperbolic equation derived from the equation of a bimolecular reaction and the law of mass action, B = (Bmax × [F])/(KD + [F]), where B is the amount of ligand bound at equilibrium, Bmax is the maximum number of binding sites, [F] is the concentration of free ligand, and KD is the ligand dissociation constant. The experiments were performed three times in triplicate, except for the mutants P654S and P654S/S657C, for which experiments were performed four times; the mean ± S.D. of the individual KD and Bmax values were calculated and are reported in the Table 1. Statistical significance was determined using the two-tailed t test (Prism 3.0; GraphPad Software).
Binding properties of WT rmGlu5a and mutants
Saturation binding isotherms of [3H]MPEP were performed on membrane preparations from HEK-293 cells transiently transfected with the WT and point-mutated receptors as described under Materials and Methods. With exception of the mutants P654S and P654S/S657C, which are from four independent experiments, the values for other mutants are mean ± S.D. of the KD and Bmax, calculated from three independent experiments, each performed in triplicate. The mutations that affected the [3H]MPEP binding affinity are in bold. Statistical significance was determined using the two-tailed t test.
Intracellular Ca2+ Mobilization Assay
HEK-293 cells, which were grown to 80% confluence in high-glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and 100 μg/μl penicillin/streptomycin, were transfected with the wild-type or mutant mGlu5 receptor cDNAs in pcDNA3.1 using LipofectAMINE Plus reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instruction. Six hours after transfection, the DNA-transfection mixture was removed, and the cells were maintained in high-glucose Dulbecco's modified Eagle's medium with reduced l-glutamine (1 mM final) and 10% dialyzed fetal calf serum. Twenty-four hours after transfection, the cells were harvested and seeded at 6 × 104 cells/well in the poly-d-lysine-treated, 96-well, black/clear-bottomed plates (BD Biosciences, Palo Alto, CA). Forty-eight hours after transfection, the cells were loaded for 1 h at 37°C with 4 μM Fluo-4 acetoxymethyl ester (Molecular Probes, Eugene, OR) in loading buffer (1× Hanks' balanced saline solution and 20 mM HEPES). The cells were washed five times with loading buffer to remove excess dye and intracellular calcium mobilization, [Ca2+]i was measured using a fluorometric imaging plate reader (Molecular Devices, Menlo Park, CA) as described previously (Porter et al., 1999). The antagonist potency of MPEP on the wild-type or mutant mGlu5 receptors was determined using 10 nM quisqualate as agonist. The antagonists were applied 5 min before the application of the agonist. Responses were measured as peak increase in fluorescence minus basal, normalized to the maximal stimulatory effect induced by 10 nM quisqualate. Inhibition curves were fitted according to the Hill equation: y = 100/(1 + (x/IC50)nH), where nH is the slope factor as determined using Prism 3.0 (GraphPad Software). The mean ± S.E.M. values of the five to eight measurements from a minimum of three independent transfections were calculated. The relative efficacy (Emax) values of quisqualate and glutamate were calculated as fitted maximum of the dose-response curve of each mutated receptor expressed as a percentage of fitted maximum of the wild-type dose-response curve from cells transfected and assayed on the same day.
Residue Numbering Scheme. The position of each amino acid residue in the 7TMD of mGlu receptor is identified both by its sequence number (including the signal peptide) and by the generic numbering system proposed by Ballesteros and Weinstein (1995), which is shown as superscript. The amino acids in the extracellular loop EC2 are labeled `45′ to indicate their location between the helix 4 and 5. The highly conserved cysteine, thought to be disulfide bonded, was given the index number 45.50 (Swiss-Prot: opsd_bovin C187), and the residues within the EC2 are then indexed relative to the “50” position.
Molecular Modeling
Alignment. An alignment of the seven transmembrane helices of rmGlu5 toward the transmembrane helices of bovine rhodopsin (Protein Data Bank code 1f88) were obtained with help of our in-house program Xsae (C. Broger, unpublished work) using a modified version of CLUSTAL V (Higgins, 1992). Sequences were obtained from Swiss-Prot: mgr5_rat, P31424. The sequence of bovine rhodopsin was read directly from the rhodopsin structure (Protein Data Bank code 1f88).
Model Building. All modeling calculations were made on a Silicon Graphics Octane with a single MIPS RISC R12000 processor (SGI, Mountain View, CA) using our in-house modeling package Moloc (Gerber and Muller, 1995; Gerber, 1998) (http://www.moloc.ch). An initial C-α model of rat mGlu5 was built by fitting the aligned rmGlu5 sequence lacking the extracellular domain on the bovine rhodopsin template C-α structure. Loops were optimized with the Moloc C-α force field. In the next step, a full-atom model was generated. φ and ψ Angles were obtained for aligned amino acids from the rhodopsin template. χ Angles were also adopted from the bovine rhodopsin structure where possible or in case of nonidentical amino acids generated by using the most probable value applying the Ponder-Richards method (Ponder and Richards, 1987). An energy calculation of the initial full peptide structure revealed regions with bad van der Waals (vdW) contacts of amino acid side chains that were subsequently improved by manually adjusting the relevant χ angles. Repulsive vdW interactions were removed manually where necessary. The model was refined by keeping all backbone atoms in fixed positions and allowing only the side chains to move.
In the following optimization step, only C-α atoms were kept in a fixed position and all other atoms were allowed to move. In a third round of optimization, no atoms were kept stationary but constraints were applied to C-α atoms. The quality of the model was then checked with Moloc internal programs. MPEP was manually docked into the 7TMD region taking cis-retinal as template for location. Where necessary, nonconserved amino acid side chains in the rmGlu5 model were rotated such that no vdW conflicts with the antagonist occurred. All amino acid side chains reaching within 6 Å of the antagonist were subsequently included in a round of optimization.
Results
Alignment of 7TM Domains of the rmGlu Receptors toward Rhodopsin. To elucidate the binding mode of MPEP, an alignment of the seven transmembrane helices of the whole rmGlu family toward the transmembrane helices of bovine rhodopsin (Protein Data Bank code 1f88) was made. The inverse agonist of rhodopsin, cis-retinal, was used as a template for the location of MPEP. Amino acids, which were found 6.0 Å away from retinal in the X-ray crystal structure of rhodopsin (Palczewski et al., 2000; Teller et al., 2001), were considered likely candidates to affect binding of MPEP. The alignment of these amino acids of the rmGlu family toward the rhodopsin is shown in Fig. 1. From this preselection, amino acids in the TM3, -5, -6, -7, and EC2 regions were chosen for mutational studies.

Alignment of amino acids residues of 7TMD and the EC2 loop in the rmGlu sequences relative to bovine rhodopsin. The amino acids shown were found near the inverse agonist cis-retinal in the X-ray crystal structure of the bovine rhodopsin (6 Å, Protein Data Bank code 1f88) and were considered as guidance for mutational studies. The Ballesteros-Weinstein numbering scheme of the amino acids is given to facilitate the comparison with other GPCRs (see Materials and Methods). The conserved residues in each TM of rhodopsin, which is assigned to 50, are shown in the bottom row. Asterisks indicate identical residues between mGlu and rhodopsin. Colons indicate identical residues among mGlus. The critical residues involved in the cis-retinal-binding pocket of rhodopsin (Palczewski et al., 2000; Teller et al., 2001), EM-TBPC-binding pocket of rmGlu1α (Malherbe et al., 2003), and MPEP-binding pocket of rmGlu5a (this work) are shown in bold.
Generation of Point Mutations and the [3H]MPEP Binding.Fig. 2 shows the location of 13 amino acids chosen for the mutation studies. The choice of four residues, Pro-654, Ser-657, Ala-809, and Met-801, were based on the earlier report by Pagano et al. (2000). The choice of the nine new mutations in the present investigation were based on the alignment of the 7TMD of rmGlu5 toward bovine rhodopsin as described above and also our earlier study with the mGlu1 7TMD complexed with EM-TBPC (Malherbe et al., 2003). Therefore, 16 point mutations and one double-mutation in rat mGlu5a were introduced in the 7TMD region by site-directed mutagenesis. Saturation binding analyses were performed on membranes isolated from the HEK-293 transfected with the wild-type (WT) and mutated receptors using 0.04 to 100 nM concentrations of [3H]MPEP. The dissociation constants (KD) and the maximum binding sites (Bmax) derived from the saturation isotherms are given in Table 1. The mutations S657C, Y658F, N733A, and V788M did not significantly affect the [3H]MPEP binding affinity compared with the WT rmGlu5a receptor (Table 1). The mutation R647A led to an increase in binding affinity that was statistically significant (P < 0.05; two-tailed t test). However, four mutations, Y658V, W784A, F787A, and A809V, abolished [3H]MPEP binding to undetectable levels. The other mutations, P654S, P654S/S657C, L743V, L743A, T780A, W784F, and Y791A, led to statistically significant decreases of 40.0-, 9.0-, 3.9-, 4.7-, 4.6-, 3.3- and 4.9-fold, respectively, in binding affinity (Table 1). Although the mutation P654S led to a dramatic reduction in binding affinity by 40.0-fold, it was a surprise that the double-mutation P654S/S657C had a less marked effect on the binding affinity (KD values increased only by 9.0-fold). We observed that the Bmax values were reduced in the majority of the mutated receptors compared with the WT.

Amino acid sequence alignment of the TM3-TM7 region of the rmGlu1α with the rmGlu5a using the Pileup program. Asterisks indicate identical residues between mGlu1 and -5. The TM domains are boxed. The residues that have been mutated in the current work are shown in bold. EC, extracellular loop; IC, intracellular loop.
Effect of Mutations on MPEP-Mediated Inhibition of Quisqualate-Evoked [Ca2+]iResponse. In HEK-293 cells transiently transfected with the WT and mutated rmGlu5a receptors, quisqualate and glutamate elicited a concentration-dependent increase in intracellular free calcium [Ca2+]i, as monitored using the Ca2+-sensitive dye Fluo-4 and a fluorometric imaging plate reader. The EC50 and relative Emax values, calculated from concentration-response curves of quisqualate and glutamate in the cells expressing WT and mutated receptors, are given in Table 2. For the mutations R647A, Y791A, and L743V, reductions in the relative efficacy of quisqualate and glutamate were observed (Table 2), a fact consistent with the decreased Bmax values of these mutated receptors (Table 1). Nevertheless, this was not always the case; some mutations showed a decrease in Bmax but no decrease in Emax. We should mention, too, the large but not significant increase in Emax values of the mutation F787A for which no binding was detected. The basal activity of GPCRs is generally modulated by expression levels. Because of the large variability in the expression levels of the mutated receptors, as seen in Table 1, we do not attempt to address the issue of constitutive activity in the present report. However, we note the possibility that the basal activity of the receptors could be altered by these mutations, which may manifest itself as an alteration in Emax values compared with WT receptors. If one considers the inherent variability in a readout of a functional activity from a transient transfection and the differences in expression levels, all other mutated receptors elicited concentration-response curves for glutamate and quisqualate with EC50 and relative Emax values (Table 2) that were near those of WT mGlu5 receptor, always assayed at the same time. Thus the mutations seemed to have no significant effect on the agonist recognition site and the receptors were functionally active. However, coapplication of MPEP at various concentrations with 10 nM quisqualate (a concentration that gave 60 to 80% of maximum agonist response) in the cells expressing WT rmGlu5a resulted in a concentration-dependent inhibition of quisqualate-evoked [Ca2+]i response with IC50 = 13.0 nM. The concentration-dependent inhibition of quisqualate (10 nM)-evoked increases in [Ca2+]i by MPEP in the cells expressing various mutated receptors is shown in Fig. 3, A-F, and their derived IC50 and Hill slope (nH) values in Table 3.
The effect of mutations on mGlu5-mediated of agonist-induced [Ca2+]i response
EC50, Hill coefficient (nH), and relative efficacy (Emax) values for the quisqualate- and glutamate-induced [Ca2+]i response in the HEK-293 cells transiently transfected with the rmGlu5a WT and mutated receptors. For quisqualate, the data are presented as mean ± S.E. of five to eight dose-response measurements; for glutamate, they represent mean ± S.E. of two to five dose-response measurements from two or three independent transfections.

Effect of allosteric antagonist on quisqualate-evoked [Ca2+] in WT and mutated receptors. A to F, concentration-dependent inhibition of 10 nM quisqualate-stimulated increases in [Ca2+]i by MPEP as assayed using the Ca2+-sensitive dye Fluo-4 and a fluorometric imaging plate reader in HEK-293 cells transiently transfected with the rmGlu5a WT and mutated receptors. Responses are normalized to the first control response. Each curve represents the mean of five to eight dose-response measurements from a minimum of three independent transfections.
Effect of mutations on inhibition of quisqualate-induced [Ca2+]i response by MPEP
IC50 and Hill coefficient (nH) values for the inhibition by MPEP of quisqualate (10 nM)-evoked [Ca2+]i response in the HEK-293 cells transiently transfected with the rmGlu5a WT and mutated receptors. Data are presented as means ± S.E. of the five to eight dose-response measurements from a minimum of three independent transfections. The affected mutants are shown in boldface type.
The antagonist inhibition of quisqualate-induced [Ca2+]i response was not affected by the mutants S657C, Y658F, N733A, and V788M. In cells expressing four mutants, Y658V, W784A, F787A, and A809V, which did not bind [3H]MPEP, MPEP was not able to efficiently inhibit quisqualate-evoked [Ca2+]i response and thus resulted in the large increases in IC50 values (54.0-, 21.0-, 10.0-, and 45.0-fold, respectively). In good agreement with the binding experiments, the quisqualate-induced functional response with the point-mutant P654S was inhibited by MPEP with a marked increase in the IC50 value by 15.0-fold, whereas the double-mutant P654S/S657C again exhibited a less marked increase in the IC50 value, by 8.2-fold.
The mutants W784F, M801T, L743A, and T780A, which showed decreased radioligand binding affinities, also exhibited moderate increases in the IC50 values (2.0-, 2.0-, 5.3-, and 9.2-fold, respectively) compared with WT. Interestingly, the mutant Y791A exhibited a greater influence on the functional potency of MPEP (IC50 value increased by 22.4-fold) than that of binding affinity (KD is increased by 4.9-fold). The mutant R647A, which caused an increase in binding affinity, also resulted in an increase in MPEP functional potency IC50 value (3 versus 13 nM) relative to WT. Of note, however, was the apparent incomplete inhibition of quisqualate-induced response (∼80%; see Fig. 3B). In general, there was a good agreement between the binding and functional results.
Discussion
The novel allosteric site located within the 7TMD of group I mGlu receptors represents an important region for selective pharmacological modulation of receptor function. As we have shown previously for the interaction of mGlu1 7TMD with the selective mGlu anatgonist EM-TBPC, the X-ray crystal structure of rhodopsin could be applied as a template for the 7TMD region of family 3 GPCRs (Malherbe et al., 2003). Therefore, we have used the 7TM domains of rhodopsin as a model for the mGlu5-7TMD to gain further insight into the mechanism of action of MPEP, a potent negative modulator of mGlu5 receptors. cis-Retinal, the inverse agonist of rhodopsin receptors, served as a template for the location of MPEP within the TM domains. Thirteen amino acids of the TM regions near cis-retinal were selected as candidates for mutation studies. Of the 16 point mutations and the single double mutation that are located in TM3, -5, -6, and -7 and EC2 of rmGlu5a, we observed that the Y6583.40V, W7846.48A, F7876.51A, and A8097.47V mutations resulted in complete loss of the [3H]MPEP binding affinity and also blocked the MPEP inhibition of quisqualate-induced intracellular Ca2+ mobilization.
In the TM3 region, two residues, Tyr-6583.40 and Pro-6543.36, were found to play a critical role in MPEP-binding pocket. Because the conversion of tyrosine 658 (a residue conserved in all mGlus) to a valine abolished the [3H]MPEP binding whereas its replacement to a phenylalanine had no effect on affinity and potency, we speculate that the forces involved in the interaction between Tyr-6583.40 and MPEP are via π/π interactions. Interestingly, mutagenesis of Arg-6473.29 (a residue conserved in all mGlus) to alanine increased the binding affinity and potency of MPEP. In an earlier study, Pagano et al. (2000), using radioligand [3H]MMPEP to probe the MPEP binding pocket of human mGlu5 receptor, demonstrated that the three nonconserved residues Pro-6553.36, Ser-6583.39, and Ala-8107.47 are important molecular determinants for the binding of the [3H]M-MPEP. Furthermore, they have shown that two residues, Pro-6553.36 and Ser-6583.39, play an essential role in the selectivity and binding affinity of MPEP for hmGlu5 receptor. The double mutations P6553.36S and S6583.39C in hmGlu5 that converted these residues to homologous residues of hmGlu1 (Ser-668 and Cys-671) led to complete loss of [3H]M-MPEP binding. Now, we have found in our study that the mutation A8097.47V had a detrimental effect on binding affinity of [3H]MPEP, and the mutation P6543.36S led also to a dramatic reduction in binding affinity by 40.0-fold and potency by 15.0-fold, whereas the mutation S6573.39C had no significant effect on the affinity or potency of MPEP. Interestingly, in our model, the 3-methoxy group of [3H]M-MPEP used by Pagano and colleagues would be in the vicinity of residues Pro-655 and Ser-658. Steric hindrance caused by 3-methoxy group might explain this discrepancy between the results observed by Pagano et al. (2000) for [3H]M-MPEP and the results from present study with [3H]MPEP.
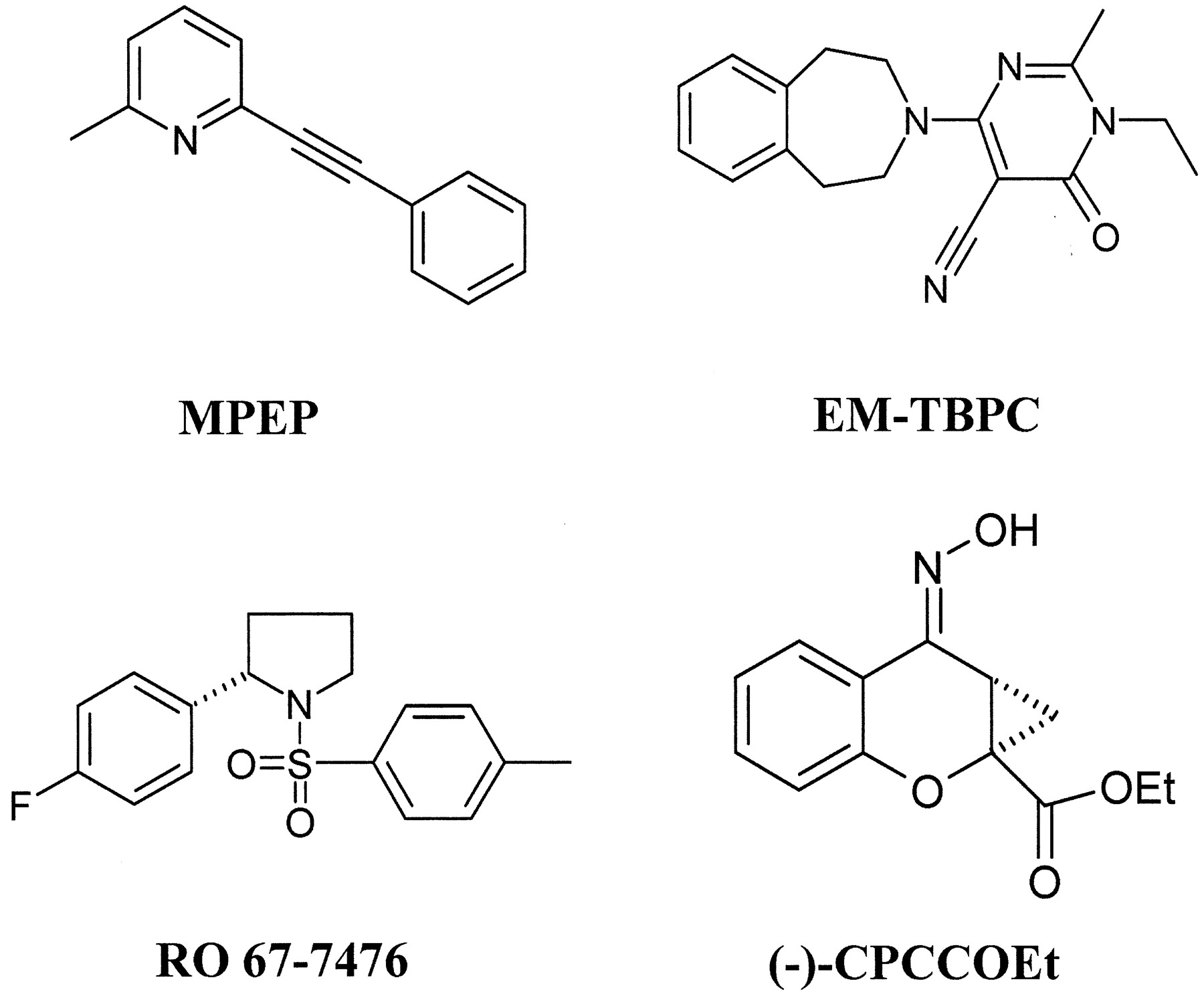
In the TM5 region, the amino acid Leu-7435.47, which is conserved among all rat and human mGlus (except for rat mGlu1, which carries a valine at this position), was another residue that seemed to play an important role in the binding of [3H]MPEP to mGlu5 receptors. Mutagenesis of Leu-7435.47 to alanine decreased the binding affinity of antagonist by 4.7-fold and its potency by 5.3-fold. Interestingly, we have shown previously that Val-7575.47 is a critical residue for the enhancing effect of the positive allosteric modulator of rat mGlu1, RO 67-7476 (Knoflach et al., 2001) and is similarly involved in the EM-TBPC-binding pocket of rmGlu1 (Malherbe et al., 2003). Therefore, even though the compounds RO 67-7476, EM-TBPC, and MPEP are from different chemical series (Fig. 4), this result may indicate that the amino acid (5.47) occupies a strategic position to gate the effect of positive and negative allosteric modulators.

Structures of some group I mGlu receptor allosteric modulators. (-)-CPCCOEt, 7-(hydroxyimino)cyclopropan[b]chromen-1a-carboxylic acid ethyl ester.
In the TM6 region, Trp-7846.48, Phe-7876.51, Thr-7806.44 (three highly conserved residues in all mGlus), and Tyr-7916.55 (groups I mGlu has tyrosine, groups II and III mGlus have phenylalanine at this position) were identified as important residues for the MPEP-binding pocket. Interestingly, the Y7916.55A mutation had a much higher influence on MPEP potency (IC50 increased by 22.4-fold) than that of binding affinity (KD increased by 4.9-fold). In Table 4, we compare the critical residues involved in the MPEP-binding site of rmGlu5 with those of cis-retinal-binding pocket of rhodopsin (Palczewski et al., 2000; Teller et al., 2001; Sakmar et al., 2002) or in other previously reported modulators of group I mGlu receptors (Litschig et al., 1999; Pagano et al., 2000; Knoflach et al., 2001; Malherbe et al., 2003) or in inverse agonists of human 5-hydroxytryptamine type 4 receptor (Joubert et al., 2002). As we reported previously for the EM-TBPC-binding pocket of rmGlu1 receptors, there is again a striking conservation in the position of critical residues. We found that eight residues, Pro-6543.36, Tyr-6583.40, Leu-7435.47, Thr-7806.44, Trp-7846.48, Phe-7876.51, Tyr-7916.55, and Ala-8097.47, are crucial in MPEP-binding site of rmGlu5 receptor. Interestingly, in TM6 region, the conserved residues Trp-7846.48, Phe-7876.51, and Tyr-7916.55 that are shown here to form MPEP-mGlu5 contact sites are homologous with those found to be the contact sites of EMTBPC with mGlul receptor (Malherbe et al., 2003). To visualize these mutation data, we have constructed a three-dimensional model of the 7TMD of the mGlu5 receptor in complex with MPEP using the atomic coordinates of bovine rhodopsin (Protein Data Bank code 1f88). Fig. 5 shows the amino acids in the TM region that affect the binding affinity and the potency of MPEP, suggesting a possible binding mode of this allosteric modulator.
Comparison of ligand-binding pocket of mGlu5 allosteric antagonist with those of cis-retinal-binding pocket of rhodopsin or in other previously reported modulators of group I mGlu receptors or in inverse agonist of h5HT4 receptor
The 18 residues located at a distance of 4.5 Å from cis-retinal in the 3D structure of rhodopsin are shown (Palczewski et al., 2000; Teller et al., 2001; Sakmar et al., 2002). The generic numbering system proposed by Ballesteros and Weinstein (1995) was used to compare residues in the 7TMD of the different GPCRs. The residues that have been reported to be located in the binding-sites are [3H]M-MPEP. of hmGlu5a (Pagano et al., 2000); [3H]EM-TBPC of rmGlu1 α (Malherbe et al., 2003); (−)-CPCCOEt of hmGlu1b (Litschig et al., 1999); RO 67-7476 of rmGlu1α (Knoflach et al., 2001); and inverse agonist of h5HT4 receptor (Joubert et al., 2002).

Molecular modeling of the rmGlu5 complexed with MPEP. Homology model of rmGlu5 based on the high-resolution structure of bovine rhodopsin. Three-dimensional side view of the 7TM binding pocket of the rmGlu5 receptor with a suggested binding mode of MPEP. Cα-traces are represented as colored ribbons: TM1, cyan; TM2, yellow; TM3, red; TM4, blue; TM5, orange; TM6, gray; and TM7, magenta. Residues shown (one-letter code) are known to be involved in binding of MPEP. Color coding of R647, P654, Y658, L743, T780, W784, F787, Y791, A809 (blue) and MPEP (magenta) atoms: oxygen, red; nitrogen, blue; carbon, white.
An emerging view on the activation mechanism of GPCRs is that a TM network of intramolecular interactions constrains the receptor in its inactive state. The binding of agonist disrupts the constraining interaction and induces a switch-like movement in the relative positions of two or more helices that results in the activation of GPCRs (Ballesteros et al., 2001a,b). The residue Trp-2656.48 of rhodopsin (Trp-7846.48 of rmGlu5 or Trp-7986.48 of rmGlu1), which is highly conserved in family 1 GPCRs and also in all mGlus, is known to act as a switch for the transition of the rhodopsin between different allosteric states. Several different studies, including spin-labeling and cross-linking (Farrens et al., 1996; Sheikh et al., 1996; Borhan et al., 2000), have demonstrated the requirement for a rigid-body movement of the TM6 helix away from the TM3 helix at the cytoplasmic side of the membrane upon rhodopsin activation and that the Trp-2656.48 serves to transmit the chromophore motion to TM6 helix. Similarly, in the three-dimensional structure of rhodopsin, the indole side chain of Trp-2656.48 comes within about 3.8 Å of the cis-retinal C20 (Palczewski et al., 2000; Teller et al., 2001). We propose that MPEP, via its interactions with a network of the aromatic residues, including Phe-6583.40 in the TM3 helix and Trp-7986.48, Phe-7876.51 and Tyr-7916.55 in the TM6 helix, prevents the movement of the TM6 helix relative to the TM3 helix, a step that is required for receptor activation, and consequently stabilizes the inactive conformation of mGlu5 receptor. For the family 1 GPCRs, it is well documented that the TM6 helix plays an essential role in the receptor activation (Ballesteros et al., 2001a,b). The fact that both MPEP and EM-TBPC have homologous contact sites with their respective receptors in the TM6 helix (a region highly conserved among all mGlu receptors) points at a common mechanism of inhibition shared by both antagonists. Furthermore, it has been shown that MPEP acts as an inverse agonist; it inhibited the agonist-independent (constitutive) activity in cells transiently over-expressing mGlu5 receptor (Pagano et al., 2000). Moreover, a recent report by Joubert et al. (2002) demonstrated that three residues of human 5-hydroxytryptamine type 4 receptor, Asp-1003.32, Trp-2726.48, and Phe-2756.51, are important molecular determinants for the effects of inverse agonists. Two of these residues are homologous to the critical residues Trp-7986.48 and Phe-7876.51 that we identified in the MPEP-binding pocket of rmGlu5. A similar observation was also noted previously for the EM-TBPC-binding pocket of rmGlu1 (Malherbe et al., 2003). Thus, it is plausible that two residues, Trp-7986.48 and Phe-7876.51, are part of network involved in the inverse agonist activity of MPEP.
In conclusion, in the present study, we demonstrate that as for mGlu1 receptors (Malherbe et al., 2003), rhodopsin-based homology modeling can be successfully used as a template to predict the critical residues involved in the allosteric binding-site crevice of mGlu5 receptors, another representative member of the family 3 GPCRs. Dysfunction of mGlu receptors could contribute to the pathology of major neurological diseases such as Alzheimer's and Parkinson's diseases as well as depression, schizophrenia, anxiety, and pain (Bordi and Ugolini, 1999). The knowledge of the molecular determinants that form the basis for the interaction between negative allosteric modulators and receptor activation could have an important utility for the design of therapeutic compounds for selective pharmacological modulation of mGlu receptor function.
Acknowledgments
We thank Prof. S. Nakanishi (Kyoto University, Kyoto, Japan) for the rmGlu5 receptor cDNA clone, Mr. Klaus Christensen for assistance in performing transfections, and Dr. Will Spooren for the critical reading of the manuscript.
Footnotes
-
ABBREVIATIONS: mGlu, metabotropic glutamate; GPCR, G-protein-coupled receptor; CNS, central nervous system; 7TMD, seven-transmembrane domain; TM, transmembrane; MPEP, 2-methyl-6-(phenylethynyl)-pyridine; M-MPEP, 2-methyl-6-(3-methoxyphenyl)ethynyl-pyridine; EMTBPC, 1-ethyl-2-methyl-6-oxo-4-(1,2,4,5-tetrahydro-benzo[d]azepin-3-yl)-1,6-dihydro-pyrimidine-5-carbonitrile; EC, extracellular loop; HEK, human embryonic kidney; VdW, van der Waals; WT, wild type; RO 67-7476, (S)-2-(4-Fluoro-phenyl)-1-(toluene-4-sulfonyl)-pyrrolidine.
- Received May 1, 2003.
- Accepted July 1, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}