


Abstract
We recently reported a novel class of compounds, represented by 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CD-PPB), that act as positive allosteric modulators (potentiators) of metabotropic glutamate receptor (mGluR) subtype 5. Studies of CDPPB analogs revealed that some compounds in this series serve also as positive allosteric modulators of mGluR1. Although CDPPB is selective for mGluR5 relative to other mGluR subtypes, several CDPPB analogs also showed 2.5-fold potentiation of glutamate-induced calcium transients in cells expressing mGluR1 at 10 μM, with 4-nitro-N-(1,4-diphenyl-1H-pyrazol-5-yl)benzamide (VU-71) being selective for mGluR1. In previous studies, we found that two structural classes of mGluR5-selective allosteric potentiators, including CDPPB, share a common binding site with the allosteric mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine. Negative allosteric modulators of mGluR1, regardless of structural class, have been reported to bind to a common allosteric antagonist site on this receptor. However, neither the novel CDPPB analogs nor previously identified allosteric mGluR1 potentiators [e.g., (S)-2-(4-fluorophenyl)-1-(toluene-4-sulfonyl)pyrrolidine (Ro 67-7476), ethyl diphenylacetylcarbamate (Ro 01-6128), and butyl (9H-xanthene-9-carbonyl)carbamate (Ro 67-4853)] displaced the binding of [3H]1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-2-phenyl-1-ethanone (R214127), a high-affinity radioligand for the allosteric antagonist site on mGluR1 at concentrations several orders of magnitude higher than those required to induce allosteric potentiation of mGluR1 responses. These data suggest that allosteric potentiators of mGluR1 act at a site that is distinct from that of allosteric antagonists of mGluR1. Site-directed mutagenesis revealed that valine at position 757 in transmembrane V of mGluR1a is crucial for the activity of multiple classes of allosteric mGluR1 potentiators.
In the mammalian central nervous system, glutamate is the major excitatory neurotransmitter, exerting its effects through activation of two major classes of glutamate receptors. These include cation channels, termed ionotropic glutamate receptors, and G protein-coupled receptors, termed metabotropic glutamate receptors (mGluRs) (Conn and Pin, 1997). The mGluRs are members of G protein-coupled receptor family 3, which consist of a large bilobed N-terminal extracellular domain containing the orthosteric agonist binding site, a seven-transmembrane domain, and a C-terminal intracellular domain (Gasparini et al., 2002; Kew, 2004). Based on sequence homology, pharmacology, and signal transduction, the eight known mGluR subtypes have been classified into three groups: group I (mGluR1 and -5), group II (mGluR2 and -3), and group III (mGluR4, -6, -7, and -8) receptors (Conn, 2003; Kew, 2004). The group I receptors couple to Gαq and phospholipase C, whereas group II and group III mGluRs couple to Gαi/o (Conn and Pin, 1997; Schoepp et al., 1999). Group I mGluRs have been implicated as potential therapeutic targets in various neurological disorders, including pain (Fisher et al., 2002; Adwanikar et al., 2004), anxiety (Roppe et al., 2004), Parkinson's disease (Conn et al., 2005), epilepsy (Nagaraja et al., 2005), schizophrenia (Epping-Jordan et al., 2005; Kinney et al., 2005), cognitive disorders (Campbell et al., 2004; Moghaddam, 2004), and drug abuse (Tessari et al., 2004; Rasmussen et al., 2005).
In the past decade, research efforts have been focused on the development of compounds that act as allosteric modulators of specific mGluR subtypes. Positive allosteric modulators, or allosteric potentiators, offer several potential advantages over orthosteric (glutamate-like) agonists, including greater receptor subtype selectivity, maintenance of activity-dependent receptor function, and the potential for reduced receptor desensitization (Conn, 2003). Using high-through-put screening assays, a number of novel positive allosteric modulators of group I mGluRs have been identified. To date, two chemical classes of allosteric mGluR1 potentiators have been reported: benzenesulfonylpyrrolidine derivatives, for which the prototypical compound is Ro 67-7476, and carbamic esters, a class that includes Ro 01-6128 and Ro 67-4853 (Knoflach et al., 2001).
3-Cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CD-PPB) is a selective positive modulator of mGluR5 (Lindsley et al., 2004; Kinney et al., 2005). We have synthesized a series of CDPPB analogs to investigate whether alterations in the chemical structure CDPPB would alter its pharmacological function or subtype selectivity (de Paulis et al., 2006). We now report that chemical modifications of CDPPB result in compounds that have lost their selectivity for mGluR5 and act as positive allosteric modulators of mGluR1. Allosteric antagonists of mGluR1, regardless of structural class, bind to a common antagonist site as evidenced by displacement of [3H]R214127, a high-affinity radioligand for this site (Lavreysen et al., 2003; Zheng et al., 2005). It is noteworthy that the present novel allosteric potentiators of mGluR1 together with three previously identified allosteric potentiators of mGluR1 (Ro 67-4853, Ro 01-6128, and Ro 67-7476) did not bind to this allosteric antagonist site at concentrations several orders of magnitude higher than those to potentiate mGluR1 activity. Finally, we identified a single point mutation that eliminates the activity of each of the structurally distinct mGluR1 potentiators. Together, our data suggest that all known allosteric mGluR1 potentiators interact at a single site that is distinct from that of multiple classes of negative allosteric modulators of mGluR1.
Materials and Methods
Materials. All CDPPB analogs were synthesized and characterized as described previously (de Paulis et al., 2006) and identified by the numbers taken from de Paulis et al. (2006). Unlabeled R214127 was synthesized according to Mabire et al. (2005). [3H]R214127 (25 Ci/mmol) was custom labeled by American Radiolabeled Chemicals (St. Louis, MO) from the corresponding bromo analog (Mabire et al., 2005). [3H]Quisqualate (31 Ci/mmol) was obtained from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Ro 67-4853, Ro 01-6128, and racemic Ro 67-7476 were synthesized as described previously (Knoflach et al., 2001). 3′-(((2-Cyclopentyl-6,7-dimethyl-1-oxo-2,3-dihydro-1H-inden-1-yl)oxy)methyl)biphenyl-4-carboxylic acid was synthesized as described previously (Galici et al., 2006). l-Quisqualic acid, l-glutamate, PHCCC, and l-(+)-2-amino-4-phosphonobutyric acid were obtained from Tocris Cookson Inc. (Ellisville, MO). Methotrexate was obtained from Calbiochem (San Diego, CA). The Calcium 3 assay kit was obtained from Molecular Devices (Sunnyvale, CA). Probenecid and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis, MO). All tissue culture reagents were obtained from Invitrogen (Carlsbad, CA). Timedmated pregnant Sprague-Dawley rats were obtained from Charles River Laboratories, Inc. (Wilmington, MA). Unifilter-96 GF/B plates and MicroScint-20 were obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). BioCoat poly-d-lysine 96-well culture plates were obtained from BD Biosciences Discovery Labware (Bedford, MA). QuikChange site-directed mutagenesis kit and Pfu Ultra high-fidelity DNA polymerase were obtained from Stratagene (La Jolla, CA). Complimentary oligonucleotides were obtained from Operon Biotechnologies (Huntsville, AL).
Cell Culture and Transfections. Baby hamster kidney (BHK) cells stably expressing the rat mGlu1a receptor were generously provided by Dr. Betty Haldeman (Zymogenetics, Seattle, WA). Cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 5% heat-inactivated fetal bovine serum (FBS), 2 mM GlutaMAX I, antibiotic-antimycotic (100 units of penicillin, 100 μg of streptomycin, and 0.25 μg of amphotericin B), 1 mM sodium pyruvate, 20 mM HEPES, and 250 nM methotrexate. Rat mGluR2 and the promiscuous G protein, Gqi5, were cotransfected into HEK293A cells using Lipofectamine 2000 (Invitrogen) and were grown in DMEM containing 10% heat-inactivated FBS, 2 mM GlutaMAX I, antibiotic-antimycotic, 0.1 mM nonessential amino acids, and 20 mM HEPES. Chinese Hamster Ovary (CHO) cells stably expressing the human (h) mGluR2 were transiently transfected with Gqi5 and CHO cells stably expressing the human mGluR4/Gqi5 were grown in DMEM containing 10% heat-inactivated dialyzed FBS, 2 mM GlutaMAX I, antibiotic-antimycotic, 1 mM sodium pyruvate, 20 mM HEPES, 5 nM methotrexate, and 20 μg/ml l-proline. All recombinant cell lines were plated at a seeding density of 7 to 8 × 105 cells/well, in clear-bottomed, poly-d-lysine-coated 96-well plates. Cells were then incubated in glutamate/glutamine-free medium overnight at 37°C in an atmosphere of 95% air, 5% CO2, with the exception of BHK cells stably expressing mGluR1a, which were maintained in regular medium.
Secondary astrocytic cultures were derived from neocortices of Sprague-Dawley rat pups (2-4 days old) and were prepared as described previously (Peavy et al., 2001). In brief, cells were harvested and maintained in growth medium containing DMEM, 10% heat-inactivated FBS, 2 mM GlutaMAX I, 20 mM HEPES, and antibiotic-antimycotic in tissue culture flasks. The medium was changed the following day, and cell cultures were maintained at 37°C for 1 week in 95% air, 5% CO2. Cells were shaken at 37°C overnight (280-310 rpm) to remove other types of glial cells. After shaking, the cells were trypsinized and plated at a seeding density of 3 × 105 cells/well in clear-bottomed, poly-d-lysine-coated 96-well plates and maintained in the same manner. One day after seeding, the medium was replaced with fresh medium containing G5-supplement (1:100 dilution). After the 3rd day in culture, cells were incubated overnight (16-24 h) in glutamine-free growth medium containing 10% dialyzed FBS. The following day, the cells were used in the calcium mobilization assay.
Functional Calcium Mobilization Assay. Cells were loaded with calcium indicator dye (Calcium 3 assay kit) at 37°C for 1 h. Dye was removed and replaced with the appropriate volume of assay buffer containing 1× Hanks' balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid, pH 7.4. CDPPB analogs were dissolved in 100% DMSO and then serially diluted into assay buffer containing 0.1% bovine serum albumin for a 5× stock. The stock solution was added to the assay plate to a final DMSO concentration of 0.1%. Glutamate and l-(+)-2-amino-4-phosphonobutyric acid were prepared to 10× stock solution in assay buffer before addition to assay plates. Calcium mobilization was measured using the FLEX Station II (Molecular Devices).
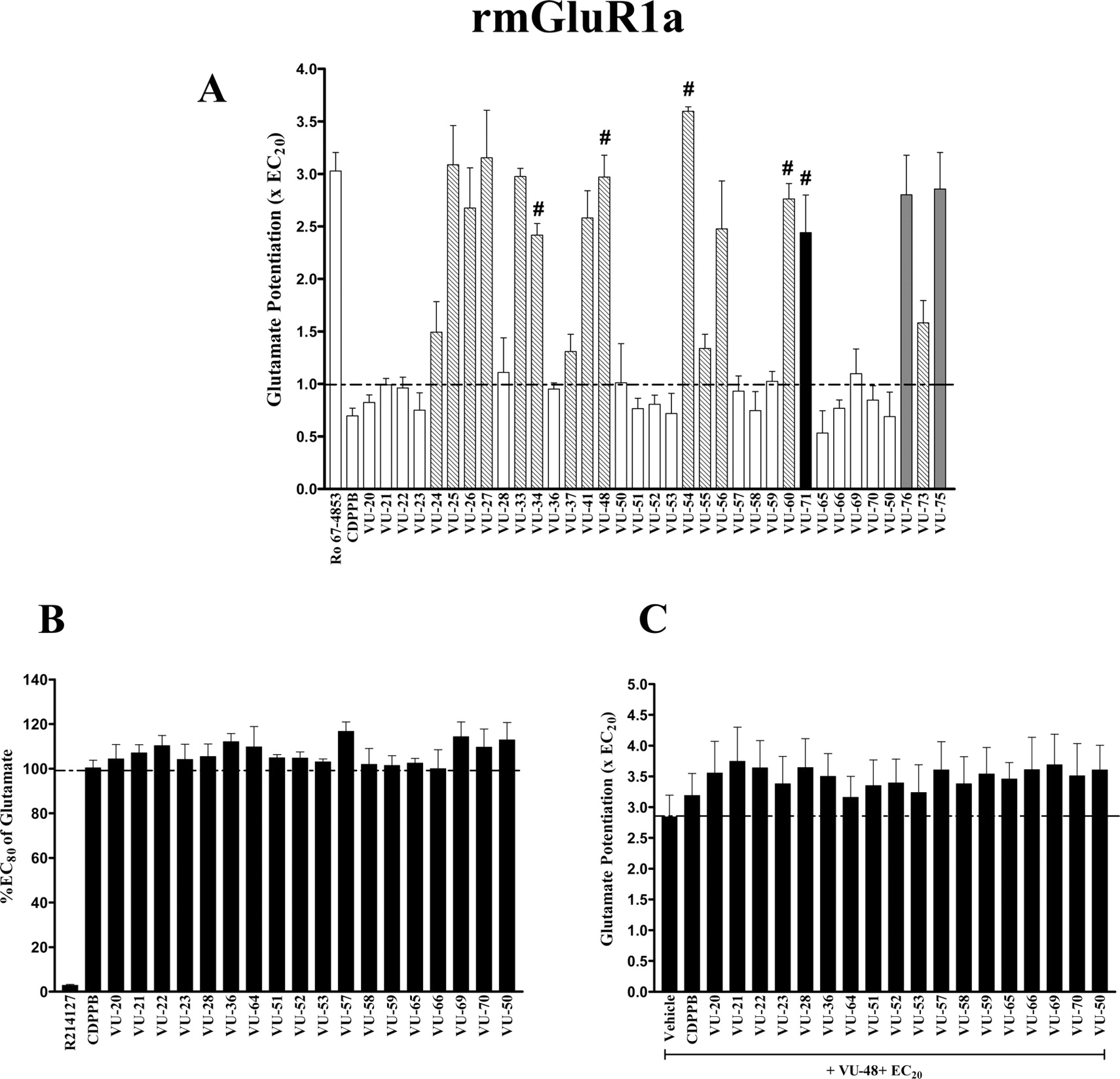
A series of CDPPB analogs were tested for allosteric glutamate-potentiating activity at rat mGluR1a (A). Calcium mobilization was measured using FLEXstation II where 10 μM glutamate was used as positive control. Cells were preincubated with test compounds (10 μM) for 5 min before the addition of a submaximal concentration (EC20) of glutamate. The black column indicates a compound (VU-71) that was a selective mGluR1 potentiator, whereas the hatched columns represent compounds that were potentiating glutamate at both mGluR1a and mGluR5. Compounds that showed little or no potentiating activity were further examined for allosteric antagonist activity at mGluR1a (B). Cells were preincubated with test compounds (10 μM) for 5 min before the addition of a near maximum concentration (EC80) of glutamate. R214127 (200 nM) was used as a positive control. To identify neutral ligands at an allosteric site on mGluR1 (C), cells were preincubated for 1 min with test compounds followed by 4-min incubation with compound VU-48, identified in the present study as an allosteric potentiator of mGluR1 and mGluR5. The recorded signal was normalized as percentage of the maximum response (EC100) and presented as potentiation above control level (EC20), indicated by the broken line. Five representative compounds, indicated by # that showed potentiation activity approximately 2.5-fold above the EC20 value, were selected for further characterization. Bar graphs illustrate the mean ± S.E.M. of at least three independent experiments, each performed in triplicate.
For experiments aimed at identifying allosteric potentiators or antagonists, cells were preincubated with test compounds for 5 min before the addition of a submaximal concentration (EC20) or a near maximum concentration (EC80) of agonist, respectively. For the experiment designed to identify neutral allosteric ligands for mGluR1 and mGluR5, cells were incubated for 1 min with test compounds followed by 4-min incubation with the allosteric potentiator VU-48. Cells were activated for 1 min with an EC20 concentration of glutamate. The recorded signal amplitude was normalized as a percentage of the maximal response to 10 μM glutamate, and potentiation above control (EC20) was determined.
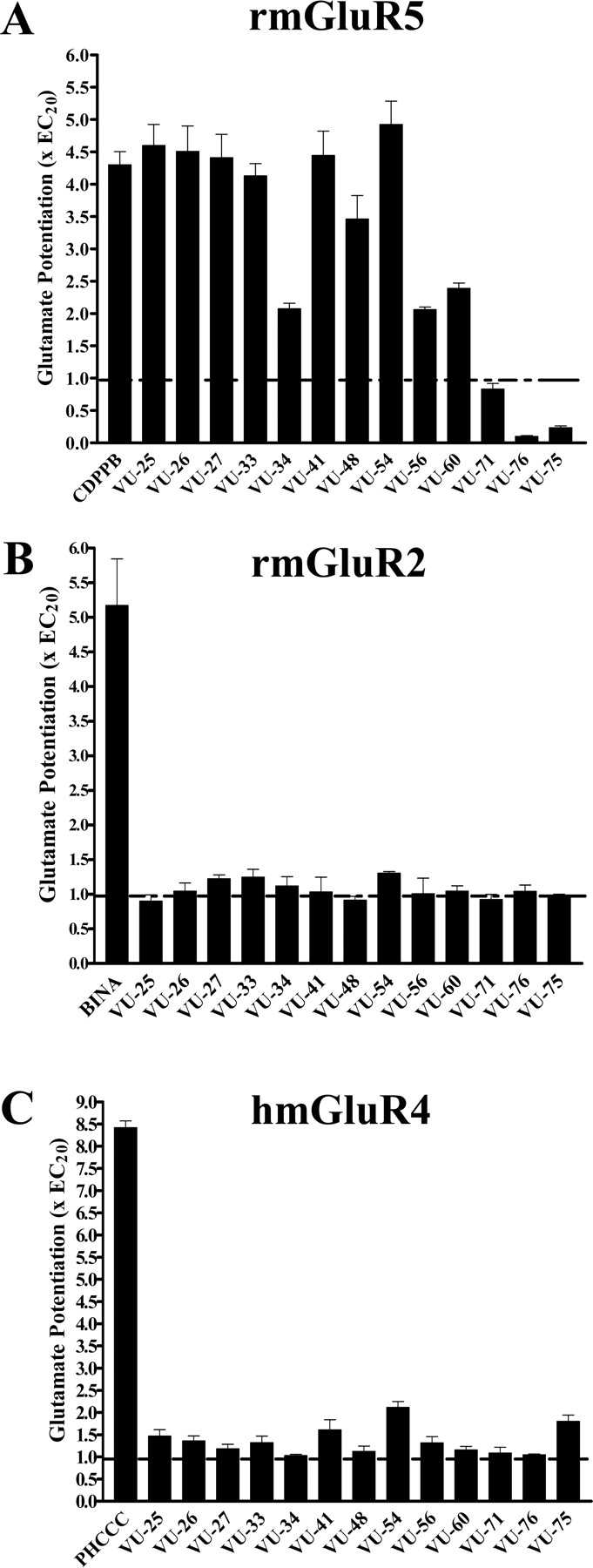
A subset of 13 allosteric potentiators of mGluR1 was tested for their selectivity on mGluR5 expressed in rat cortical astrocytes (A). The selective allosteric potentiators CDPPB (10 μM), 3′-(((2-cyclopentyl-6,7-dimethyl-1-oxo-2,3-dihydro-1H-inden-5 yl)oxy)methyl)biphenyl-4-carboxylic acid (BINA) (100 nM) (Galici et al., 2006), and PHCCC (10 μM) of mGluR5, mGluR2, and mGluR4, respectively, were used as positive controls. None of these compounds demonstrated potentiation activity on mGluR2 (B) or mGluR4 (C) with the possible exception of compounds VU-41, VU-54, and VU-75, which exhibited approximately 2-fold potentiating activity of mGluR4 when tested at 10 μM. Data were obtained and presented as described in Fig. 1.
Membrane Preparation and Radioligand Binding Studies. Membranes were prepared from BHK cells stably expressing rat mGluR1a. In brief, confluent cells were washed once with ice-cold phosphate-buffered saline. Cells were then harvested with a cell scraper and resuspended in ice-cold binding buffer containing 50 mM Tris-HCl, 1.2 mM MgCl2, and 2 mM CaCl2, pH 7.4, and were processed using a Polytron homogenizer (Kinematica, Basel, Switzerland) for 2 s. The homogenate was centrifuged at 20,000g for 20 min at 4°C. This last step was repeated twice, with homogenization between centrifugations for 10 and 5 s, respectively. The final pellets were resuspended and homogenized using a glass homogenizer (Dounce) and were stored in aliquots at -80°C until use. Protein concentrations were measured using the Bio-Rad Protein Assay kit (Bio-Rad, Hercules, CA) using serum albumin (Pierce Chemical, Rockford, IL) as the standard. After thawing, the membranes were resuspended and homogenized in ice-cold buffer as described above using a glass homogenizer. Binding experiments were prepared as described previously (Lavreysen et al., 2003) with a minor modification. Compounds were dissolved in 100% DMSO, diluted into assay buffer to a 5× stock, and added to the assay for a final DMSO concentration of 0.1%. All binding reactions were performed in 96-well deep plates in a final volume of 100 μl. For [3H]R214127 binding, assay mixtures containing 10 μg of membrane protein, appropriate concentrations of test compounds, and 2.5 nM [3H]R214127 were incubated for 30 min at 4°C. Nonspecific binding was determined in the presence of 1 μM R214127. For [3H]quisqualate binding, assay mixtures containing 20 μg of membrane protein, appropriate concentrations of test compounds, and 10 nM [3H]quisqualate were incubated for 1 h at room temperature. Nonspecific binding was determined in the presence of 1 mM glutamate. The binding equilibrium was terminated by rapid filtration through Unifilter-96 GF/B filter plates (presoaked with ice-cold binding buffer), and the filter plates were washed three times with ice-cold binding buffer using a 96-well Brandel harvester (Brandel Inc., Gaithersburg, MD). Filter plates were dried and filled with 30 μl of MicroScint-20, and the radioactivity was recorded by TopCount NXT microplate scintillation and luminescence counter (PerkinElmer Life and Analytical Sciences).
Site-Directed Mutagenesis. The cDNA encoding rat mGluR1a in the pGTh backbone (Grinnell et al., 1991) was generously provided by Dr. M. Baez (Eli Lilly & Co., Indianapolis, IN). Point mutations were generated using the QuikChange site-directed mutagenesis kit according to the manufacturer's instructions (Stratagene). Complimentary oligonucleotides were designed to contain the desired mutation(s), as well as a novel restriction site used for screening purposes, which does not alter the amino acid sequence. Sense and antisense oligonucleotides, based on the following sequences, were used to introduce the single mutation (V757L) and a novel NarI site (5′-GGTGTAGTGGCGCCTTTGGGTTACAATGGACTC-3′), or the double mutation (T815M, A818S) in combination with a novel BsmBI site, (5′-AAGATCATCACTATGTGCTTCAGCGTCTCCCTCAGTGTGACG-3′), into the mGluR1a sequence. Polymerase chain reaction amplification was performed using Pfu Ultra high-fidelity DNA polymerase. Final constructs were verified by sequencing at Vanderbilt University DNA sequencing facility (Nashville, TN) using a DNA analysis system (Applied Biosystems, Foster City, CA).
Results
CDPPB Analogs Positively Modulate Recombinant mGluR1a Activity in a Calcium Mobilization Assay. CDPPB is the prototypical member of one of four known structural classes of mGluR5 allosteric potentiators. Thus far, each of the mGluR5 potentiators that have been characterized is selective for mGluR5 relative to other mGluR subtypes. We recently synthesized a range of novel compounds based on the CDPPB scaffold (de Paulis et al., 2006). CDPPB is selective for mGluR5 relative to other mGluR subtypes. Because of the homology between mGluR1 and mGluR5, we sought to determine whether CDPPB analogs with activity at mGluR1 could be identified and whether these analogs might exhibit a range of activities at mGluR1a in a manner similar to that observed for other allosteric modulators of mGluR5 (O'Brien et al., 2003; Rodriguez et al., 2005). To test this hypothesis, compounds were analyzed for their ability to alter glutamate-induced calcium mobilization in BHK cells stably expressing rat mGluR1. We found that some compounds in this class act as allosteric potentiators of mGluR1 in addition to mGluR5 (Fig. 1A). Thus, unlike CDPPB, several compounds in the series showed potentiation of the response of mGluR1 to an EC20 concentration of glutamate. Because members within a single structural class of ligands at the MPEP-sensitive allosteric site on mGluR5 can have a range of activities from allosteric antagonists to potentiators as well as neutral allosteric ligands (O'Brien et al., 2003; Rodriguez et al., 2005), we wanted to determine whether those compounds that showed no potentiating effects on mGluR1 have antagonist or neutral allosteric activity on mGluR1. To identify allosteric antagonist activity on mGluR1, these compounds were tested for their ability to inhibit the effect of an EC80 concentration of glutamate (Fig. 1B). The response to an EC80 concentration of glutamate was not attenuated by any analog, suggesting that none of the compounds tested have allosteric antagonist activity. In contrast, the selective allosteric mGluR1 antagonist R214127, at 200 nM, markedly blocked the glutamate-induced calcium response. Compounds that showed no potentiator or antagonist effects were also examined to determine whether they act as neutral allosteric site ligands (Fig. 1C). Again, none of these analogs exhibited neutral allosteric activity, because they failed to inhibit the allosteric potentiation response to VU-48, a potent but unselective allosteric mGluR1 potentiator identified in the present study.

A, allosteric potentiation of mGluR5 response by VU-48 was not inhibited by the selective mGluR1 potentiator VU-71. Rat cortical astrocytes expressing mGluR5 were preincubated for 1 min with VU-71, followed by 4-min incubation with 10 μM VU-48. Cells were then stimulated with EC20 concentration of glutamate for 1 min. Bar graphs illustrate the means of three independent experiments, where error bars represent S.E.M. B, VU-75 and VU-76 concentration dependently inhibited an EC80 concentration of glutamate. Rat cortical astrocytes were preincubated with various concentrations of VU-75 or VU-76 for 5 min, and an EC80 concentration of glutamate was added. The fluorescence responses were normalized as a percentage of the EC80 concentration of glutamate (10 μM) and are reported as means ± S.E.M. of three individual experiments performed in triplicate.
Thirteen of the compounds that elicited the most robust potentiating response at mGluR1 at 10 μM were chosen to test their ability to potentiate glutamate-induced response mediated by mGluR5 as well as by representative group II (mGluR2) or group III (mGluR4) receptors. The majority of these compounds also potentiated the activation of mGluR5 by a submaximal concentration of glutamate (Fig. 2A) but had no effect in the absence of glutamate (data not shown). No potentiation was seen with VU-71, whereas compounds VU-75 and VU-76 were found to be antagonists at mGluR5. The selective mGluR2 potentiator 3′-(((2-cyclopropyl-6,7-dimethyl-1-oxo-2,3-dihydro-1H-inden-1-yl)oxy)methyl)biphenyl-4-carboxylic acid) (Galici et al., 2006) and the selective mGluR4 potentiator PHCCC (Maj et al., 2003) induced robust potentiation of mGluR2 (Fig. 2B) and mGluR4 (Fig. 2C), respectively. In contrast, all CDPPB analogs were devoid of potentiating activity in cells expressing mGluR2 (Fig. 2B) or mGluR4 (Fig. 2C), with the possible exception of compounds VU-41, VU-54, and VU-75, which exhibited approximately 2-fold potentiating activity of mGluR4 when tested at 10 μM (Fig. 2C).
Several CDPPB analogs showed no allosteric potentiating activity at mGluR5 (de Paulis et al., 2006). These compounds could be completely inactive at mGluR5 or could act as allosteric antagonists or neutral allosteric site ligands at this receptor. Therefore, we tested the effects of each of these compounds in studies aimed at determining whether they have mGluR5 antagonist or neutral activity (Fig. 3, A and B). We found that compound VU-71 was devoid of potentiating activity at mGluR5 (Fig. 2A) and did not exhibit neutral allosteric activity at mGluR5 (Fig. 3A), whereas compounds VU-73 and VU-76 were confirmed as antagonists of mGluR5 (Fig. 3B). The results indicate that, although a number of compounds in this structural class were identified as positive modulators at both mGluR1 and mGluR5 (Fig. 1A, hatched columns), one compound, VU-71, was found to be a selective mGluR1a potentiator (Fig. 1A, black column). In addition, compounds acting as both antagonists of mGluR5 and potentiators of mGluR1 (Fig. 1A, gray columns) were discovered. Furthermore, other compounds that had been previously characterized as allosteric potentiators of mGluR5 (de Paulis et al., 2006) showed no potentiating effects at mGluR1 (Fig. 1A, open columns).

Chemical structures of representative novel allosteric potentiators of mGluR1 and mGluR5.

Novel CDDPB analogs VU-34, VU-48, VU-54, VU-60, and VU-71 have no agonist activity on mGluR1 when added alone but shift the concentration-response curve of glutamate approximately 2- to 3-fold to the left. Cells were preincubated with a fixed concentration (10 μM) of each compound for 5 min before the addition of a range of concentrations of glutamate. The fluorescence response was normalized as a percentage of the maximal response to 10 μM glutamate and is presented as the mean of three individual experiments performed in triplicate. Error bars are S.E.M.

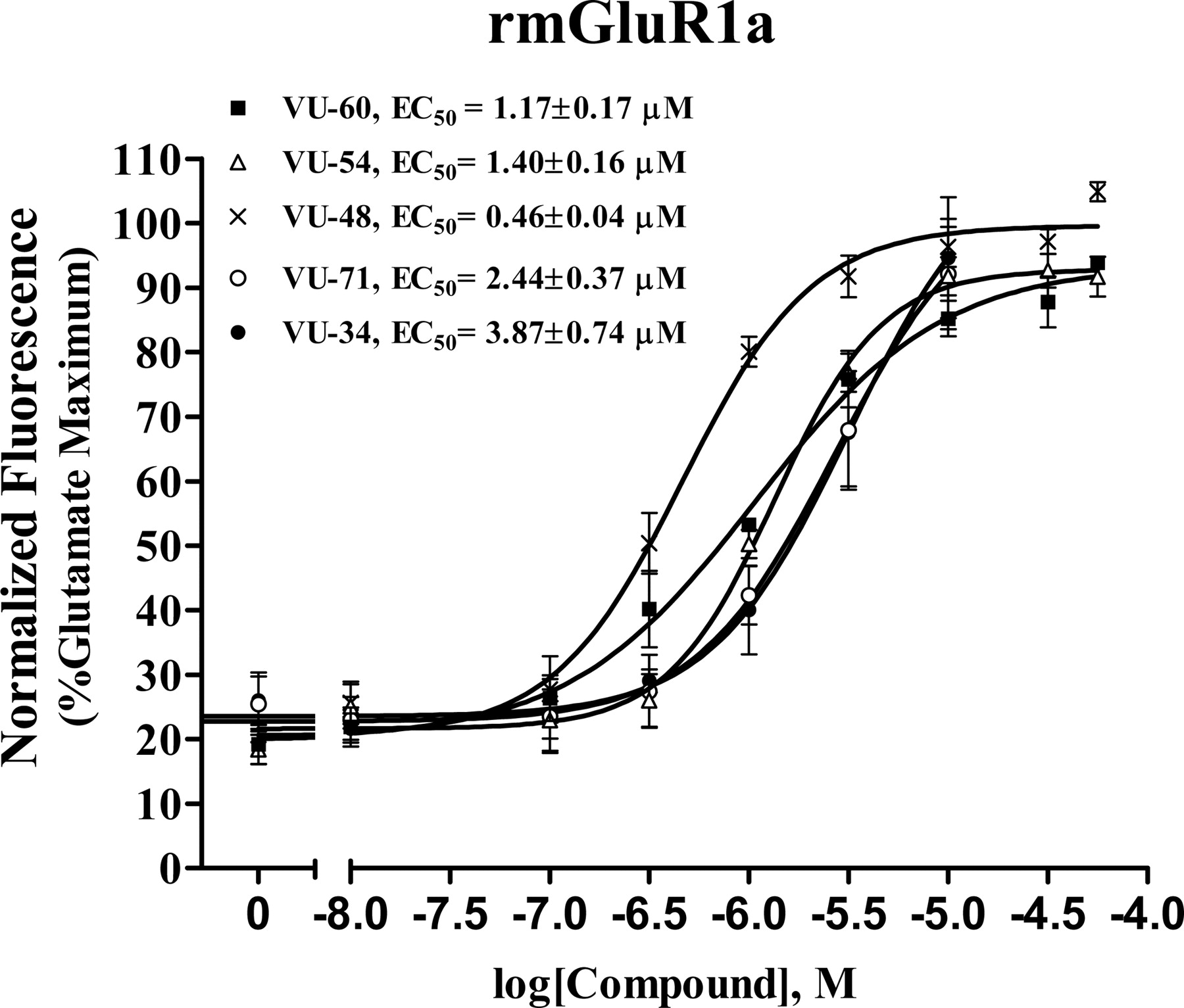
Concentration-response curves of VU-34, VU-48, VU-54, VU-60, and VU-71 in the presence of an EC20 concentration of glutamate in BHK cells stably expressing mGluR1a. These compounds potentiated threshold responses to glutamate in the calcium mobilization assay 3-fold, with EC50 values ranging from 0.47 to 3.87 μM. The fluorescence responses were normalized as a percentage of the maximum response to 10 μM glutamate and are presented as the mean of at least three individual experiments performed in triplicate. Error bars are S.E.M.
Five of 13 compounds that exhibited robust potentiation at mGluR1 (10 μM) were selected (indicated by # in Fig. 1A) for further characterization of their interaction with mGluR1a. The chemical structures of these compounds are shown in Fig. 4. Each of these compounds (10 μM) shifted the concentration-response curve of glutamate approximately 2- to 3-fold to the left (Fig. 5). In addition, these compounds produced a concentration-dependent potentiation of the response of mGluR1a to the EC20 concentration of glutamate (200-300 nM). The maximum potentiation of EC20 glutamate response was approximately 3-fold, with EC50 values ranging from 0.5 to 3.9 μM (Fig. 6). We have shown previously that the activity of CDPPB analogs at mGluR5 can be drastically reduced by having two methoxy groups in the benzamide moiety (de Paulis et al., 2006) [i.e., VU-34 (Fig. 4) and its 3,4-diOMe isomer, VU-33]. Compounds VU-33 and VU-34 potentiated glutamate response 3.0- and 2.4-fold, respectively. The compound with the highest activity was VU-48. It has a similar structure as CDPPB, except having a bromo atom in the ortho-position of the 1-phenyl ring and a para-nitro group instead of the meta-cyano group in the benzamide ring (Fig. 4). It is noteworthy that the positional isomer VU-65 of CDPPB was inactive at mGluR1 (Fig. 1A), as was the para-nitro analog VU-66, but the corresponding 4-phenyl analog VU-71 was active with an EC50 of 2.4 ± 0.4 μM. This suggests that the structural requirement for positive allosteric modulating activity at mGluR1 is different from that of mGluR5. The three compounds with the highest activity (VU-48, VU-54, and VU-60) were selected for further analysis. To confirm the noncompetitive nature of the binding of these compounds at mGluR1, the ability of these compound to displace the radiolabeled orthosteric agonist [3H]quisqualate was evaluated. When assessed up to a concentration of 100 μM, none of the test compounds displaced 10 nM [3H]quisqualate binding (Fig. 7). These data suggest that none of these compounds have affinity for the orthosteric agonist binding site.

Novel mGluR1 potentiators VU-48, VU-54, and VU-60 do not alter [3H]quisqualate binding. Membranes were prepared from BHK cells stably expressing mGluR1a and were incubated with 10 nM [3H]quisqualate, in a final volume of 100 μl for 1 h at room temperature in the presence of varying concentrations of quisqualate or CDPPB analogs. Bound and free radioligand were separated by vacuum filtration through Unifilter-96 GF/B filter plates. Nonspecific binding was determined with 1 mM glutamate. Binding of 10 nM [3H]quisqualate was displaced by unlabeled quisqualate but not by CDPPB analogs. The present data are from three independent experiments performed in triplicate. Error bars are S.E.M.
Positive Allosteric Modulators of mGluR1 Interact at a Site That Is Distinct from the Binding Pocket of Negative Allosteric Modulators of mGluR1. Previous studies have shown that both allosteric potentiators of mGluR5 (e.g., CDPPB) and allosteric antagonists (e.g., MPEP) interact at a common site (O'Brien et al., 2003; Kinney et al., 2005). In addition, a recent study revealed that all known allosteric mGluR1 antagonists (for review, see Mabire et al., 2005) bind to a common allosteric site on mGluR1. Based on these findings, we sought to determine whether the novel CDPPB analogs that are positive allosteric modulators of mGluR1 interact with this allosteric antagonist site. To address this question, we measured the ability of CDPPB analogs to displace the binding of the high-affinity radioligand [3H]R214127 in membranes prepared from BHK cells expressing mGluR1. As expected, unlabeled R214127 potently displaced binding of the radioligand at this site (Fig. 8). However, none of the selected mGluR1 potentiators, VU-48, VU-54, and VU-60, displaced [3H]R214127 binding from the mGluR1 antagonist site (Fig. 8).

Novel mGluR1 potentiators VU-48, VU-54, and VU-60 do not affect [3H]R214127 binding. Membranes were prepared from BHK cells stably expressing mGluR1a and were incubated with 2.5 nM [3H]R214127 in a final volume of 100 μl for 30 min at 4°C in the presence of varying concentrations of R214127 or CDPPB analogs. Bound and free radioligand was separated by vacuum filtration through Unifilter-96 GF/B filter plates. Nonspecific binding was determined in the presence of 1 μM R214127. Binding of 2.5 nM [3H]R214127 was displaced by unlabeled R214127 but not by the CDPPB analogs. The graphs present means from three independent experiments performed in triplicate. Error bars are S.E.M.
The finding that the novel mGluR1 potentiators did not bind to the allosteric site shared by known allosteric mGluR1 antagonists raises the question of whether other mGluR1 allosteric potentiators bind to the allosteric antagonist site of mGluR1. To address this question, we synthesized the allosteric potentiators Ro 67-4853, Ro 01-6128, and racemic Ro 67-7476 as described by Knoflach et al. (2001) and subjected them to the same tests as the CDPPB analogs. Consistent with previous studies (Knoflach et al., 2001), Ro 67-4853, Ro 01-6128, and racemic Ro 67-7476 induced a robust concentration-dependent increase in the response of mGluR1a to glutamate (Fig. 9A). The maximum potentiation of glutamate-induced calcium release was approximately 3- to 5-fold with EC50 values of 10.7 ± 1.2, 104.2 ± 10.3, and 60.1 ± 3.4 nM, respectively (Fig. 9A). It is noteworthy that none of these mGluR1 potentiators were effective at displacing the binding of the allosteric antagonist [3H]R214127 to membranes expressing mGluR1a (Fig. 9B) at concentrations several orders of magnitude higher than those required for allosteric potentiation of mGluR1. One compound, Ro 67-7476, did induce 40% displacement [3H]R214127 binding at a concentration of 100 μM. However, this is greater than 1000 times the concentration required to potentiate mGluR1 responses. This, coupled with the lack of discernible binding of the other five mGluR1 potentiators suggests that the allosteric potentiators do not act by competitive binding to the same site as that occupied by allosteric antagonists. Consistent with this, we also performed functional studies in which we found that R214127 reduces the maximal potentiator response to representative potentiators (VU-48 and Ro 67-7476) rather than inducing a parallel shift in the potentiator concentration-response relationship (data not shown).
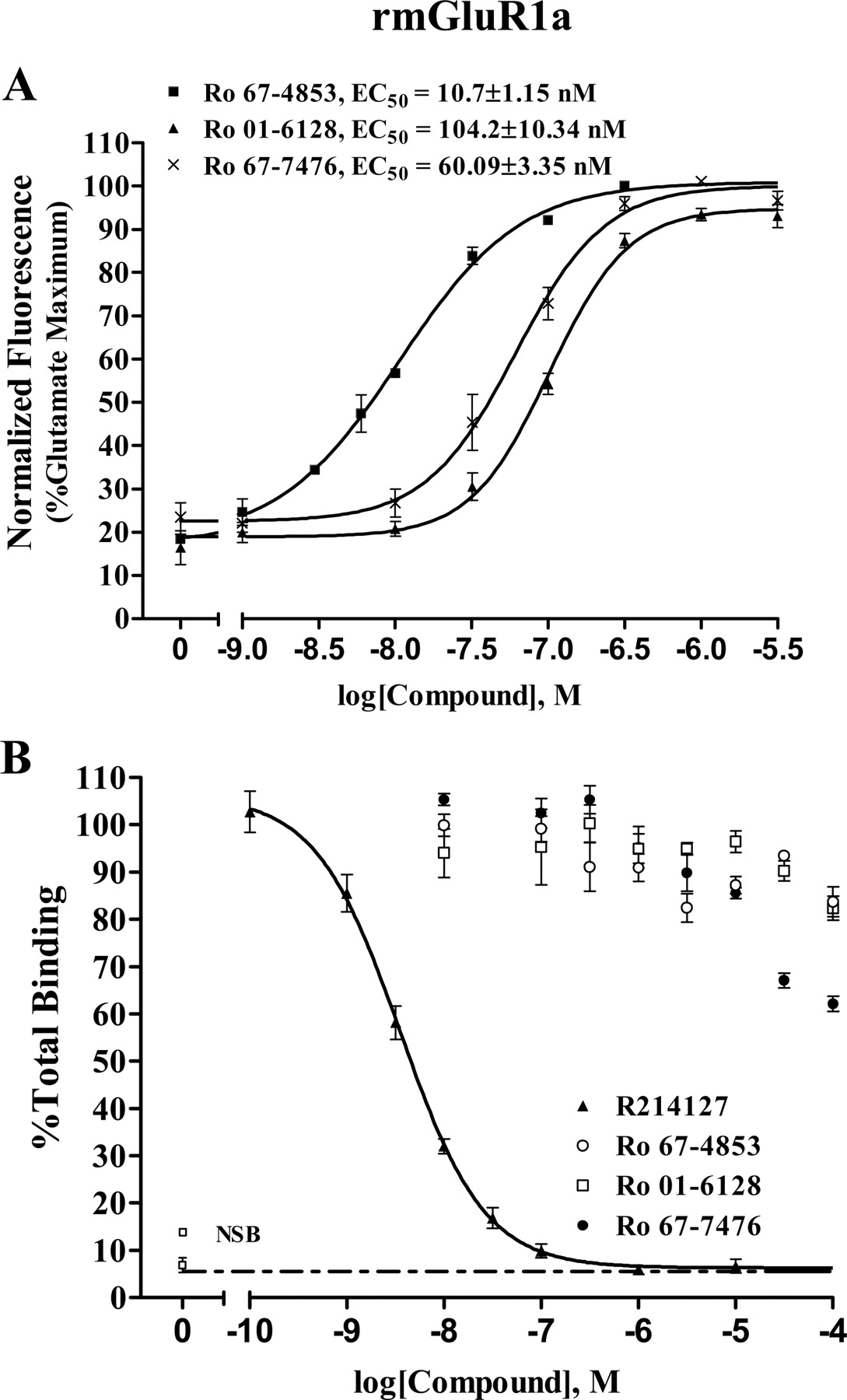
Activity of three previously identified allosteric potentiators of mGluR1 was assessed in a calcium mobilization assay and radioligand binding study. A, Ro 67-4853, Ro 01-6128, and racemic Ro 67-7476 potentiated threshold responses to an EC20 concentration of glutamate approximately 4- to 5-fold as measured by calcium mobilization in BHK cells stably expressing mGluR1a. The EC50 values were 10.7 ± 1.2, 104.2 ± 10.3, and 60.1 ± 3.4 nM for Ro 67-4853, Ro 01-6128, and racemic Ro 67-7476, respectively. The fluorescence responses were normalized as a percentage of the maximal response to 10 μM glutamate and are presented as the means of at least three independent experiments performed in triplicate. Error bars are S.E.M. B, effect of allosteric potentiators of mGluR1 on [3H]R214127 binding. These compounds did not affect [3H]R214127 binding, with the exception of Ro 67-7476, which showed 40% displacement at 100 μM. Membranes were prepared from BHK cells stably expressing mGluR1a and were incubated with 2.5 nM [3H]R214127, in a final volume of 100 μl for 30 min at 4°C in the presence of varying concentrations of R214127 or allosteric mGluR1 potentiators. Bound and free radioligand were separated by vacuum filtration through Unifilter-96 GF/B filter plates. Nonspecific binding was determined in the presence of 1 μM R214127. Binding of 2.5 nM [3H]R214127 was displaced by unlabeled R214127 but not by the allosteric potentiators. The data represent four independent experiments performed in triplicate. Error bars are S.E.M.
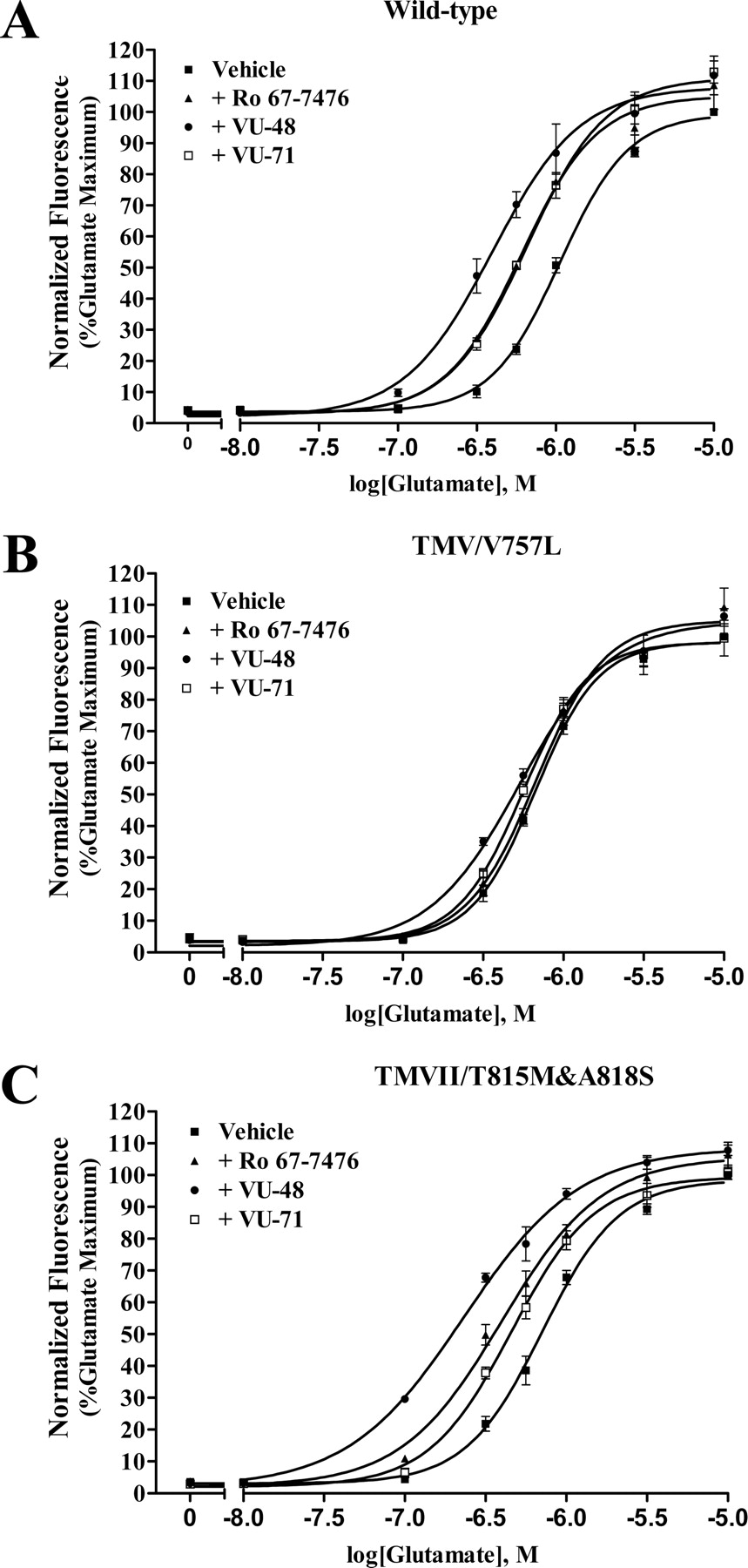
Activity of novel mGluR1 allosteric potentiators and Ro 67-7476 is dependent on Val757 in TM V of mGluR1a. A to C, effect of novel allosteric mGluR1 potentiators (VU-48 and VU-71) and Ro 67-7476 on glutamate-induced calcium mobilization in HEK293A cells transiently transfected with the wild-type mGluR1a, or a mutant mGluR1a, containing either a single point mutation, V757L, or the double mutation, T815M and A818S. These compounds cause a parallel leftward shift of the glutamate concentration-response curve in cells expressing wild-type mGluR1a (A) and double mutant mGluR1a (T815M and A818S) (C). However, the parallel leftward shift of the glutamate-concentration curve was not observed with the V757L mutant mGluR1a (B). The fluorescence responses were normalized as a percentage of the maximal response to 10 μM glutamate and are presented as means of four independent experiments performed in triplicate. Error bars are S.E.M.
Mutation of Residue 757 in TM Domain V from Valine to Leucine Abolishes Potentiation Activities of Allosteric Potentiators Derived from CDPPB Analogs. The novel compounds described here represent a third structural class of positive allosteric modulators of mGluR1, with racemic Ro 67-7476 and Ro 01-6128 representing the other two classes. The finding that none of the CDPPB analogs or previously identified allosteric modulators of mGluR1 were able to displace the binding of [3H]R214127 led us to postulate that positive allosteric modulators act at a common allosteric site that is distinct from the allosteric antagonist site. To test this hypothesis, we measured the ability of representative allosteric potentiators in the new class to potentiate a mutant form of mGluR1a that previously has been shown to be insensitive to Ro 67-7476. Knoflach et al. (2001) reported that valine-757, located in TM V of rat mGluR1, is critical for the activity of Ro 67-7476. This is thought to be responsible for the specificity of Ro 67-7476 for rat versus human mGluR1 (Knoflach et al., 2001). For these experiments, we chose VU-48 and VU-64 as representative compounds because VU-48 is selective for both mGluR1 and mGluR5 as well as having the most robust activity of the new compounds and VU-64 is selective for mGluR1. In addition, we included racemic Ro 67-7476 as a positive control. Each of the allosteric potentiators, VU-48 (10 μM) and VU-64 (10 μM), and racemic Ro 67-7476 (100 nM), induced parallel leftward shifts of the glutamate concentration-response curve in HEK293A cells, transiently expressing wild-type mGluR1a (Fig. 10A). However, mutation of residue valine-757 in mGluR1a to leucine abolished the potentiating activity of these compounds (Fig. 10B). Consistent with previous report (Knoflach et al., 2001), we confirmed that Ro 67-7476 is inactive at human mGluR1 and found that VU-71 is also inactive at the human receptor. However, we were surprised to find that VU-48 was active at human mGluR1 (data not shown). Thus, this single amino acid difference may not fully explain the species specificity of these compounds.
In contrast to the V757L mutation, a double mutation of two amino acids in TM VII, Thr815 and Ala818, previously shown to be critical for the activity of the allosteric mGluR1 antagonist 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester (Knoflach et al., 2001), did not alter the allosteric potentiating activity of the novel compounds or racemic Ro 67-7476 (Fig. 10C).
Discussion
In recent years, a number of positive and negative allosteric modulators of mGluRs have been identified. Particular progress has been made with mGluR5, where highly selective allosteric modulators of have been reported to exhibit a range of pharmacological activities, including positive, negative, and neutral activity (O'Brien et al., 2003; Rodriguez et al., 2005). Several classes of positive allosteric modulators of mGluR5 have been reported, including 3,3′-difluorobenzaldazine (DFB) (O'Brien et al., 2003), N-[4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl]-2-hydroxybenzamide (O'Brien et al., 2004), CDPPB (Lindsley et al., 2004; Kinney et al., 2005), and (S)-(4-fluorophenyl)-[3-(3-(3-(4-fluorophenyl)-[1,2,4]oxadiazol-5-yl)-piperidin-1-yl]methanone (Epping-Jordan et al., 2005). It is noteworthy that DFB and CDPPB can bind to an allosteric site that is also occupied by the prototypical allosteric mGluR5 antagonist MPEP (O'Brien et al., 2003; Kinney et al., 2005; de Paulis et al., 2006). In addition, members of the DFB series exhibit a range of pharmacological activities at mGluR5, including positive allosteric modulation, negative allosteric modulation, and neutral activity (O'Brien et al., 2003; Rodriguez et al., 2005). Based on this, there is an emerging view that most known allosteric modulators of mGluR5 bind to the same site where they have a range of activities analogous to activities of traditional orthosteric receptor agonists, antagonists, and inverse agonists at a common orthosteric site.
The most important and surprising finding in the present studies is that structural analogs of CDPPB can act as allosteric potentiators of mGluR1 and that these compounds do not seem to elicit their effects by binding to the previously identified allosteric site labeled by [3H]R214127 that is shared between all known mGluR1 antagonists (Lavreysen et al., 2003; Mabire et al., 2005; Zheng et al., 2005). We previously reported that CDPPB and the majority of its analogs bind to the MPEP site, as demonstrated by the displacement of [3H]3-methoxy-5-(2-pyridinylethynyl)pyridine binding (Chen et al., 2005; Kinney et al., 2005; de Paulis et al., 2006). Because CDPPB and its analogs bind to the same allosteric site on mGluR5 as MPEP and other mGluR5 allosteric antagonists, we anticipated that these compounds would be likely to bind to the homologous allosteric site on mGluR1a that is labeled with [3H]R214127 and has been extensively characterized using site-directed mutagenesis (Pagano et al., 2000; Malherbe et al., 2003). All negative allosteric mGluR1 modulators that have been discovered to date act at this site (Lavreysen et al., 2003; Kohara et al., 2005; Mabire et al., 2005; Zheng et al., 2005). These compounds belong to several different structural classes and were discovered by multiple independent groups (for review, see Mabire et al., 2005). This has led to the view that there is a common allosteric site on mGluR1, which provide the dominant binding pocket for allosteric modulators of this receptor. Based on this, it was surprising to find that VU-48, VU-54, and VU-60 did not bind to this site and that the previously described allosteric potentiators of mGluR1, Ro 67-4853 and Ro 01-6128, were also without activity. One mGluR1 potentiator, Ro 67-7476, slightly (40%) displaced [3H]R214127 binding at 100 μM. However, the concentrations of compounds in this series required for binding to this site are at least 1000 to 10,000 times those required for allosteric potentiation of mGluR1. This compares to approximately 10- to 20-fold differences between potencies of DFB and CDPPB analogs as allosteric potentiators at mGluR5 and for binding to the MPEP site. This weak binding of one of the allosteric potentiators is highly unlikely to be responsible for allosteric potentiation of mGluR1 because this would require potentiation at concentrations that do not appreciably occupy the receptor.
Consistent with the radioligand binding studies, site-directed mutagenesis revealed that a single point mutation eliminates the activity of multiple allosteric potentiators. Thus, positive allosteric modulators of mGluR1 seem to act at a site that is distinct from that of the known allosteric antagonists. Obviously, the effect of this mutation may be coincidental, and it might be possible to develop compounds with affinities for each of these sites that have overlapping activities. However, it is also noteworthy that none of the CDPPB analogs in the present study have allosteric antagonist or neutral allosteric activity at mGluR1. Given the relatively broad range of compounds tested in which changes were made in each major portion of the CDPPB scaffold, this suggests that it may not be possible to develop compounds in this series that have a range of activities by acting at the site involved in allosteric potentiation.
It is important to note that it is not yet entirely clear that binding of CDPPB to the MPEP site of mGluR5 is responsible for the allosteric potentiator activity of this compound. It is conceivable that CDPPB binding to this site is coincidental and is not directly responsible for its allosteric potentiator activity. In fact, at least one allosteric modulator of mGluR5 has been identified that does not interact with the MPEP binding site (O'Brien et al., 2004). This compound, N-[4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]-phenyl]-2-hydroxybenzamide, is a robust allosteric potentiator of mGluR5 and has functional activity that is very similar to CDPPB but cannot displace [3H]3-methoxy-5-(2-pyridinylethynyl)pyridine binding. This suggests that there are multiple allosteric sites on mGluR5 that could contribute to the activity of mGluR5 allosteric potentiators (O'Brien et al., 2004). In the future, it will be important to systematically study the relationship of binding of CDPPB to this site and its allosteric potentiator activity.
The finding that structural modifications of CDPPB can yield mGluR1 potentiators is interesting in light of the finding that allosteric modulators are often highly selective for specific mGluR subtypes. However, although subtype selectivity is common, it is clear that allosteric sites are likely to be conserved across multiple mGluR subtypes and contain similar pharmacophores. For example, although MPEP and its analogs are selective allosteric antagonists for mGluR5, some compounds in this series also have weak allosteric potentiating activity at mGluR4 (Mathiesen et al., 2003). Furthermore, the recently described mGluR4 allosteric potentiator (-)-PHCCC (Maj et al., 2003; Marino et al., 2003) has mGluR1 antagonist activity and is a close structural analog of the selective mGluR1 allosteric antagonist 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester (Litschig et al., 1999). In addition, as discussed above, extensive mutagenesis studies suggest that MPEP and allosteric antagonists of mGluR1 bind to a homologous site in their respective receptors (Pagano et al., 2000; Malherbe et al., 2003). This suggests that allosteric modulators are likely to act at similar binding pockets across mGluR subtypes. Because these sites may not bind a common endogenous ligand, as is the case for the orthosteric site, it is possible that there is less evolutionary pressure for conservation of the allosteric binding pocket across mGluR subtypes.
In conclusion, we report a novel series of positive allosteric modulators of mGluR1, belonging to a different structural class than those of previously reported. A positional pyrazole isomer of CDPPB, with a p-nitro instead of an m-cyano group (i.e., compound VU-471), is a selective allosteric potentiator of mGluR1 with low micromolar activity. The structural requirement of CDPPB analogs for positive modulating activity at mGluR1 is different from that found at mGluR5, suggesting that other, more potent and selective mGluR1 modulators can be discovered. Furthermore, we found that members of different structural classes of positive allosteric modulators of mGluR1 interact at a site distinct from that of known negative allosteric modulators of mGluR1.
Acknowledgments
We thank Yongqin Zhang for making the Gqi5 construct and Jennifer Edl for developing the mGluR4 stable cell lines.
Footnotes
-
This work was supported by grants from National Institute of Mental Health, National Institute of Neurological Disorders and Stroke, National Alliance for Research on Schizophrenia and Depression, and the Stanley Foundation. Vanderbilt is a site in the National Institutes of Health-supported Molecular Libraries Screening Center Network.
-
ABBREVIATIONS: mGluR, metabotropic glutamate receptor; Ro 67-7476, (S)-2-(4-fluorophenyl)-1-(toluene-4-sulfonyl)pyrrolidine; Ro 01-6128, ethyl diphenylacetylcarbamate; Ro 67-4853, butyl (9H-xanthene-9-carbonyl)carbamate; CDPPB, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide; R214127, 1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-2-phenyl-1-ethanone; PHCCC, N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide; DMSO, dimethyl sulfoxide; BHK, baby hamster kidney; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; HEK, human embryonic kidney; CHO, chinese hamster ovary; VU-33, 3,4-dimethoxy-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide; VU-34, 3,5-dimethoxy-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide; VU-41, N-(1-(2-bromophenyl)-3-phenyl-1H-pyrazol-5-yl)benzamide; VU-48, 4-nitro-N-(1-(2-bromophenyl)-3-phenyl-1H-pyrazol-5-yl)benzamide; VU-54, N-(6-methyl-3-pyridinyl)-1,3-diphenyl-5-amino-1H-pyrazole; VU-60, N-(4-nitrocinnamoyl)-1,3-diphenyl-5-amino-1H-pyrazole; VU-65, 3-cyano-N-(1,4-diphenyl-1H-pyrazol-5-yl)benzamide; VU-66, 4-nitro-N-(1,3-diphenyl-1H-pyrazol-4-yl)benzamide; VU-75, N-(2-phenylimidazo[1,2-a]pyridin-3-yl)benzamide; VU-76, 3-cyano-N-(2-phenylimidazo[1,2-a]pyridin-3-yl)benzamide; VU-71, 4-nitro-N-(1,4-diphenyl-1H-pyrazol-5-yl)benzamide; MPEP, 2-methyl-6-(phenylethynyl)pyridine; TM, transmembrane; DFB, 3,3′-difluorobenzaldazine.
- Received December 19, 2005.
- Accepted April 27, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}