


Abstract
Execution of apoptotic program in mitochondria is associated with accumulation of cardiolipin peroxidation products required for the release of proapoptotic factors into the cytosol. This suggests that lipid antioxidants capable of inhibiting cardiolipin peroxidation may act as antiapoptotic agents. Etoposide, a widely used antitumor drug and a topoisomerase II inhibitor, is a prototypical inducer of apoptosis and, at the same time, an effective lipid radical scavenger and lipid antioxidant. Here, we demonstrate that cardiolipin oxidation during apoptosis is realized not via a random cardiolipin peroxidation mechanism but rather proceeds as a result of peroxidase reaction in a tight cytochrome c/cardiolipin complex that restrains interactions of etoposide with radical intermediates generated in the course of the reaction. Using low-temperature and ambient-temperature electron paramagnetic resonance spectroscopy of H2O2-induced protein-derived (tyrosyl) radicals and etoposide phenoxyl radicals, respectively, we established that cardiolipin peroxidation and etoposide oxidation by cytochrome c/cardiolipin complex takes place predominantly on protein-derived radicals of cytochrome c. We further show that etoposide can inhibit cytochrome c-catalyzed oxidation of cardiolipin competing with it as a peroxidase substrate. Peroxidase reaction of cytochrome c/cardiolipin complexes causes cross-linking and oligomerization of cytochrome c. With nonoxidizable tetraoleoyl-cardiolipin, the cross-linking occurs via dityrosine formation, whereas bifunctional lipid oxidation products generated from tetralinoleoyl-cardiolipin participate in the production of high molecular weight protein aggregates. Protein aggregation is effectively inhibited by etoposide. The inhibition of cardiolipin peroxidation by etoposide, however, is realized at far higher concentrations than those at which it induces apoptotic cell death. Thus, oxidation of cardiolipin by the cytochrome c/cardiolipin peroxidase complex, which is essential for apoptosis, is not inhibited by proapoptotic concentrations of the drug.
Mitochondria play a central role in the execution of apoptotic program realized through intrinsic mechanisms and extrinsic pathways in type II cells (Scaffidi et al., 1998). It is well-documented that one of the early mitochondrial responses to proapoptotic stimuli is the generation of reactive oxygen species (ROS) (Raha and Robinson, 2001). Whereas the overall significance of ROS production in apoptosis has been established by its inhibition by different antioxidant enzymes and free radical scavengers (Nomura et al., 1999; Genova et al., 2003), specific ROS-dependent mechanisms of apoptosis are still elusive. We have demonstrated recently that cytochrome c acts as a redox catalyst in oxidizing a mitochondria-specific phospholipid, cardiolipin, thus facilitating the accumulation of cardiolipin hydroperoxides (CL-OOH) required for the release of proapoptotic factors from mitochondria into the cytosol (Kagan et al., 2004, 2005). This suggests that free radical scavengers, particularly lipid antioxidants, capable of inhibiting cardiolipin oxidation may affect apoptotic responses of cells.
Etoposide, a widely used antitumor drug and a topoisomerase II inhibitor, induces apoptosis accompanied by mitochondrial ROS production (Gorman et al., 1997; Pham and Hedley, 2001). At the same time, we and others have reported that etoposide is an effective scavenger of lipid radicals propagating lipid peroxidation (Kalyanaraman et al., 1989; Kagan et al., 2001; Tyurina et al., 2004), and as such, it should suppress apoptosis. Thus, both pro- and antiapoptotic properties may be realized during etoposide-induced apoptosis.
During apoptosis, cardiolipin oxidation can proceed predominantly through an enzymatically catalyzed mechanism whereby cytochrome c/cardiolipin complex acts as a cardiolipin-specific oxygenase inserting hydroperoxide groups in one or more of the four polyunsaturated fatty acid residues of cardiolipin [similar to cyclooxygenase-catalyzed oxygenation of arachidonic acid (Egan et al., 1976)]. On the other hand, the cytochrome c/cardiolipin complex can be involved only in catalytic initiation of cardiolipin oxidation followed by a nonenzymatic propagation of free radical cardiolipin oxidation, similar to well-known mechanisms of peroxidation of polyunsaturated lipids in biomembranes (Kagan, 1988). Moreover, different reactive intermediates of the peroxidase reaction (heme-centered compounds I and II and amino acid radicals formed by oxidation of protein residues) can be involved in lipid oxidation. Given the important role of cardiolipin oxidation in apoptosis (Garcia Fernandez et al., 2002; Ott et al., 2002; Petrosillo et al., 2003; Zamzani and Kroemer, 2003), characterization of the molecular mechanism of this reaction is important for the regulation of apoptosis and the development of targeted antiapoptotic therapeutic interventions.
Lipid antioxidant action of etoposide yields etoposide phenoxyl radicals that have very low reactivity toward lipids but relatively high reactivity toward ascorbate, glutathione, and protein cysteines (Kagan et al., 2001). Thus, etoposide is an excellent and specific lipid antioxidant that does not protect other important biomolecules against oxidation (Kagan et al., 1994; Yalowich et al., 1996). Hence, probing lipid oxidation pathways of apoptosis with etoposide is specific and is not likely to interfere with the other redox-sensitive mechanisms of apoptosis (Tyurina et al., 2004). In the current work, we attempted to discriminate between the two mechanisms of cardiolipin oxidation by monitoring the generation of CL-OOH and cytochrome c-derived radicals in model systems and correlating them with the development of etoposide-triggered apoptosis in cells. We now demonstrate that cardiolipin oxidation during apoptosis probably is not realized via a random cardiolipin peroxidation mechanism but rather proceeds as a result of peroxidase reaction in a tight cytochrome c/cardiolipin complex that restrains interactions of etoposide with radical intermediates generated in the course of the reaction. We further show that etoposide can inhibit cytochrome c-catalyzed oxidation of cardiolipin competing with it as a peroxidase substrate. These inhibitory effects of etoposide, however, are realized at far higher concentrations than those at which it induces apoptotic cell death. Thus, oxidation of cardiolipin by cytochrome c/cardiolipin peroxidase complex, which is essential for apoptosis, is not inhibited by proapoptotic concentrations of the drug.
Materials and Methods
Chemicals. Etoposide (demethylepipodophyllotoxin-ethyledieneglucopyranoside), microperoxidase 11, cytochrome c, SDS, butylated hydroxytoluene, diethylenetriaminepentaacetic acid (DTPA), EGTA, catalase, porcine pancreas-derived phospholipase A2, and digitonin were purchased from Sigma-Aldrich (St. Louis, MO). Amplex Red (N-acetyl-3,7-dihydroxyphenoxazine) reagent was obtained from Molecular Probes (Eugene, OR). HPLC-grade solvents, fetal bovine serum, RPMI 1640 medium with phenol red, RPMI 1640 medium without phenol red, phosphate-buffered saline (PBS), and cocktail protease inhibitor were purchased from Invitrogen (Carlsbad, CA). Caspase Glow 3/7 detection kit was purchased from Promega (Madison, WI). 1-Palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC), 1-palmitoyl-2-arachidonoyl-sn-glycero-3-[phospho-l-serine] (PAPS), 1,1′,2,2′-tetralinoleoyl-cardiolipin (TLCL), and 1,1′,2,2′-tetraoleoyl-cardiolipin (TOCL) were obtained from Avanti Polar Lipids, Inc. (Albaster, AL). Azo-initiator 2,2′-azo-bis(2,4-dimethylvaleronitrile) (AMVN) was from Wako Chemicals U.S.A. (Richmond, VA). GelCode SilverSNAP Stain Kit II was purchased from Pierce (Rockford, IL), NO donor (Z)-1-[N-3-aminopropyl)-N-(n-propyl)amino]diazen-1-ium-1,2-diolate (PAPANONOate) was from Cayman Chemical Co. (Ann Arbor, MI).
Cell Culture. HL-60 human promyelocytic leukemia cells (American Type Culture Collection, Manassas, VA) were grown in RPMI 1640 medium with phenol red supplemented with 12.5% heat-inactivated fetal bovine serum at 37°C in a humidified atmosphere (5% CO2 plus 95% air). Cells from passages 45 to 50 were used for the experiments. The density of the cells at collection time was 0.5 × 106 cell/ml. HL-60 cells were incubated with either AMVN (500 μM) or etoposide (25 μM) or both together in fetal bovine serum-free RPMI 1640 medium without phenol red for 2 h at 37°C.
Caspase-3/7 activity was measured using a luminescence Caspase-Glo 3/7 assay kit (Promega). In brief, 50 μl of cell homogenates (20 μg of protein) was mixed with 50 μl of Caspase-Glo reagent and incubated at room temperature for 1 h. At the end of incubation, luminescence of each sample was measured using a plate reading chemiluminometer ML1000 (Dynatech Laboratories, Chantilly, VA). Activity of caspase-3/7 was expressed as luminescence arbitrary units per milligram of protein.
Cytochrome c Release. Cells were resuspended in lysis buffer containing 250 mM sucrose, 20 mM HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 0.05% digitonin for 3 min on ice. Samples were then centrifuged at 8000g for 5 min at 4°C. Supernatants were collected as the cytosol fraction. Aliquots of 40 μg of cytosol fraction were examined on 12% SDS-PAGE with anti-cytochrome c antibody (1:2000) (BD PharMingen, San Jose, CA).
Preparation of Liposomes. Small unilamellar liposomes containing 50% PC and 50% TOCL or TLCL were produced as described previously (Tyurina et al., 2004; Kagan et al., 2005). Individual phospholipids stored in chloroform were dried under N2. PBS containing 100 μM DTPA was added to obtain the phospholipid concentration of 200 μM, and the lipid mixture was vortexed and sonicated for 3 min under nitrogen on ice. All liposomes were used immediately after preparation. For the measurements of EPR spectra of etoposide phenoxyl radical at room temperature, liposomes were prepared in N2 buffer and stored under N2.
Oxidation of Phospholipids in Liposomes. Liposomes were incubated with etoposide (1, 3, 5, 10, 25, 50, and 100 μM) for 15 min on ice. Samples were treated with 250 μM AMVN or with cytochrome c (4 μM) and H2O2 (100 μM) for 1 h at 37°C in a water bath. H2O2 was added four times (every 15 min) during the incubation period. Catalase (2 U) was added to eliminate residual H2O2. In some experiments, liposomes (1.5 mM DOPC/TLCL at a ratio of 1:1) were preincubated with etoposide (7.5-700 μM) and then were treated with cytochrome c (30 μM) and H2O2 [100 μM, H2O2 was added four times (every 15 min) during the incubation period] for 1 h at 37°C in a water bath. Lipids were then extracted in chloroform/methanol (2:1, v/v) solvent. The extracted lipids were used to determine phosphorus content and measure the amount of lipid hydroperoxides generated as documented below.
Two-Dimensional High-Performance Thin-Layer Chromatography. The extracted lipids (either from cells or liposomes) were dried under nitrogen and separated by two-dimensional high-performance thin-layer chromatography (2D-HPTLC) on silica G plates. The plates were first developed with a solvent system consisting of chloroform/methanol/28% ammonium hydroxide (65:25:5, v/v). After drying the plates with a forced-air blower to remove the solvent, the plates were developed in the second dimension with a solvent system consisting of chloroform/acetone/methanol/glacial acetic acid/water (50:20:10:10:5, v/v). The phospholipids were visualized by exposure to iodine vapors and identified by comparison with migration of authentic phospholipid standards. The spots identified by iodine staining were scraped, and the silica was transferred to tubes. Lipid phosphorus was determined by the submicro method as described previously (Tyurina et al., 2004). The identity of each phospholipid was established by comparison with the Rf values measured for authentic standards.
Assay for Phospholipid Peroxidation. Lipid extracts were dried under nitrogen and then incubated with 100 μl of buffer containing 25 mM NaH2PO4, 0.5 mM EGTA, and 1 mM calcium, pH 8.0, 10 μl of 5 mM SDS, and 1 μl of phospholipase A2 for 30 min at room temperature. Two microliters of 1 mM HCl and 1 mM EGTA was added to adjust the pH of the samples. Amplex red (1.5 μl of 50 μM) and microperoxidase 11 (1 μg/μl) solution was added and incubated on ice for 40 min. Reaction was stopped using 100 μl of stop reagent (solution of 10 mM HCl and 4 mM butylated hydroxytoluene in ethanol). The samples were centrifuged at 15,000g for 5 min, and the supernatant was used for HPLC analysis. Aliquots (5 μl) were injected into a C-18 reverse phase column (Eclipse XDB-C18, 5 μm, 150 × 4.6 mm). The column was eluted by mobile phase composed of 25 mM NaH2PO4, pH 7.0/methanol (60:40 v/v) with 1 ml/min flow rate. Lipid phosphorus was determined by a micro method. The resorufin (an Amplex Red oxidation product) fluorescence was measured at 590 nm after excitation at 560 nm using a Shimadzu LC-100AT vp HPLC system equipped with fluorescence detector (model RF-10Axl) and autosampler (model SIL-10AD vp).
Mass Spectrometry of TLCL Oxidation Products. Electrospray ionization mass spectrometry of lipids was performed by direct infusion into a triple quadrupole mass spectrometer TQ70 (Finnigan, San Jose, CA). Sheath flow was adjusted to 5 μl/min, and the solvent consisted of chloroform/methanol (1:2, v/v). The electrospray probe was operated at a voltage differential of -3.5 kV in the negative ion mode. Mass spectra for doubly charged cardiolipin species were obtained by scanning in the range of 400 to 950 m/z every 1.5 s and summing individual spectra. Source temperature was maintained at 70°C.
EPR Measurements. EPR spectra were recorded on a JEOLREIX spectrometer with 100 kHz modulation (JEOL, Kyoto, Japan). Measurements at room temperature were performed under N2 conditions in gas-permeable Teflon tubing (0.8 mm internal diameter, 0.013 mm thickness) obtained from Alpha Wire Corporation (Elizabeth, NJ). The tubing was filled with 60 μl of sample, folded doubly, and placed in an open 3.0 mm internal diameter EPR quartz tube. Etoposide phenoxyl radical spectra were recorded at 3350 G, center field; 50 G, sweep width; 10 mW, microwave power; 0.5 G, field modulation; 103, receiver gain; 0.03 s, time constant; and 4 min, scan time. The time course of etoposide radical EPR signals was obtained by repeated scanning of the part of the spectrum (3350 G, centered field; 5 G, sweep width; other instrumental conditions were the same).
The time course of etoposide radical EPR signals was used to determine the rate of etoposide radical production. Recombination rate constant of etoposide radicals was estimated at kR = 1.2 × 10-3 M-1 s-1 from the observation of the decay of EPR signal of etoposide radicals obtained by fast oxidation of etoposide in the presence of an excess of H2O2 and horseradish peroxidase. In the experiments described further, concentration of etoposide radicals did not exceed cmax = 1 μM. At these concentrations, the lifetime of etoposide radical t½ = 1/(kR × cmax) ∼ 340 s was greater than the time of the experiment (240 s). Therefore, to compute the etoposide oxidation rate, we assumed that etoposide radicals were accumulated without recombination during the course of the experiment, and the rate of etoposide radical production was proportional to the increase of EPR signal amplitude.
For low-temperature EPR measurements of radicals (protein-derived or etoposide radicals), samples (150 μl) were placed into a Teflon tube (3.7 mm internal diameter) and immediately immersed in liquid nitrogen. Frozen samples were removed from the tube and stored in liquid nitrogen. EPR spectra of the frozen samples were detected at 77 K under the following conditions: 3230 G, centered field; 100 G, sweep width; 5 G, field modulation; 1 mW, microwave power; 0.1 s, time constant; and 4 min, time scan. Dependence of relative magnitude (percentage of the maximal magnitude) of EPR signals of radicals on square root from microwave power (in milliwatts) was presented as saturation curves. The spin-lattice relaxation time was determined by fitting the experimental curve of radical signal saturation to the theoretical one as described previously (Castner, 1959).
For low-temperature EPR measurements of heme-nitrosylated cytochrome c, ferro-cytochrome c (100 μM) was incubated with liposomes in the presence of NO donor PAPANONOate (500 μM) under N2 at 37°C for 15 min. The reaction was stopped by freezing the samples in liquid nitrogen. Spectra were recorded at 77 K under the following instrumental settings: 3200 G, center field, 500 G, scan range, 5 G, modulation amplitude; 10 mW, microwave power; 0.1 s, time constant; 4 min, scan time; and 5 × 102, receiver gain.
Fluorescence Measurements of Dityrosine Formation. After incubating of 10 μM cytochrome c with 250 μM TOCL or TLCL liposomes in the presence of 100 μMH2O2 in presence or absence of etoposide for 1 h at 37°C, dityrosine fluorescence in digested samples was determined as described previously (Giulivi et al., 2003).
PAGE Electrophoresis of Cytochrome c/CL Complexes. DOPC/CL liposomes (200 μM, at a ratio of 1:1) were incubated with cytochrome c (4 μM) in the presence of 100 μMH2O2. Electrophoresis was performed in 12.5% PAGE. Gels were stained by silver using GelCode SilverSNAP Stain Kit II (Pierce). For Coomassie blue staining of the gels, the amounts of liposomes and cytochrome c were increased up to 1.5 mM and 30 μM, respectively. To quantitatively evaluate cytochrome c aggregation, densitometry of stained protein bands on gels using Fluor-S MultiImager (Bio-Rad, Hercules, CA) was performed. Protein bands from Coomassie blue-stained gels were excised and cut into smaller pieces. Destaining, digestion with trypsin, and extraction of peptides was performed as described in Jimenez et al. (1998). Dityrosine fluorescence was measured as described above.
Statistics. The results are presented as mean ± S.E. values from at least three experiments, and statistical analysis were performed by either paired or unpaired Student's t test of one-way analysis of variance. The statistical significance of difference was set at p < 0.05.
Results
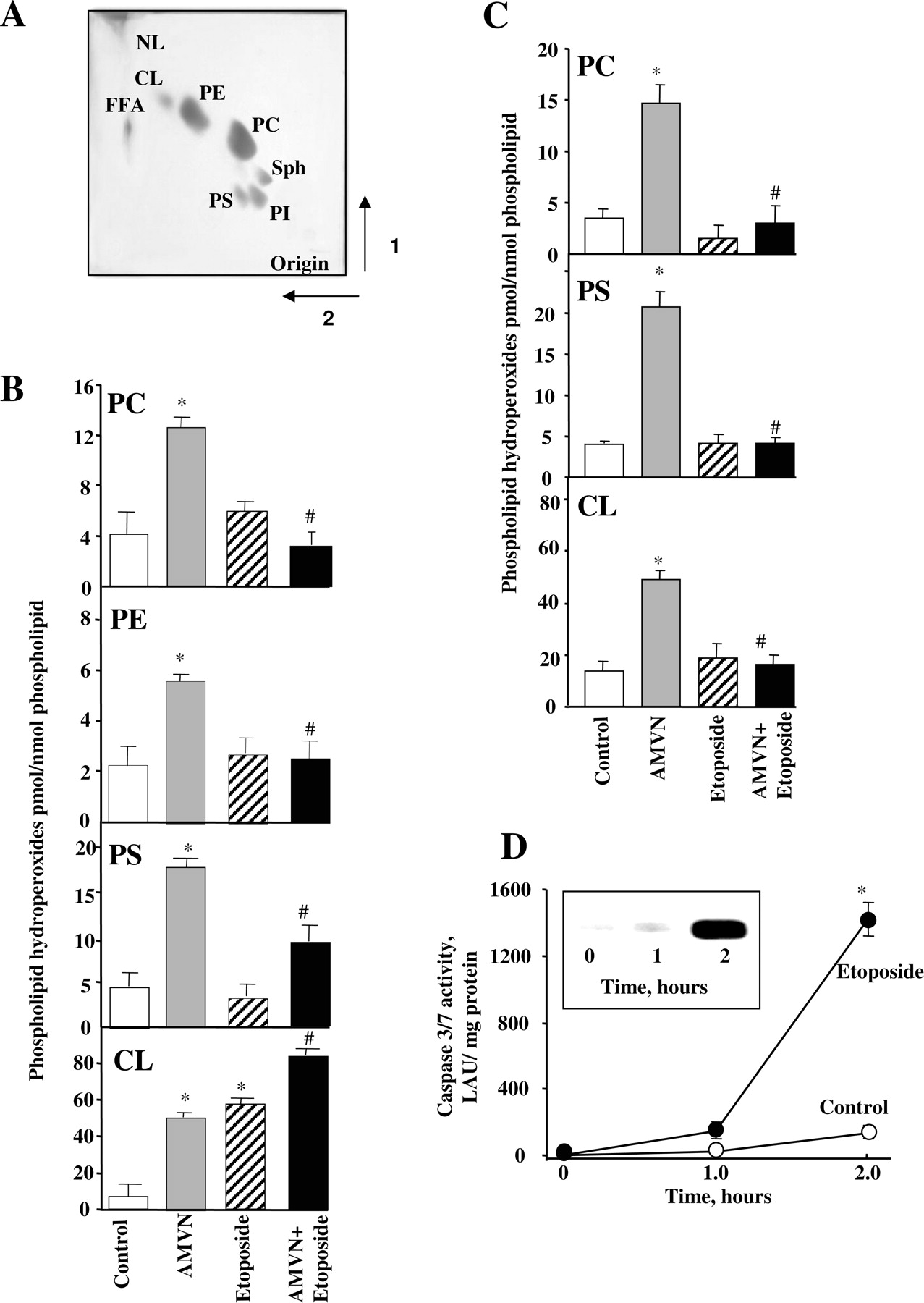
Effect of Etoposide on Phospholipid Oxidation in HL-60 Cells. We assessed lipid peroxidation in HL-60 cells treated with the following (for 2 h at 37°C): 1) a lipophilic azo-initiator of peroxyl radicals, AMVN (500 μM); 2) etoposide (25 μM); and 3) a combination of AMVN plus etoposide. Phospholipids were extracted and separated by 2D-HPTLC. A typical HPTLC separation profile is shown in Fig. 1A. Phosphatidylcholine represented approximately one half of the total phospholipids with phosphatidylethanolamine (PE) being the next most abundant phospholipid class (approximately 20%). In addition, the phospholipids in the order of their abundance—phosphatidylinositol > sphingomyelin > phosphatidylserine > cardiolipin—were detectable on the HPTLC plates. There were no significant differences in the pattern of distribution of phospholipid classes between treated and untreated HL-60 cells (data not shown). Spots corresponding to different phospholipids were scraped, and the amounts of phospholipid hydroperoxides generated in HL-60 cells were determined using our newly developed fluorescence HPLC/Amplex Red-based assay (Kagan et al., 2005). We found that AMVN induced oxidation of all major classes of phospholipids, including phosphatidylcholine, PE, phosphatidylserine, and cardiolipin (Fig. 1B). Etoposide alone caused a strong oxidation of cardiolipin, whereas it did not induce oxidation of other more abundant phospholipid classes, which was in line with our results published previously (Tyurina et al., 2004). Moreover, when applied in a combination with AMVN, etoposide suppressed the AMVN-induced oxidation of all phospholipids—phosphatidylcholine, PE, and phosphatidylserine—with a notable exception of cardiolipin. In fact, the AMVN-induced oxidation of cardiolipin was markedly enhanced by etoposide. These results indicate that there are profound differences in mechanisms of phospholipid oxidation by etoposide and AMVN in HL-60 cells that are probably associated with the apoptosis-specific pathways. Therefore, we next compared the effects of AMVN, etoposide, and a combination of AMVN plus etoposide on oxidation of phospholipids in liposomes prepared from readily oxidizable polyunsaturated phospholipids, PAPC, PAPS, and TLCL. Similarly to cells, AMVN induced oxidation of all phospholipids in liposomes (Fig. 1C). As expected, etoposide did not cause any oxidation of liposomal phospholipids. In contrast to HL-60 cells, however, etoposide equally protected all phospholipids, including cardiolipin, against AMVN-induced oxidation in liposomes (Fig. 1C). Because in these experiments, relatively low etoposide concentrations (25 μM) were used that were sufficient to induce apoptosis in HL-60 cells (Fig. 1D), these results clearly demonstrate that apoptosis-associated peroxidation of cardiolipin is not effectively quenched by etoposide. It is noteworthy that the conditions of AMVN exposure of HL-60 cells were well-suited for the induction of apoptosis. Therefore, both random oxidation of phospholipids by a radical initiator and apoptosis-specific oxidation of cardiolipin were triggered by AMVN. The ability of etoposide to induce cardiolipin oxidation in HL-60 cells both in the presence and absence of AMVN is related to its proapoptotic properties. Execution of apoptotic program is accompanied by the generation of ROS and lipid oxidation (Raha and Robinson, 2001; Kagan et al., 2004), which is catalyzed by cytochrome c molecules bound to cardiolipin-containing mitochondrial membranes (Kagan et al., 2005). Thus, involvement of specific cytochrome c-dependent mechanisms in cardiolipin oxidation during apoptosis may be responsible, at least in part, for a decreased sensitivity of cardiolipin oxidation to inhibitory effects of radical scavengers like etoposide. Therefore, we further tested cardiolipin oxidation in the presence and absence of etoposide in a cellfree system containing model cardiolipin membranes and cytochrome c molecules.

Effect of etoposide and AMVN on peroxidation of phospholipids in HL-60 cells and in liposomes. A, a typical 2D-HPTLC of lipids extracted from HL-60 cells; 50 μg of phospholipids was used in the analysis. NL, neutral lipids; CL, cardiolipin; PE, phosphatidylethanolamine; PC, phosphatidylcholine; SPH, sphingomyelin; PI, phosphatidylinositol; PS, phosphatidylserine; FFA, free fatty acids. B and C, quantification of lipid peroxidation in HL-60 cells and liposomes. B, HL-60 cells (2 × 106/ml) or liposomes (C) composed of PAPC, PAPS, and TLCL were incubated with either AMVN (500 μM) or etoposide (25 μM) or AMVN (500 μM) plus etoposide (25 μM) for 2 h at 37°C. At the end of the incubation, lipids were extracted and separated by 2D-HPTLC. Phospholipid hydroperoxides were quantified by fluorescence HPLC using Amplex Red protocol. Data presented as mean ± S.E. (n = 3); *, p < 0.001 versus control; #, p < 0.001 versus AMVN. D, etoposide induced caspase-3/7 activation and cytochrome c release (inset) in HL-60 cells. Cells were incubated in the presence of etoposide (25 μM) for 2 h at 37°C. At the end of the incubation, caspase-3/7 activity and cytochrome c release were measured by luminescence Caspase 3/7 kit and Western blotting, respectively. Data are presented as mean ± S.E. (n = 3); *, p < 0.01 versus control.
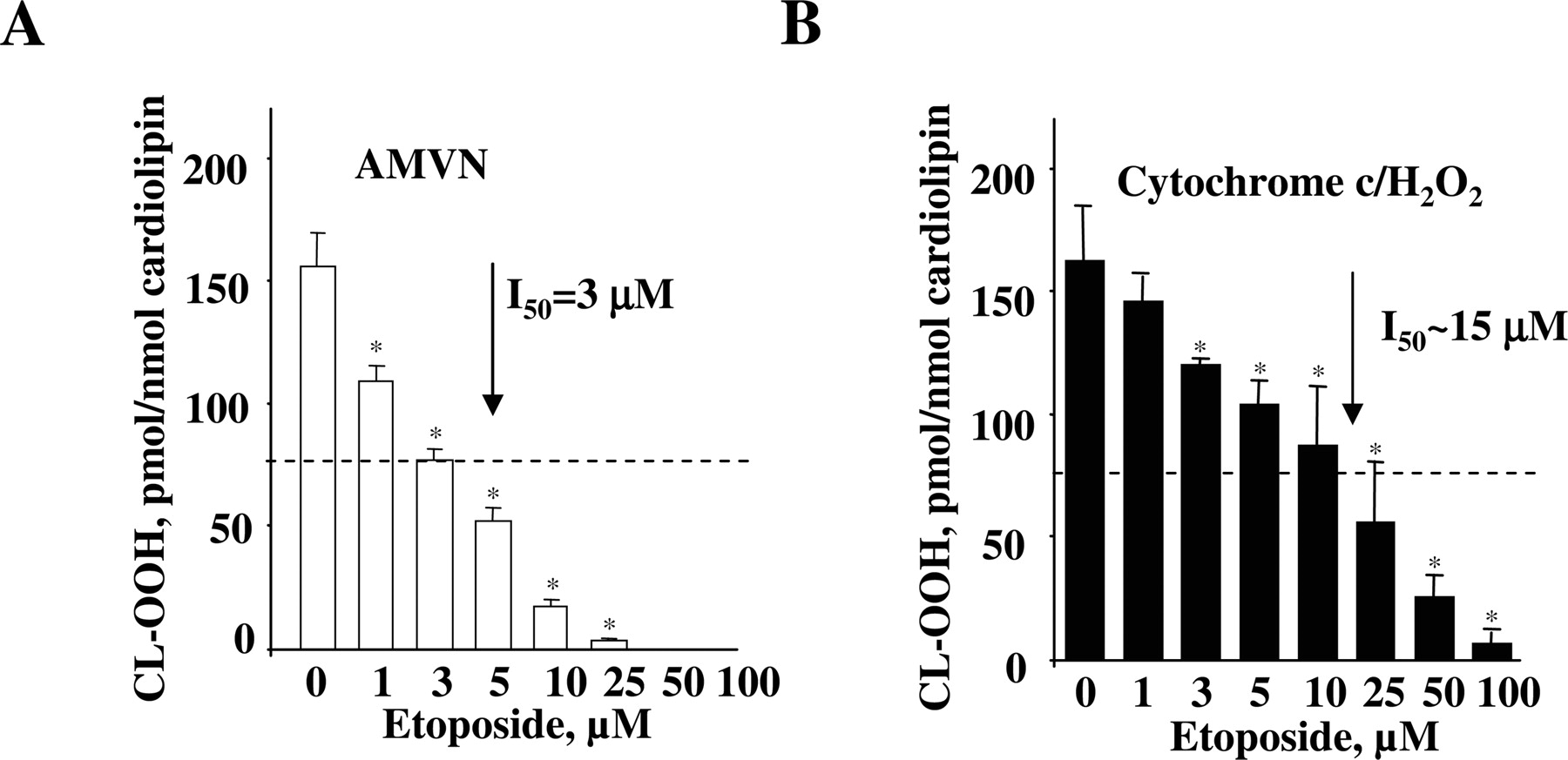
Effect of Etoposide on Cardiolipin Oxidation in a Model System. We compared the effectiveness of etoposide in inhibiting cardiolipin oxidation induced by a free radical generator AMVN with its ability to suppress cytochrome c-catalyzed cardiolipin oxidation. To this end, we used liposomes containing DOPC and TLCL at a ratio of 1:1. TLCL was a natural choice because it is the most abundant cardiolipin species found in mammalian mitochondria (Schlame and Rustow, 1990). Liposomes were incubated in PBS (containing 100 μM DTPA to prevent nonspecific metal-catalyzed oxidation) for 1 h at 37°C with 250 μM AMVN, which generates peroxyl radicals at constant rate at a given temperature (Niki, 1990). At the end of the incubation, phospholipids were extracted, resolved by 1-dimensional HPTLC to separate etoposide from the phospholipids, and the content of CL-OOH was determined using the Amplex Red fluorescence HPLC-based assay (Kagan et al., 2005). We found that incubation of liposomes with AMVN resulted in the accumulation of up to 155.8 ± 13.0 pmol of CL-OOH per nmol of cardiolipin (∼1.55 nmol/ml). This corresponds well with the total amount of radicals generated by AMVN (1.22 nmol/ml) during this period of time (3600 s) given that its decomposition rate is 1.36 × 10-6 [AMVN] M s-1 (Niki, 1990). The addition of etoposide to the incubation system resulted in a significant decrease of cardiolipin oxidation, the magnitude of which was proportional to etoposide concentration. Half-maximal inhibition (I50) was achieved at ∼3 μM etoposide, whereas nearly complete inhibition was obtained at 25 μM etoposide (Fig. 2A). These results demonstrate that etoposide is an effective radical scavenger, as demonstrated previously by us in other systems (Kagan et al., 2001).
We next evaluated the potency of etoposide as an inhibitor of enzymatic cytochrome c/cardiolipin-catalyzed oxidation. We chose the conditions (incubation of TLCL liposomes in PBS for 1 h at 37°C with cytochrome c (4 μM) and H2O2 (100 μM, added every 15 min) yielding approximately the same cardiolipin oxidation level (162 ± 38 pmol of CL-OOH/nmol of cardiolipin) as the one generated by the AMVN system (compare Fig. 2, A and B). Whereas etoposide protected TLCL against enzymatic cytochrome c oxidation, its potency was approximately 5-fold lower, with I50 = 15 μM (Fig. 2B). The lower antioxidant activity of etoposide in the cytochrome c/H2O2 system compared with AMVN-induced cardiolipin oxidation is not entirely unexpected and may be related to its hindered interactions with reactive intermediates of cytochrome c/cardiolipin peroxidase complex [compound I, compound II, protein-derived (tyrosyl) amino acid radicals] compared with stochastic interaction with lipid peroxyl radicals during AMVN-induced TLCL oxidation. To gain a better understanding of etoposide participation in cytochrome c/cardiolipin peroxidase reaction, we applied EPR spectroscopy to detect radical intermediates generated by the cytochrome c/cardiolipin complexes such as etoposide phenoxyl radicals and protein-derived (tyrosyl) radicals (Kalyanaraman et al., 1989).
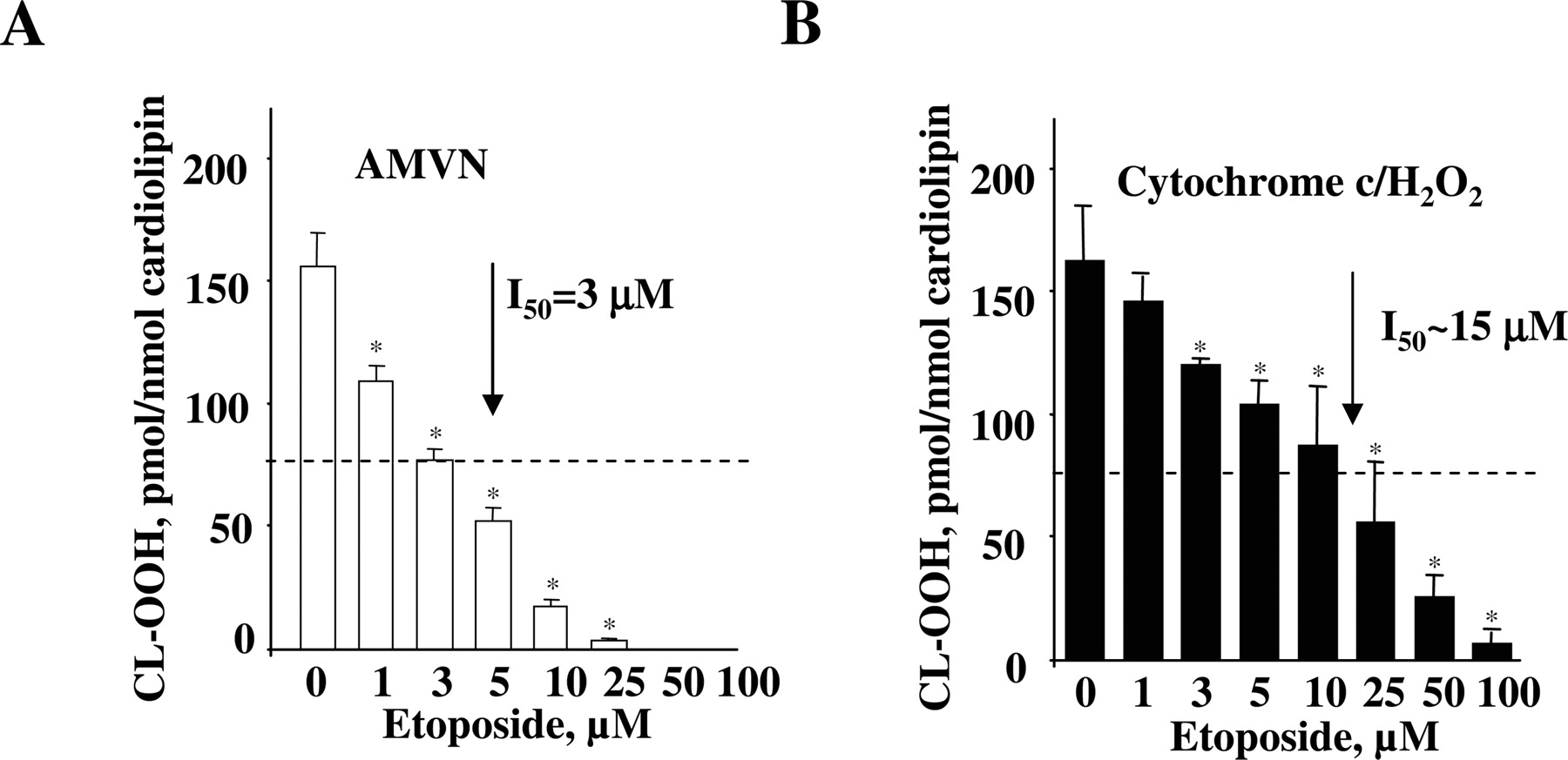
Effect of etoposide on cardiolipin peroxidation induced by cytochrome c/cardiolipin complex in the presence of H2O2 or a lipid-soluble azo-initiator of peroxyl radicals, AMVN. DOPC/TLCL (200 μM, at a ratio of 1:1) liposomes were incubated in PBS at pH 7.4, containing 100 μM DTPA with AMVN (250 μM) (A) or in the presence of cytochrome c (4 μM) and H2O2 (100 μM, four times every 15 min) (B) for 60 min at 37°C. At the end of the incubation, lipids were extracted, and hydroperoxides of cardiolipin were quantified by fluorescence HPLC using Amplex Red protocol. The degree of lipid peroxidation at different etoposide concentrations is presented as pmol CL-OOH/nmol cardiolipin. The control group represents liposomes without etoposide treatment. The data are presented as the mean ± S.E. (n = 3); *, p < 0.001 versus control.
EPR Measurements of Etoposide Phenoxyl Radical Formation. Both nonenzymatic and enzymatic (e.g., peroxidases-catalyzed) one-electron oxidation of etoposide yields its phenoxyl radicals with characteristic signals in the EPR spectra (Kagan et al., 1999). Therefore, incubation of TLCL liposomes with cytochrome c, etoposide, and H2O2 (under N2) produced a characteristic EPR signal of etoposide phenoxyl radical (Fig. 3Aa). This signal was not observed in the absence of H2O2 (data not shown). The magnitude of the signal increased over time, and its growth was approximately linear over the initial 5 min of incubation (Fig. 3Ab). Assuming that changes in the magnitude of the signal are proportional to the etoposide radical production rate (see Materials and Methods for details), we assessed the rate of etoposide radical production at different concentrations of the drug in the presence of TLCL. Peroxidase activity of cytochrome c/TLCL can use both etoposide and TLCL as substrates resulting in TLCL hydroperoxide and etoposide radical formation, respectively. As shown on Fig. 3B, etoposide radical production increased and then saturated at etoposide concentrations of greater than 100 μM. This may be due to the ability of etoposide to outcompete TLCL as a peroxidase substrate. We performed the same series of experiments with monounsaturated TOCL that does not undergo cytochrome c/H2O2 catalyzed peroxidation (Fig. 3B, inset). In this case, saturation of the etoposide radical production was achieved at significantly lower etoposide concentrations (∼50 μM). The half-maximal rate of etoposide oxidation was reached at concentrations of approximately 5 μM in the presence of TOCL compared with ∼15 μM for TLCL (Fig. 3B). These results indicate that cytochrome c is an effective catalyst of TLCL oxidation, resulting in the requirement of high etoposide concentrations to block TLCL oxidation, which is in line with the results of Fig. 2.
To further characterize the involvement of protein radicals in lipid peroxidation induced by cytochrome c/TLCL complex, we performed low-temperature EPR studies of cytochrome c/cardiolipin complexes in the presence of H2O2 and etoposide.
EPR Measurements of Protein-Derived (Tyrosyl) Radical Formation. Catalytic activation of peroxidases is known to form highly reactive oxidizing intermediates—compounds I and II (Lassmann et al., 1991). The production of the latter intermediate is also associated with the generation of another oxidizing species, an amino acid-centered (often tyrosyl) radical detectable by EPR (Tsai et al., 1999; Svistunenko, 2005). Viewed from a thermodynamic standpoint, any of these reactive intermediates of cytochrome c/cardiolipin peroxidase complex can oxidize etoposide to generate its phenoxyl radical (Koppenol, 1990; Buettner, 1993). EPR spectra of protein-derived (tyrosyl) radicals of cytochrome c at 77 K induced by high concentrations of oxidants have been reported (Lassmann et al., 1991; Chen et al., 2004a,b). As shown in Fig. 4A, a characteristic low-temperature EPR signal of (tyrosyl) radical with peak-trough width of 16 G and g = 2.005 was readily detectable in cytochrome c/TOCL and cytochrome c/TLCL systems immediately upon the addition of H2O2. This signal was only barely detectable from cytochrome c plus H2O2 in the absence of TOCL or TLCL or from the mixture of cytochrome c with PC liposomes plus H2O2 (data not shown). The magnitude of the (tyrosyl) radical signal from cytochrome c/cardiolipin complex (Fig. 4Ab) was approximately 30 ± 7% lower for readily peroxidizable TLCL than for nonoxidizable TOCL (Fig. 4A). It is likely that the difference is due to partial quenching of the (tyrosyl) radical by the involvement of the radical in oxidation of TLCL.
Addition of etoposide to either TOCL or TLCL complexes of cytochrome c (Fig. 4Ac) resulted (in the presence of H2O2)in approximately 2-fold increase of the magnitude of the EPR signal and change of its shape (g = 2.005 with a peak and trough width of 11.5 G). This seemingly “unexpected” growth of the signal may be explained by superposition of the protein-derived (tyrosyl) radical signal and the etoposide phenoxyl radical signal. In fact, because both of them have similar g-factors (2.005 and 2.005, respectively) and half-widths (16 and 11.5 G, respectively), their simultaneous formation is likely to cause an increase of the overall EPR signal.

Etoposide-phenoxyl radicals production in the presence of cytochrome c/TOCL and cytochrome c/TLCL complexes. Liposomes (100 μM DOPC and 100 μM TOCL or TLCL) were incubated with etoposide for 15 min on ice. Then, cytochrome c (4 μM) was incubated with liposomes for 1 min at room temperature, and the time course of EPR signal of etoposide radical was recorded for 4 min starting less than 1 min after the addition of 100 μMH2O2 (N2 conditions). A, EPR spectrum of etoposide phenoxyl radical (top spectrum, a), the underscored part of the spectrum was scanned repeatedly to measure the kinetics of etoposide radical formation (bottom spectrum, b). B, changes of the magnitude of etoposide radical EPR signal during 4-min measurements (see A, bottom spectrum) versus total etoposide concentration used in the experiment; DOPC/TOCL (□) or DOPC/TLCL (▪) liposomes. Inset, oxidation of TOCL and TLCL (hydroperoxide content) at 0 min (□) and 60 min (▪) after incubation with cytochrome c and H2O2. The data illustrated are from one representative experiment of a total of four experiments (with duplicate measurements for each etoposide concentration).
To verify that reduced magnitude of the EPR signal detectable in the presence of TLCL did not relate to diminished generation of tyrosyl radicals, we measured cytochrome c nitrosylation in the presence of TLCL and TOCL using low-temperature EPR spectroscopy. We found that incubation of cytochrome c with TOCL or TLCL in the presence of an NO donor, PAPANONOate, results in the appearance of EPR spectra characteristic for heme-nitrosylated cytochrome c (Fig. 4B). Under the experimental conditions used, the magnitude and shape of the EPR spectra were essentially the same. No significant spectra were observed in the absence of cardiolipins. Thus, interaction of cytochrome c with cardiolipins caused similar unfolding of the protein and similar accessibility of the cytochrome c heme to NO.
If oxidation of etoposide to its phenoxyl radical occurred on the protein-derived (tyrosyl) radical of cytochrome c/cardiolipin complex, one would expect to detect a decrease of its magnitude (due to its reduction by etoposide). Given very similar parameters for both of the signals—the protein-derived (tyrosyl) radical and etoposide phenoxyl radical—the reduction of tyrosyl radical by etoposide probably does not change the overall EPR signal magnitude. The observed large increase of the signal is in line with a longer lifespan of etoposide phenoxyl radicals (100-1000 s under our experimental conditions) compared with tyrosyl radicals (Tsai et al., 1999; Chen et al., 2004a). Therefore, steady-state concentrations of phenoxyl radicals are higher, resulting in a greater magnitude of the EPR signal. This is also supported by our results on power saturation of etoposide phenoxyl radical and protein-derived (tyrosyl) radical EPR signals (Fig. 4C). The etoposide phenoxyl radical EPR signal saturated at a significantly lower power of magnetic field than protein-derived (tyrosyl) radical signal. Power saturation of radical EPR signal depends strongly on the distance between a radical and metal ion (Lassmann et al., 1991). To quantitate tyrosyl and phenoxyl radical saturation parameters, we evaluated the spin-lattice relaxation time that is sensitive to the radical interaction with paramagnetic metals in two different systems: cytochrome c/TOCL + H2O2 and cytochrome c/TOCL + etoposide + H2O2. We found that the spin-lattice relaxation time for the tyrosyl radicals and phenoxyl radicals were substantially different and estimated as T1 = (1.2 ± 0.3) × 10-5 s and (4.3 ± 0.5) × 10-5 s, respectively.
Formation of Dityrosines in Cytochrome c Peroxidase Reaction. A protein-derived tyrosyl radical formed during peroxidase reaction of cytochrome c/cardiolipin complex is relatively long-lived and can isomerize and combine with another tyrosyl radical with subsequent enolization (Giulivi et al., 2003). As a result, a stable, covalent, carboncarbon bond is generated, yielding 1,3-dityrosine. The latter is distinguished by the intense 420-nm fluorescence emission, measurable upon excitation at 315 nm (Malencik et al., 1996). Several oxidizing systems were found to produce dityrosines during oxidant exposure of both purified proteins in vitro and intact cells. We found a markedly increased fluorescence characteristic of dityrosines from cytochrome c/TOCL complex incubated in the presence of H2O2. The fluorescence response was quenched by etoposide in a concentration-dependent manner (Fig. 5A). Assuming that no dityrosines are formed at high etoposide concentrations (250 μM) and the fluorescence level at these conditions can be taken as a background, the half-maximal inhibition of dityrosine fluorescence quenching was achieved by the etoposide concentration of ∼5 μM. This is consistent with the concentration dependence of etoposide radical formation shown on Fig. 3B. It is noteworthy that no characteristic dityrosine fluorescence was observed from cytochrome c/TLCL complexes treated with H2O2 in the presence or absence of etoposide. This suggests that protein-derived tyrosyl radicals do not form dimers in the presence of readily oxidizable TLCL but rather participate in lipid peroxidation.

Low-temperature EPR measurements of phenoxyl radicals. A, EPR spectra of H2O2-induced phenoxyl radicals for cytochrome c/TOCL (a), cytochrome c/TLCL (b), or cytochrome c/TOCL in the presence of 500 μM etoposide (c). Liposomes (5 mM, DOPC; 5 mM TOCL or TLCL) were incubated with cytochrome c (200 μM) for 1 min at room temperature, then H2O2 (1 mM) was added. Reaction was stopped after 20 s by freezing the samples in liquid nitrogen. B, EPR spectra of nitrosylated cytochrome c in the presence of DOPC/TOCL (a) or DOPC/TLCL (b) liposomes. Ferrocytochrome c (100 μM) and DOPC/TLCL or DOPC/TOCL liposomes (2.5 mM at a ratio of 1:1) were incubated in 20 mM phosphate buffer, pH 7.4, containing DTPA (100 μM) in the presence of PAPANONOate (500 μM) under N2 for 15 min at 37°C. The reaction was stopped by freezing the samples in liquid nitrogen, and EPR spectra were recorded at 77 K. C, power saturation curves of etoposide phenoxyl radical (○) and proteinderived tyrosyl radical (○) EPR signals. Saturation curves are the same for cytochrome c/TOCL and cytochrome c/TLCL complexes.
Detection of CL/Cytochrome c Oligomerization Products by PAGE. Formation of dityrosine cross-links between cytochrome c molecules should be associated with the accumulation of different protein oligomers detectable on PAGE gels under denaturing conditions (Chen et al., 2004b). Indeed, when cytochrome c/TOCL complexes were incubated with H2O2, dimers, trimers, tetramers of cytochrome c, and larger aggregates, which did not enter the gel because of their very high molecular weight, were observed—along with the monomeric species (Fig. 5B). Etoposide effectively inhibited oligomerization of cytochrome c/TOCL complexes, particularly the formation of very high molecular weight aggregates, thus preserving the monomeric form of cytochrome c (Fig. 5, C and D).

Electrophoresis of cytochrome c/cardiolipin complexes. Liposomes (DOPC/TOCL or DOPC/TLCL) were incubated with cytochrome c (30 and 4 μM for Coomassie blue and silver staining, respectively) (cardiolipin/cytochrome c at a ratio of 25:1) in PBS with 100 mM DTPA at 37°C for 1 h, and 100 mM H2O2 was added every 15 min; 15 μl of the mixture was added to gel wells. Electrophoresis was performed using 12.5% SDS-PAGE with 5% stacking gel. A, effect of etoposide on the formation of dityrosine cross-links in the incubation system TOCL/cytochrome c + H2O2 compared with TLCL/cytochrome c + H2O2. Fluorescence at 420 nm (λex 315 nm) was measured in the presence of varying concentrations of etoposide (0 -250 μM), TOCL (□), and TLCL (▪). Data are mean ± S.E. (n = 3); *, p < 0.005 versus control (no etoposide). Inset, dityrosine fluorescence of cytochrome c oligomeric cross-links formed in the presence of TOCL or TLCL after separation and elution from PAGE. Protein bands from Coomassie blue-stained gels were excised and cut into smaller pieces. Destaining, digestion with trypsin, and extraction of peptides were performed. Dityrosines fluorescence of cytochrome c/TLCL in the presence of H2O2 and cytochrome c/TOCL in the presence of H2O2 was measured as described under Materials and Methods. Data are presented as dityrosine fluorescence intensity normalized to the content of protein in corresponding PAGE bands (as determined by densitometry of Coomassie blue-stained gels). Data are mean ± S.E. (n = 3). B, typical SDS-PAGE gels stained with Coomassie blue. C, integral density of monomeric cytochrome c bands of these samples (B). D, effect of etoposide of the formation of cytochrome c monomers (a) and aggregates (b) induced by H2O2 in the presence of TOCL or TLCL [gels were stained with GelCode SilverSNAP Stain Kit II (Pierce)]. Data are presented as the percentage of control (pure cytochrome c sample); mean ± S.E. (n = 3); *, p < 0.005 versus control.
When ∼8-times higher concentration of cytochrome c (30 μM) was used, a much higher concentration of etoposide (∼350 μM; Fig. 5B, inset) was needed to prevent dityrosine formation in experiments with TOCL. We additionally studied the effects of etoposide on TLCL peroxidation in the same concentration range as for the assessments of dityrosine formation. We incubated TLCL/DOPC liposomes (1.5 mM) in the presence of cytochrome c (30 μM; note that the cytochrome c concentration in these was also higher) and H2O2 (100 μM added four times every 30 min during the incubation) and measured TLCL oxidation by Amplex Red procedure. Using different concentrations of etoposide (7.5-750 μM), we found that under these conditions, I50 for etoposide inhibition of cardiolipin peroxidation was ∼150 μM (Fig. 6A). Furthermore, we verified the protective effect of high concentrations of etoposide (750 μM) against TLCL peroxidation using electrospray ionization mass spectrometry. Significant accumulation of monohydroperoxy/monohydroxy derivatives of TLCL was observed after the incubation in the absence of etoposide (m/z 724.2 + 8 = 732.2; 724.2 + 16 = 739.7; 724.2 + 24 = 748.2; 724.2 + 32 = 755.9; 724.2 + 40 = 763.8; 724.2 + 48 = 771.8; 724.2 + 56 = 779.7; 724.2 + 64 = 787.9; and 724 + 72 = 795.7). This effect was markedly reduced by etoposide (Fig. 6B). These observations indicate that protein tyrosyl radicals are formed even at high (>100 μM) concentrations of etoposide that are sufficient to prevent lipid peroxidation, and etoposide exerts it antioxidant activity by interacting with tyrosyl radicals rather than with hemecentered compound I- and II-reactive intermediates.
The cytochrome c/TLCL complexes did not display the formation of di-, tri-, or tetraoligomers of cytochrome c upon exposure to H2O2. Instead, only very high molecular weight aggregates that did not enter the gel accumulated (Fig. 5A). The dityrosine fluorescence of cytochrome c oligomeric crosslinks formed in the presence of TOCL and TLCL after separation by PAGE. We found that cytochrome c/TOCL aggregates displayed higher levels of characteristic dityrosine fluorescence than aggregates formed during the incubation of cytochrome c/TLCL complexes with H2O2 (Fig. 5A, inset). These results suggest that oxidative oligomerization of cytochrome c in the presence of TLCL probably involves not only dityrosine cross-linking, typical of oligomerization in the presence of TOCL, but additional mechanisms, possibly including cross-linking by bifunctional secondary products of lipid peroxidation (e.g., dialdehydes) (Kagan, 1988). A weaker silver-staining of very high molecular weight aggregates was observed in samples treated with etoposide. This was confirmed quantitatively by densitometry of protein bands performed using Fluor-S Multilmager (Bio-Rad). Additional bands with molecular masses in the range of 30 to 40 kDa appeared in the presence of etoposide. Because the formation of dityrosine cross-links is realized through carboncarbon bonds, the tyrosine hydroxy functionalities remain available for multiple cross-linking. As a result, very high molecular weight aggregates may be produced. Our results suggest that etoposide was able to inhibit this multiple crosslinking and the production of high molecular weight aggregates. However, lower molecular weight oligomers were still formed in detectable amounts in the presence of etoposide as documented by the gels.
Etoposide was able to significantly prevent accumulation of these high molecular weight cross-links (Fig. 5C, inset). Thus, two different types of oligomerization products were formed from complexes of cytochrome c with either nonperoxidizable TOCL or readily peroxidizable TLCL. The former resulted from the production of dityrosine cross-links and exerted characteristic fluorescence, whereas the latter were cross-linked by secondary lipid peroxidation products that did not exert characteristic dityrosine fluorescence. Etoposide was able to substantially inhibit the aggregation of cytochrome c complexes with both TOCL and TLCL, although substantial formation of lipid-protein aggregates in TLCL experiments was still observed at etoposide concentrations as high as 100 μM, indicating that lipid and protein radicals formed as a result of the peroxidase reaction in the tight cytochrome c/cardiolipin complex are protected against antioxidant activity of etoposide molecules.

Mass spectrometry analysis of TLCL oxidation induced by cytochrome c/H2O2 in liposomes. TLCL/DOPC liposomes (1.5 mM at the ratio of 1:1) were treated with cytochrome c (30 μM) and H2O2 (100 μM was added four times every 30 min of incubation) in the absence and presence of etoposide (7.5-750 μM). Lipids were extracted, and the accumulation of oxidized TLCL was quantified by fluorescence HPLC using Amplex Red protocol (A) and analyzed by mass spectrometry (B). The degree of cardiolipin peroxidation at different etoposide concentrations is presented as picomoles of CL-OOH per nanomoles of cardiolipin. The control group represents liposomes without etoposide treatment. The data are presented as mean ± S.E. (n = 3); *, p < 0.05 versus control. Note: the significant accumulation of monohydroperoxy/monohydroxy derivatives of TLCL molecular species (B) as observed after incubation liposomes with cytochrome c and H2O2 was the following: m/z 724.2 + 8 = 732.2; 724.2 + 16 = 739.7; 724.2 + 24 = 748.2; 724.2 + 32 = 755.9; 724.2 + 40 = 763.8; 724.2 + 48 = 771.8; 724.2 + 56 = 779.7; 724.2 + 64 = 787.9; and 724 + 72 = 795.7). This effect was markedly reduced by etoposide (750 μM). Mass spectra prototypical of three independent experiments are presented.
Discussion
Generation of ROS and lipid peroxidation represent two essential events in the execution of apoptotic program, as evidenced by the ability of free radical scavengers and overexpressed antioxidant enzymes to suppress apoptosis (Nomura et al., 1999; Genova et al., 2003). Their specific mechanisms of action in apoptosis are still not completely understood. We have described recently a new catalytic role of cytochrome c as a specific cardiolipin-oxygenase acting at early mitochondrial stages of apoptosis to generate CL-OOH required for the release of proapoptotic factors into the cytosol (Kagan et al., 2005). This implies that lipid antioxidants capable of suppressing cardiolipin oxidation also should be effective as antiapoptotic agents. Indeed, several studies have demonstrated antiapoptotic properties of major lipid-soluble antioxidants such as vitamin E (Buettner, 1993) and ubiquinol (Kelso et al., 2001). We and others have reported high efficiency of etoposide as a specific lipid antioxidant (Tyurina et al., 2004) which, in contrast to the above prediction, is also known as a potent inducer of apoptosis. Therefore, we were anxious to discern why etoposide readily induces apoptosis, notwithstanding its known antioxidant properties. In particular, we analyzed how the apparent inability of etoposide to prevent apoptosis but, conversely, to trigger it may be related to the mechanism of mitochondrial cardiolipin oxidation catalyzed by cytochrome c. We were surprised to find that cardiolipin was oxidized preferentially by etoposide at concentrations at which it induced apoptosis. In fact, the level of cardiolipin oxidation was higher than that induced by a pro-oxidant inducer of apoptosis, AMVN (which does not elicit antioxidant properties). Oxidation of other more abundant phospholipids was markedly lower in the case of etoposide-induced apoptosis compared with AMVN-induced apoptosis. Thus, etoposide was effective as a scavenger of phospholipid radicals, but this scavenging activity could not be realized toward radical intermediates of cardiolipin oxidation. These findings indicated that oxidation of cardiolipin during apoptosis was executed through a different mechanism compared with oxidation of other phospholipids.
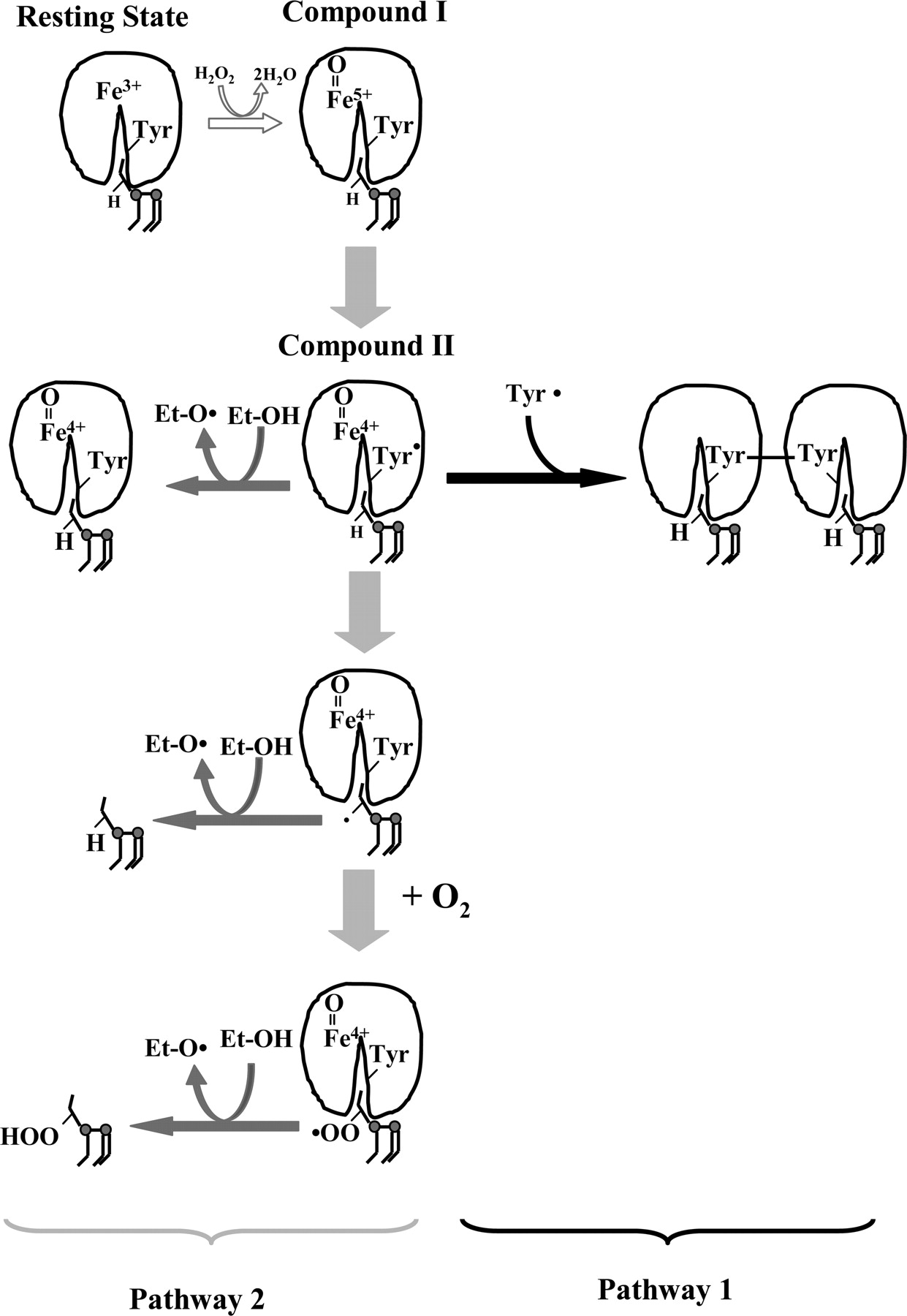
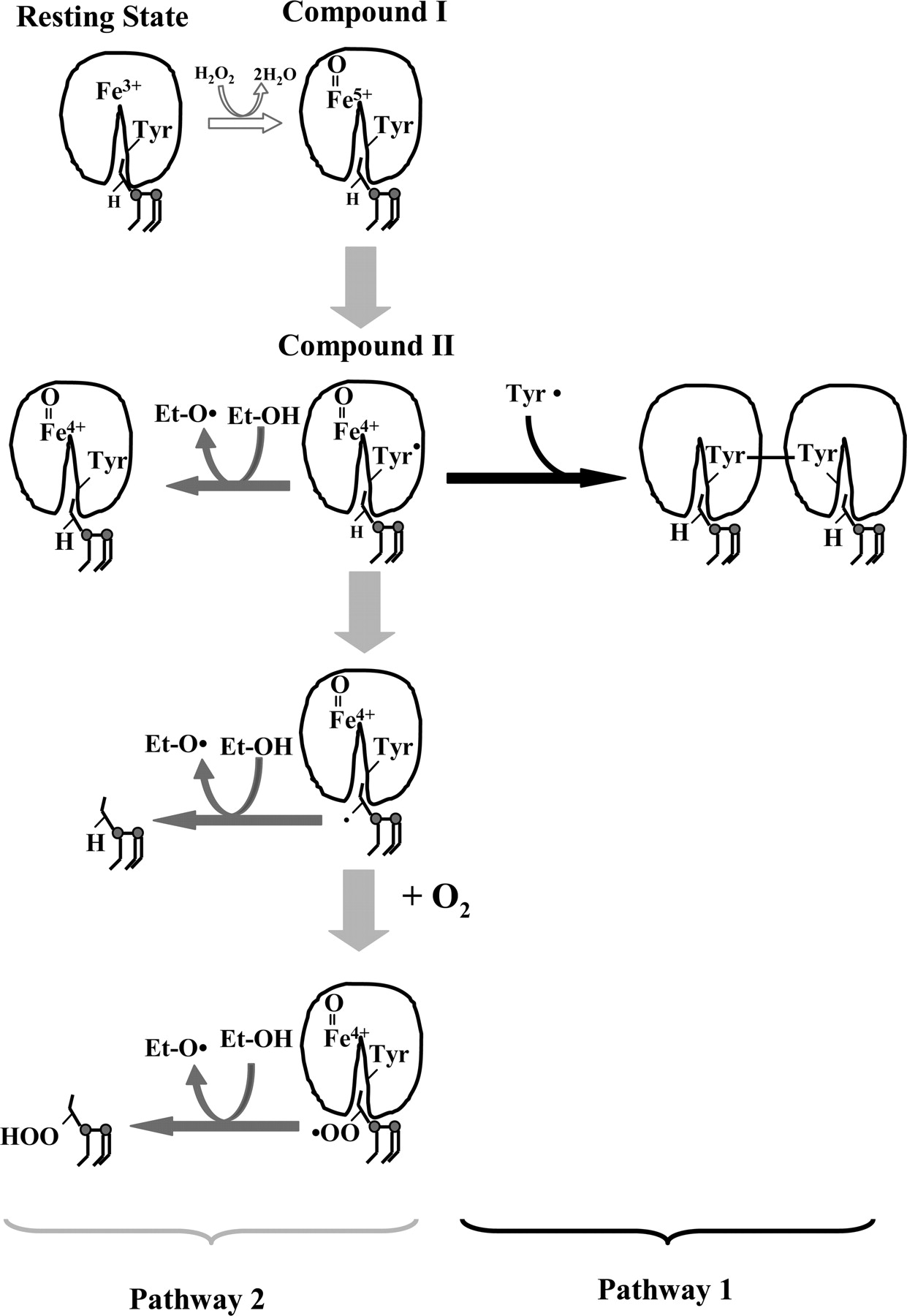
Upon binding to an anionic phospholipid, cardiolipin, cytochrome c is converted into a peroxidase, which, in the presence of H2O2, catalyzes oxidation of cardiolipin, protein amino acids, and other oxidizable compounds. As a result of the reaction of H2O2 with the heme of cytochrome c, highly reactive intermediates compound I and compound II are formed. The scheme shows that the etoposide can interact with protein-derived tyrosyl radical and/or lipid radicals to yield etoposide phenoxyl radicals. However, in the absence of etoposide, the reaction may proceed along one of the two pathways. Compounds I and II can oxidize protein amino acids, specifically tyrosine, (pathway 1), that can dimerize and form protein oligomers or abstract hydrogen from polyunsaturated cardiolipin (e.g., TLCL), resulting in its peroxidation (pathway 2). The lipid alkyl radicals generated can form peroxyl radicals under aerobic conditions and recombine with other lipid and protein radicals, producing lipid and lipid/protein aggregates. Tyrosyl radicals are formed very effectively by compounds I and II, and oligomerization proceeds mostly via pathway 1 in the absence of lipids or in the presence of nonoxidizable lipids (TOCL); when oxidizable polyunsaturated lipids (TLCL) are available, tyrosyl radicals effectively oxidize TLCL acyl chains.
Our previous work has demonstrated that cardiolipin in mitochondria is oxidized mostly as a result of peroxidase reaction catalyzed by cytochrome c bound to cardiolipin-containing mitochondrial membranes (Kagan et al., 2005). In native cytochrome c, hexa-coordinate heme iron—with His18 and Met80 as axial ligands—cannot interact with small molecules, resulting in its very weak peroxidase activity and barely detectable EPR signal of tyrosyl radical in the presence of H2O2. Both the activity and the signal may be associated with partial unfolding of the protein and a slow oxidation of Met80 by H2O2 (Griffiths and Cooney, 2002). It is noteworthy that the magnitude of the H2O2-induced EPR signal from solubilized cytochrome c is approximately 30 times lower than the signal from the cytochrome c/TOCL complex. Several lines of evidence suggest that cytochrome c, the heme protein that normally serves as an electron carrier and which is located in the intermembrane space of mitochondria, dramatically increases its peroxidase ability when it is bound to membranes containing negatively charged lipids, particularly cardiolipin. There are also indications that cardiolipin, which constitutes approximately 25% of phospholipids in the inner mitochondrial membranes, is normally located preferentially in the inner leaflet of the inner mitochondrial membrane (Robinson et al., 1990). However, during apoptosis, cardiolipin is redistributed such that a significant fraction of it is found in the outer leaflet of the inner mitochondrial membrane and in the outer membrane (Robinson et al., 1990; Garcia Fernandez et al., 2002). Thus, physical interactions between cardiolipin and cytochrome c in apoptotic mitochondria are possible, allowing for a switch in cytochrome c's function from an electron carrier to a peroxidase with a high affinity for cardiolipin (Kagan et al., 2005). This explains the low efficiency of etoposide in inhibiting enzymatic (rather than random nonenzymatic) cardiolipin oxidation, whereby cardiolipin radical intermediates are not readily accessible to radical scavengers, similarly to radicals formed during arachidonic acid oxidation by cyclooxygenases (Lassmann et al., 1991). Indeed, our in vitro studies showed a very different protective effect of etoposide against random, free radical oxidation of cardiolipin and oxidation via peroxidation reaction catalyzed by cytochrome c bound to cardiolipin, containing liposomes. The decreased antioxidant potency of etoposide in the cytochrome c/cardiolipin/H2O2 system can be attributed to a tight complex formed by cytochrome c and cardiolipin that ensures close interactions of peroxidase reaction intermediates like compound I, compound II, and amino acid radicals with cardiolipin and prevent their inactivation by etoposide. These results also raise an important question about possible sites of etoposide interactions with radical intermediates generated by peroxidase activation of cytochrome c. Scheme 1 presents possible pathways in the cytochrome c/cardiolipin-catalyzed peroxidase reaction and interactions with etoposide.
The ability of etoposide to quench reactive species generated in the course of the peroxidase reaction and to form etoposide radicals is maximal and similar for both TLCL and TOCL, indicating that peroxidase reaction proceeded similarly for both cardiolipin species (Scheme 1).
Our results show that protein-derived radicals in the case of TLCL were quenched compared with TOCL by 30 ± 7%. This quenching may be the result of oxidation of TLCL by amino acid radicals. A weak dityrosine formation was detected in TLCL experiments, indicating that some of the protein radicals observed are inside the protein and cannot form dityrosine bridges, whereas protein surface tyrosine radicals are quenched by TLCL.
We have demonstrated that peroxidase activity of cytochrome c complexes with TOCL and TLCL was the same as evidenced by two different assays based on H2O2-induced oxidation of 2′,7′-dichlorodihydrofluorescin to 2′,7′-dichlorofluorescein and of Amplex Red to resorufin (Kagan et al., 2005). Because peroxidase activity of cytochrome c is closely associated with the generation of tyrosyl radicals, we assumed that the production of the radicals is probably equivalent in the presence of either TOCL or TLCL. Moreover, according to our estimates, the binding constants of cytochrome c with TOCL and TLCL were essentially identical (1.7 ± 1.0 × 109 M-1 and 1.6 ± 0.2 × 109 M-1, respectively) (Belikova et al., 2006). Furthermore, our low-temperature EPR spectroscopy measurements revealed similar EPR signals of heme-nitrosylated cytochrome c complexes with TOCL or TLCL, indicating the same accessibility of the cytochrome c heme to NO (Vlasova et al., 2006). Finally, the saturation behavior of tyrosyl radicals formed in the peroxidase reactions of cytochrome c complexes with TOCL and TLCL revealed the same distance from the heme iron. Combined, these results strongly suggest that a similar pattern of the protein-derived radical formation is characteristic of cytochrome c complexes with both cardiolipins. Thus, reduced magnitude of the EPR signal detectable in the presence of TLCL is unlikely because of diminished generation of tyrosyl radicals.
It has been reported previously (Chen et al., 2004a) that tyrosyl radicals formed as a result of tyrosine oxidation by peroxidase compounds I and II can be much more effective activators of lipid peroxidation than compounds I and II themselves. Cytochrome c has four tyrosine residues, some of which are located close to the heme moiety, and some are present on the surface of the protein. Interaction of cytochrome c with cardiolipin results in partial unfolding of the protein, and tyrosine residues in the complex formed may readily interact with both heme and polyunsaturated lipid acyl chains. Thus, the formation of cytochrome c/cardiolipin complexes creates conditions for effective and specific cardiolipin peroxidation that is resistant to free radical scavengers such as etoposide.
The specificity of the oxidation process can also be explained considering the redox potentials of redox species involved. Compounds I and II with redox potentials of ∼ 0.95 to 1.0 V (Buettner, 1993) are strong oxidants capable of oxidizing a wide range of substrates. Tyrosyl radical with E° = 0.7 to 0.9 V (Buettner, 1993) can oxidize TLCL (E° = 0.6 V) (Koppenol, 1990) and etoposide (E° = 0.56 V) (Wardman, 1989). Monounsaturated TOCL has a higher redox potential (E° = 0.9 V) (Wardman, 1989), and tyrosyl radicals formed by compounds I- and II-mediated oxidation of tyrosine residues cannot oxidize it.
In summary, we have shown that etoposide can suppress cytochrome c-catalyzed cardiolipin oxidation both in model systems and in cells only at rather high concentrations, exceeding those required for the induction of apoptosis. Thus, etoposide-dependent inhibition of cardiolipin oxidation is not likely to interfere with the execution of the apoptotic program and mitochondrial membrane permeabilization dependent on cardiolipin oxidation. Both cardiolipin peroxidation and etoposide oxidation by cytochrome c/cardiolipin complex predominantly involves protein-derived radicals of cytochrome c. Because nonspecific lipid antioxidants are not effective in inhibiting cardiolipin oxidation, other approaches based on the blockade of peroxidase reaction of cytochrome c/cardiolipin complexes should be developed. In this regard, mitochondria-targeted nitroxide radicals capable of acting as selective acceptors of electrons from components of respiratory chain and preventing accumulation of superoxide and H2O2 may be promising (Wipf et al., 2005). Another interesting antiapoptotic approach may be based on manipulations of cardiolipin fatty-acid composition yielding its monounsaturated molecular species resistant to peroxidation (Kagan et al., 2005).
Footnotes
-
This work was supported by National Institutes of Health grants HL70755, U19-AIO68021, CA90787, and GM64610.
-
ABBREVIATIONS: ROS, reactive oxygen species; DTPA, diethylenetriaminepentaacetic acid; AMVN, 2,2′-azo-bis(2,4-dimethylvaleronitrile); PE, phosphatidylethanolamine; PAPC, 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine; PAPS, 1-palmitoyl-2-arachidonoyl-sn-glycero-3-[phospho-l-serine]; TLCL, 1,1′,2,2′-tetralinoleoyl-cardiolipin; TOCL, 1,1′,2,2′-tetraoleoyl-cardiolipin; CL-OOH, cardiolipin hydroperoxides; EPR, electron paramagnetic resonance; PBS, phosphate-buffered saline; 2D, two-dimensional; HPTLC, high performance thin-layer chromatography; HPLC, high-performance liquid chromatography; DOPC, 1,2-dioleoylphosphatidylcholine; PAGE, polyacrylamide gel electrophoresis; PC, phosphatidylcholine; PAPANONOate, (Z)-1-[N-3-aminopropyl)-N-(n-propyl)amino]diazen-1-ium-1,2-diolate; CL, cardiolipin.
- Received January 18, 2006.
- Accepted May 11, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}