


Abstract
Sulforaphane (SFN) is a biologically active phytochemical found abundantly in broccoli. SFN has been promoted as a putative chemopreventive agent to reduce cancer, and most studies have associated its anti-cancer effects with the induction of phase II xenobiotic metabolism enzymes via activation of the Keap1/Nrf2 antioxidant response pathway. Interestingly, SFN can significantly down-regulate cytochrome P450 3A4 (CYP3A4) expression in human primary hepatocytes. CYP3A4 is responsible for the hepatic and intestinal metabolism of numerous protoxicants, pharmaceutical compounds, and endogenous sterols. Among the most important mediators of CYP3A4 expression is the nuclear hormone receptor, steroid and xenobiotic receptor (SXR; also called “hPXR”). SXR functions as a xenobiotic sensor to coordinately regulate xenobiotic metabolism via transcriptional regulation of xenobiotic-detoxifying enzymes and transporters. Here, we report that SFN is a specific antagonist of human SXR and that it inhibits SXR-mediated induction of drug clearance. SFN can bind directly to SXR, inhibit SXR coactivator recruitment, and efficiently repress SXR activities. Furthermore, SFN inhibited SXR-mediated CYP3A4 expression and CYP3A4-catalyzed midazolam clearance in human primary hepatocytes. Thus, SFN is the first identified naturally occurring antagonist for SXR (hPXR). Because induction of CYP3A4 can result in adverse drug responses (e.g., lack of efficacy), which are a major public health problem, this discovery could lead to the development of important new therapeutic and dietary approaches to reduce the frequency of undesirable inducer-drug interactions.
Sulforaphane (SFN) is one of the most biologically active phytochemicals in the human diet (Fig. 1A), and it is present at high concentrations in some cruciferous vegetables, especially broccoli (Zhang et al., 1992; Kushad et al., 1999). Epidemiological and clinical studies have indicated that diets high in cruciferous vegetables protect against a number of cancers, including non-Hodgkin's lymphoma, liver, prostate, cervical, ovarian, lung, and gastrointestinal tract (Murillo and Mehta, 2001). Numerous studies in animal models and human cells support the putative chemopreventive effects of SFN (Zhang et al., 1994; Chung et al., 2000; Conaway et al., 2002). For example, SFN treatment reduced 7,12-dimethylbenz(a)anthracene-induced mammary tumors (Zhang et al., 1994), inhibited benzo(a)pyrene-induced forestomach tumors in mice (Fahey et al., 2002), lowered the formation of colonic aberrant crypt foci in rats (Chung et al., 2000), and inhibited cell proliferation of an HT-29 colon cancer cell line (Frydoonfar et al., 2004). In addition, in a recent study with human hepatocytes in primary culture, we demonstrated that pretreatment of hepatocytes with 50 μM SFN produced more than a 90% decrease in DNA adduction of the potent hepatocarcinogen aflatoxin B1 (Gross-Steinmeyer et al., 2005).
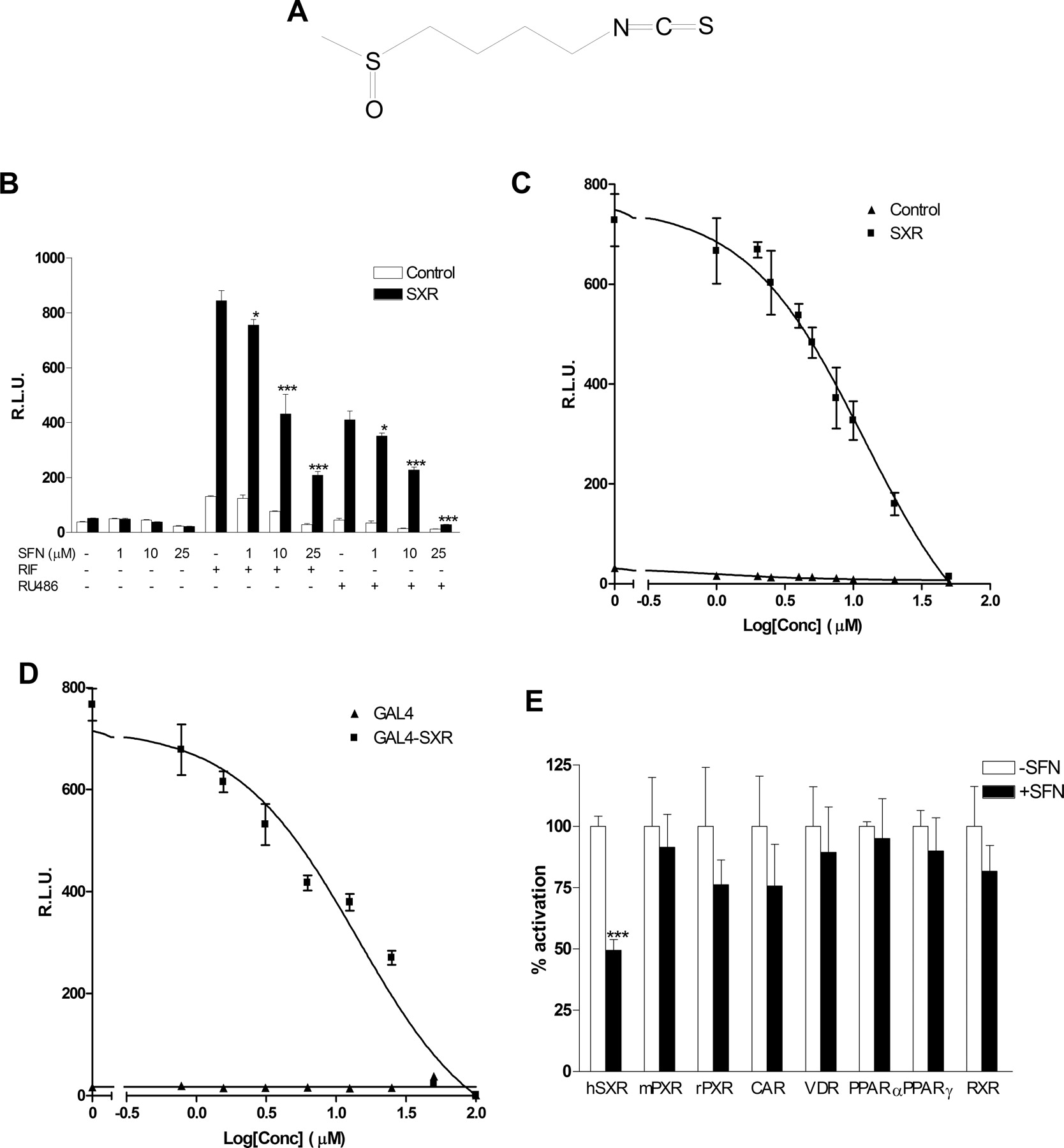
SFN efficiently inhibits SXR activity. A, structure of SFN (4-methylsulfinylbutyl isothiocyanate). B, HepG2 cells were transiently transfected with full-length SXR together with a CYP3A4-luciferase reporter and CMX-β-galactosidase transfection control plasmid. After transfection, cells were treated with control medium or medium containing 10 μM RIF or 10 μM RU486 in the absence or presence of SFN at the indicated concentrations for 24 h. Results were presented as relative luciferase units (RLUs) normalized to the β-galactosidase internal control. C, HepG2 cells were transiently transfected as described above. Cells were treated with 10 μM RIF with indicated concentrations of SFN for 24 h. D, HepG2 cells were transiently transfected with GAL4-SXR, a GAL4 reporter fused to luciferase and CMX-β-galactosidase transfection control plasmid. Cells were then treated with 10 μM RIF and SFN at the indicated concentrations for 24 h. E, HepG2 cells were cotransfected with GAL4 reporter and a series of GAL4 constructs in which the GAL4 DNA binding domain is linked to the indicated nuclear hormone receptor ligand binding domain. Cells were treated with the appropriated ligand or ligand plus 10 μM SFN. The ligands used were mouse PXR (mPXR) (10 μM PCN), rat PXR (rPXR) (10 μM PCN), vitamin D receptor (VDR) [10 nM 1,25(OH)2D3], and constitutive androstane receptor (CAR) [1 μM 6-(4-chlorophenyl)imidazo[2,1-b]-[1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime], PPARα (10 μM Wy-14,643), PPARγ (10 μM troglitazone), and retinoid X receptor (RXR) (100 nM 9-cis-retinoic acid). Results in the presence of SFN are presented as percentage of activation relative to the normalized luciferase values in the presence of ligands (100%). Statistically significant expression compared with control group (-SFN), respectively, is marked with asterisks: *, P < 0.05; **, P < 0.01; and ***, P < 0.001 (n = 3; Student's t test).
The mechanism(s) of action of the putative chemopreventive effects of SFN seems to be multifactorial. SFN can induce apoptosis and cell cycle arrest in human cancer cells (Gamet-Payrastre et al., 2000), and it is an inhibitor of histone deacetylases (Myzak et al., 2006). However, most studies have associated the anticancer effects of SFN with the induction of phase II drug metabolism enzymes, especially the glutathione transferases (GSTs) (Talalay et al., 1995). SFN activates the Keap1/Nrf2 transcriptional factor complex that can bind to the antioxidant response element and induce a series of detoxification enzymes, such as NAD(P)H:quinone oxidoreductase-1 (NQO1), certain GSTs, and UDP-glucuronosyltransferases (UGTs), and other genes involved in antioxidant response (Fahey and Talalay, 1999; Conaway et al., 2002; Gao and Talalay, 2004).
Interestingly, it has also been reported that SFN down-regulated CYP3A4 transcription and enzyme activity in cultured human hepatocytes, suggesting another mechanism that could also contribute to its anticancer effects (Maheo et al., 1997). Indeed, we subsequently confirmed that SFN consistently and dramatically reduced CYP3A4 mRNA content in human hepatocytes (Gross-Steinmeyer et al., 2005).
The nuclear hormone receptor, steroid and xenobiotic receptor (SXR) (Blumberg et al., 1998) [also known as pregnane X receptor (PXR) (Kliewer et al., 1998), pregnane-activated receptor, and NR1I2], plays a central role in the transcriptional regulation of CYP3A4 (for review, see Dussault and Forman, 2002; Kliewer et al., 2002). Here, we use PXR to refer to the rodent form, and SXR to refer to the human form, hPXR). SXR is activated by a diverse array of pharmaceutical agents, including paclitaxel (Taxol), rifampicin (RIF), SR12813, clotrimazole, phenobarbital, the herbal antidepressant St. John's wort, and peptide mimetic HIV protease inhibitors such as ritonavir (Dussault and Forman, 2002; Kliewer et al., 2002). These studies indicate that SXR functions as a xenobiotic sensor (Blumberg et al., 1998) to coordinately regulate drug clearance in the liver and intestine via transcriptional regulation of xenobiotic-detoxifying enzymes and transporters such as CYP3A4 and MDR1 (Dussault and Forman, 2002; Kliewer et al., 2002). Because SFN significantly inhibited CYP3A4 expression, we tested whether down-regulation of CYP3A4 by SFN is mediated by SXR.
CYP3A4 is among the most important enzymes of the P450 family because it contributes to the metabolism of more than 50% of clinically used drugs and a corresponding number of xenobiotic chemicals (Guengerich, 1999). Surprisingly, it exhibits marked interindividual variability in terms of its specific content and activity in the liver and small intestine, the primary sites of drug metabolism. Moreover, induction or inhibition of CYP3A4 is a common cause of adverse drug-drug interactions, which are a major public health problem in the United States. Adverse drug reactions account for 10 to 17% of the medical indications for acute hospital admission of elderly patients (Beard, 1992) and may contribute to more than 100,000 deaths in the United States each year. By some estimates, it represents the fourth to sixth leading cause of death in the United States (Wrighton and Thummel, 2000). Thus, modification of CYP3A4 expression and activity by consumption of SFN could have important implications for drug safety.
Here, we report that SFN is a specific antagonist of SXR and inhibits SXR-mediated induction of drug clearance. SFN was able to efficiently inhibit SXR-mediated transcription of the CYP3A4 gene in a concentration-dependent manner. SFN bound directly to SXR and inhibited SXR-coactivator interactions. Furthermore, SFN inhibited SXR-mediated CYP3A4 expression and CYP3A4-mediated midazolam (MDZ) clearance in human primary hepatocytes. Thus, SFN is the first identified naturally occurring antagonist for SXR. This discovery could contribute to a better understanding of the mechanism of interindividual variability in intestinal and hepatic CYP3A4-dependent drug metabolism. These findings could also lead to potentially important new therapeutic and dietary approaches to reduce the frequency of adverse drug reactions that are secondary to SXR-mediated induction of drug clearance via CYP3A4, MDR1, and other genes regulated in part by SXR. These findings also point to a novel and complementary mechanism by which SFN exerts its putative chemoprotective effects through a reduction in CYP3A4-dependent reactive metabolite formation.
Materials and Methods
Reagents and Plasmids. SFN, RIF, mifepristone (RU486), and clotrimazole were purchased from Sigma-Aldrich (St. Louis, MO); pregnenolone 16α-carbonitrile (PCN), 6-(4-chlorophenyl)imidazo[2,1-b]-[1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime, Wy-14,643, troglitazone, and 9-cis-retinoic acid were purchased from BIOMOL Research Laboratories (Plymouth Meeting, PA); and 1,25(OH)2D3 was purchased from Calbiochem (San Diego, CA). Iberin, cheirolin, erucin, and phenethyl isothiocyanate were purchased from LKT Laboratories, Inc. (St. Paul, MN). SXR, GAL4-SXR LBD, VP16-SXR, and CMX-β-gal expression vectors; SXR-dependent CYP3A4 promoter reporter (CYP3A4XREM-luciferase); and GAL4 reporter (MH100-luciferase) have been described previously (Blumberg et al., 1998; Synold et al., 2001; Zhou et al., 2004, 2006b).
Cell Culture. The human intestinal epithelial cell line, LS180 were obtained from American Type Culture Collection (Manassas, VA) and cultured in DMEM containing 10% FBS at 37°C in 5% CO2. The cells were seeded into six-well plates and grown in DMEM-10% FBS until 70 and 80% confluence. Twenty-four hours before treatment, the medium was replaced with DMEM containing 10% resin-charcoal stripped FBS. Immediately before treatment, the medium was removed; the cells were washed once with phosphate-buffered saline and then treated with compounds or dimethyl sulfoxide vehicle for appropriate times. Human primary hepatocytes were obtained from Liver Tissue Procurement and Distribution System (University of Pittsburgh, Pittsburgh, PA) as attached cells in six-well plates. The hepatocytes were maintained in hepatocyte medium (Sigma-Aldrich) for at least 24 h before treatment.
Transient Transfection and Luciferase Assay. Transfection assays were performed as described previously (Zhou et al., 2004). To test the ability of SFN to inhibit SXR or other nuclear receptors, HepG2 cells were seeded into 12-well plates overnight and transiently transfected with the control or SXR expression plasmid, together with the CYP3A4XREM-luciferase reporter and CMX-β-galactosidase transfection control plasmids using FuGENE 6 (Roche Diagnostics, Indianapolis, IN) in serum-free DMEM. Twenty-four hours post-transfection, the cells were treated with dimethyl sulfoxide as a negative control, and the known SXR ligands RIF, RU486, and clotrimazole, in the absence or presence of SFN. The cells were lysed 24 h after treatment, and β-galactosidase and luciferase assays were performed as described previously (Grun et al., 2002). Reporter gene activity was normalized to the β-galactosidase transfection controls, and the results are expressed as normalized relative luciferase units per optical density β-galactosidase per minute to facilitate comparisons between plates. -Fold induction was calculated relative to solvent controls. Each data point represents the average of triplicate experiments ± S.E.M. and was replicated in three to four independent experiments. For mammalian two-hybrid assays, HepG2 cells were transfected with GAL4 reporter; VP16-SXR; and GAL-SRC1, GAL-PBP, GAL-ACTR, or GAL-TIF2 (kindly provided by Dr. B. M. Forman, City of Hope National Medical Institute, Duarte, CA) (Synold et al., 2001). The cells were then treated with 10 μM RIF or RU486 in the presence or absence of SFN at the indicated concentration. IC50 values were calculated by curve fitting of data, assuming a competitive antagonism model, using Prism software (GraphPad Software, San Diego, CA).
Ligand Binding Assays. N-Terminal His6-tagged human SXR ligand binding domain (LBD) was expressed in Escherichia coli together with the steroid receptor coactivator-1 (SRC-1) receptor interaction domain, and scintillation proximity assays were performed essentially as described previously (Zhou et al., 2004). In brief, active protein was refolded from inclusion bodies solubilized in denaturation buffer [6 M guanidinium-HCl, 50 mM HEPES, pH 7.4, 0.2 M NaCl, 25 mM dithiothreitol, and 1% (w/v) Triton X-100] by rapid 10-fold dilution into binding buffer [50 mM HEPES, pH 7.4, 1 M sucrose, 0.2 M NaCl, 0.1 mM dithiothreitol, and 0.1% (w/v) CHAPS] followed by dialysis overnight at 4°C against binding buffer. Binding assays were performed by coating 96-well nickel chelate FlashPlates (PerkinElmer Life and Analytical Sciences, Boston, MA) with a 10-fold molar excess of protein for 1 h at 22°C in binding buffer (50 mM HEPES, pH 7.4, 200 mM NaCl, 1 M sucrose, and 0.1% CHAPS). Unbound protein was removed from the wells by washing four times with binding buffer. [3H]SR12813 (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK) was added to a final concentration of 50 nM in each well, either alone or together with competitor ligands in binding buffer as indicated. Incubation was continued for 3 h at room temperature. Total counts were measured using a TopCount scintillation counter (PerkinElmer Life and Analytical Sciences). Counts remaining after the addition of 10 μM clotrimazole were taken as nonspecific background and subtracted from all wells. All assays were performed in triplicate and reproduced in independent experiments.
Competition binding curves were determined at constant 3H-specific ligand concentrations (50 nM SR12813; Kd = 41 nM; Zhou et al., 2004) with increasing unlabeled competitor ligands over the range indicated in the figure. Data were analyzed in GraphPad Prism by nonlinear regression of a competitive one-site binding equation [Cheng and Prusoff (1973) method] to determine Ki values ± 95% confidence intervals (n = 3).
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction Analysis. Total RNA was isolated from primary hepatocytes and LS180 cells using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer-supplied protocol. Quantitative real-time PCR was performed using gene-specific primers and the SYBR Green PCR kit (Applied Biosystems, Foster City, CA) in an ABI 7900 system (Applied Biosystems). All samples were quantified using the comparative CT method for relative quantification of gene expression, normalized to glyceraldehyde-3-phosphate dehydrogenase (Livak and Schmittgen, 2001). The following primer sets were used in this study: CYP3A4 (5′-GGCTTCATCCAATGGACTGCATAAAT-3′ and 5′-TCCCAAGTATAACACTCTACACAGACAA-3′). Primer sites were in exon 13 of CYP3A4, which represents a unique region that distinguishes CYP3A4 from CYP3A5 and 3A7; MDR1 (5′-CCCATCATTGCAATAGCAGG-3′ and 5′-GAGCATACATATGTTCAAACTTC-3′); UGT1A1 (5′-TGCTCATTGCCTTTTCACAG-3′ and 5′-GGGCCTAGGGTAATCCTTCA-3′); NQO1 (5′-GGCAGAAGAGCACTGATCGTA-3′ and 5′-TGATGGGATTGAAGTTCATGGC-3′); and glyceraldehyde-3-phosphate dehydrogenase (5′-GGCCTCCAAGGAGTAAGACC-3′ and 5′-AGGGGAGATTCAGTGTGGTG-3′).
MDZ Clearance Analysis. An internal standard mixture containing 15N3-labeled MDZ metabolite 1′-hydoxymidazolam (1′-OH MDZ) was prepared by incubating 6 nmol of cytochrome P450 (using HL-122 microsomes) with 100 μg of 15N3-MDZ and 12 mg of NADPH (final concentration ∼1.5 mM) in potassium phosphate buffer (0.1 M, pH 7.4, in a final volume of 8 ml) at 37°C. After 10 min, the reaction was stopped by the addition of 8 ml of 0.1 M Na2CO3 (pH 12). The compounds were extracted twice with 20 ml of ethyl acetate, and the solvent was evaporated to dryness under a stream of nitrogen. The remaining solid was then reconstituted in 20 ml of methanol, split into two 10-ml aliquots, and stored at -80°C. To determine CYP3A4 activity, human primary hepatocytes were preincubated with 10 or 25 μM SFN for 24 h before addition of 10 μM RIF. Twenty-four hours later, cells were rinsed with media three times and then incubated with new media containing 8 μM MDZ for 6 h. The supernatant media were collected for 1′-OH MDZ formation analysis as described previously (Paine et al., 1997). In brief, Samples were spiked with 100 μl of a 1:5 dilution of the internal standard mixture, which represented ∼50 ng of 15N3-labeled 1′-OH MDZ. The metabolites were extracted with 5 ml of ethyl acetate, the solvent was removed under nitrogen, and the concentrated extracts were dissolved in 100 μl of derivatizing reagent [10% N-methyl-N-(t-butyl-dimethylsilyl)trifluoroacetamide in acetonitrile]. The samples were then transferred to autoinjector vials and were analyzed for 1′-OH MDZ by selective ion gas chromatography-negative chemical ionization mass spectrometry (MS) as described previously (Paine et al., 1997). The 1′-OH MDZ was quantified by comparing peak area ratios with standard curves prepared by the addition of known amounts of 1′-OH MDZ (0-160 pmol) and 100 μl of internal standard to phosphate buffer.
Statistical Analysis. We used a two-sample, two-tailed Student's t test to evaluate the effect of SFN treatment on the control, RIF-, or RU486-mediated inductive response in the different cultured cell systems. A P value less than 0.05 was considered to be significant. One-way analysis of variance was used when multiple comparisons were made, followed by Dunnett's t test for multiple comparisons to a control.
Results
SFN Efficiently Inhibits SXR Activity. We previously observed that SFN consistently and dramatically reduced CYP3A4 mRNA content in human hepatocytes. SXR contributes importantly to both constitutive and inducible expression of CYP3A4 (Dussault and Forman, 2002; Kliewer et al., 2002) and several other genes involved in xenobiotic disposition (e.g., MDR1). SXR is activated by a diverse array of pharmaceutical agents, including paclitaxel, RIF, RU486, SR12813, clotrimazole, phenobarbital, and hyperforin, and it enhances the transcription of its target genes (Blumberg et al., 1998; Kliewer et al., 1998, 2002). Thus, we tested the ability of SFN to inhibit ligand-mediated activation of SXR by use of transfection assays. Two different SXR ligands, RIF and RU486, were able to strongly induce SXR reporter activities in SXR-transfected cells (Fig. 1B). SFN significantly inhibited both RIF and RU486 induced reporter activities. This effect was SFN dose-dependent. Inhibition of SXR reporter activity was detected at a concentration as low as 1 μM, and, at 25 μM, SFN completely blocked RU486 induced SXR reporter activity. Moreover, RIF induction was completely blocked by 50 μM SFN (Fig. 1, B and C). Dose-response analysis revealed that the IC50 for SFN inhibition of 10 μM RIF-induced CYP3A4 promoter activity was approximately 12 μM (Fig. 1C). To further confirm that SFN inhibited SXR function, we also transfected HepG2 cells with a GAL4 reporter along with a vector expressing the SXR ligand binding domain linked to the DNA binding domain of GAL4 (GAL4-SXR). Consistent with the results obtained using the full-length SXR, SFN elicited a similar potency of inhibition of GAL4-SXR activity (Fig. 1D), with an IC50 of 14 μM.

SFN specifically binds to the purified SXR ligand binding domain. His6-SXR LBD was coexpressed with the SRC-1 receptor interaction domain and purified. The receptor complex was bound to nickel chelate FlashPlates and incubated with 50 nM of [3H]SR12813 in the presence of indicated concentration of SFN or clotrimazole. Values represent the average of triplicates ± S.E.M.
To determine whether SFN acts specifically on SXR, we evaluated the ability of SFN to inhibit ligand activation of a number of other nuclear hormone receptors, including mouse PXR, rat PXR, and human constitutive androstane receptor, vitamin D receptor, PPARα, and PPARγ. SFN (tested at 10 μM concentration) had little, if any, inhibitory effect on ligand activation of any of these other nuclear hormone receptors nor did it serve as an activating ligand (Fig. 1E). Surprisingly, although 10 μM SFN can efficiently inhibit SXR activity, it had comparatively little inhibitory effect on rodent PXR (mouse PXR or rat PXR) function. This observation is consistent with an in vivo study that found that SFN did not inhibit rat CYP3A gene expression (Hu et al., 2004). These data suggest that SFN is a species-selective antagonist of human SXR function, perhaps analogous to the known species selectivity of RIF as an effective human but not rodent SXR/PXR ligand (Blumberg et al., 1998).
SFN Can Specifically Bind to SXR. Because SFN effectively inhibited human SXR activities in transient transfection assays (Fig. 1), we hypothesized that SFN works as an antagonist of SXR. Most natural and synthetic nuclear receptor agonists or antagonists act as ligands by directly binding to the nuclear receptor ligand binding domain. Thus, we next tested whether SFN can bind directly to purified SXR protein in vitro using a sensitive scintillation proximity ligand binding assay (Zhou et al., 2004). This assay used the high-affinity SXR ligand [3H]SR12813 and recombinant histidine-6-tagged-SXR coexpressed with the SRC-1 receptor interacting domain. SFN as well as clotrimazole (positive control) displaced [3H]SR12813 from the SXR LBD in a dose-dependent manner (Fig. 2). The Ki for SFN binding to SXR was 16 μM, a value in the range of other known SXR ligands (Jones et al., 2000). In addition, the affinity was similar to the value we obtained for inhibition of SXR function in transfection experiments (Fig. 1, C and D). We infer from these results that SFN binds specifically to the ligand binding domain of SXR.
SFN Inhibits SXR Coactivator Interactions. In the absence of ligand, many nuclear receptors form a complex with corepressors that inhibit transcriptional activity of the complex through the recruitment of histone deacetylase. When a ligand binds to its nuclear receptor, a conformational change occurs, resulting in dissociation of corepressor and recruitment of coactivator proteins (Glass and Rosenfeld, 2000). Coactivator recruitment is therefore a critical part of nuclear receptor signaling pathways. Several coactivators have been shown to be important for nuclear receptor activation, including the SRC-1, transcriptional intermediary factor (TIF)2, activator of thyroid and retinoic acid receptor (ACTR), and peroxisome proliferator-activated receptor-binding protein (PBP) (Synold et al., 2001). Because our data demonstrate that SFN blocks ligand binding to SXR, it was of interest to determine whether SFN also inhibits ligand-induced recruitment of coactivators to SXR. We used the mammalian two-hybrid assay to evaluate whether SFN affects the SXR and coactivator interaction. HepG2 cells were transfected with a GAL4 reporter, a vector expressing VP16-SXR, and an expression vector for the GAL4 DNA binding domain or the GAL4 DNA binding domain linked to the receptor interaction domains of the indicated coactivators. Consistent with previous reports (Synold et al., 2001), RIF strongly promoted the specific interaction of SRC-1 and PBP (Fig. 3), but, as expected, it had no significant interaction with ACTR and TIF2 (data not shown). SFN inhibited RIF induced SRC-1 and PBP recruitment to SXR in a dose-dependent manner (Fig. 3), and again there was no interaction with ACTR and TIF2 (data not shown). Thus, although structurally distinct from previously described natural or synthetic ligands, SFN seems to antagonize SXR function via direct binding to SXR, inhibition of ligand binding, and subsequent inhibition of SXR coactivator recruitment, thereby potentially preventing ligand-mediated SXR transcriptional activation of SXR-regulated genes in a concentration-dependent manner.
SFN Inhibits SXR-Mediated CYP3A4 Expression in LS180 Cells and Human Primary Hepatocytes. Given the fact that SXR is a major regulator of CYP3A4 and our observation that SFN is an effective antagonist of human SXR, we evaluated whether SFN modulates SXR-mediated CYP3A4 gene expression in human cells that express CYP3A4. Human intestinal LS180 cells and primary hepatocytes were used for CYP3A4 gene expression analysis. LS180 cells are derived from a human colonic epithelial tumor, and they represent one of very few human-derived cell lines that have been demonstrated to have functional SXR and inducible CYP3A4 (Synold et al., 2001; Zhou et al., 2004). LS180 cells and human primary hepatocytes from two different donors were pretreated with various concentrations of SFN for 24 h before addition of 10 μM RIF or RU486. Pretreatment of cells was included in the experimental design to be consistent with previous studies that had demonstrated that SFN pretreatment lowered CYP3A4 mRNA and aflatoxin-DNA adduct formation (Gross-Steinmeyer et al., 2005). Total RNA was isolated 24 h later, and quantitative reverse transcription-PCR was performed to measure CYP3A4 gene expression. As expected, both RIF and RU486 were able to induce CYP3A4 gene expression. RIF was a more potent inducer of CYP3A4 than was RU486, consistent with previous reports (Zhou et al., 2004, 2006a). SFN caused a dose-related reduction in RIF- and RU486-mediated induction of CYP3A4 in both primary hepatocytes (Fig. 4, A and B) and LS180 cells (Fig. 4C). Consistent with the results obtained from transfection experiments (Fig. 1), SFN was able to significantly inhibit both RIF- and RU486-induced CYP3A4 expression at a 10 μM concentration, and it almost completely blocked CYP3A4 induction at 25 μM. Additional experiments with LS180 cells were conducted in which the 24-h pretreatment was eliminated, such that cells were only cotreated for 24 h with SFN and RIF. The results were not significantly different from experiments in which pretreatment with SFN was used (data not shown). Interestingly, SFN also significantly reduced the basal level of CYP3A4 expression in primary hepatocytes, as we observed previously (Gross-Steinmeyer et al., 2005). Furthermore, in LS180 cells, SFN inhibited RIF- or RU486-induced expression of MDR1 (ABCB1), a gene that is also regulated by SXR (Fig. 4C). As expected, SFN induced UGT1A1 gene expression, presumably through an SXR-independent and Nrf2-dependent pathway (Basten et al., 2002). SFN significantly induced another Nrf2 target gene, NQO1, in both LS180 cells and human primary hepatocytes (Fig. 4D). The net inductive effect of SFN on UGT1A1 gene expression is intriguing, because the gene is also regulated in part by SXR. Indeed, both RIF and RU486 slightly induced UGT1A1 expression, consistent with previous studies (Xie et al., 2003; Zhou et al., 2004). Thus, it is clear that two distinct pathways are involved in SFN-mediated gene regulation: SFN activates Nrf2 signaling pathway and induces antioxidant response element target genes such as NQO1 and UGT1A1, but it also acts as an antagonist of SXR, thereby inhibiting SXR-mediated CYP3A4 and MDR1 gene expression. SFN has also been shown to inhibit histone deacetylase(s), which could also contribute to changes in gene expression (Myzak et al., 2006).

SFN inhibits SXR coactivator interactions. HepG2 cells were transfected with a GAL4 reporter and VP16-SXR as well as expression vector for GAL4 DNA binding domain or GAL4 DNA binding domain linked to the receptor interaction domains of the indicated SXR coactivators (GAL-SRC1 and GAL-PBP). Cells were then treated with control medium or medium containing 10 μM RIF in the presence or absence of SFN at indicated concentrations. Statistically significant expression compared with control group (-SFN) is marked with asterisks: ***, P < 0.001 (n = 3; Student's t test).
SFN Suppresses Constitutive and Inducible CYP3A4-Mediated MDZ Clearance in Human Primary Hepatocytes. Midazolam is a commonly used short-acting benzodiazepine that is metabolized mainly to 1′OH-MDZ almost exclusively by CYP3A4 (Kronbach et al., 1989). MDZ has been used successfully as an in vivo and in vitro CYP3A4 probe to measure CYP3A4 metabolic activity (Paine et al., 1997). We tested whether SFN suppresses MDZ clearance in one preparation of human primary hepatocytes. Human primary hepatocytes were pretreated with 10 or 25 μM SFN for 24 h before addition of 10 μM RIF. After an additional 24 h of incubation with both RIF and SFN, cells were rinsed with phosphate-buffered saline and then incubated with 8 μM MDZ for 6 h. Supernatant was collected, and 1′OH-MDZ concentration was measured by liquid chromatography-MS. Consistent with CYP3A4 gene expression analysis, RIF effectively induced MDZ clearance approximately 3-fold, and 25 μM SFN blocked both basal and RIF-induced MDZ clearance (Fig. 5). Interestingly, SFN almost completely suppressed RIF-induced MDZ clearance at 10 μM, whereas it decreased RIF-induced CYP3A4 gene expression (mRNA) by only 50% at the same concentration.
Previous studies from our laboratory demonstrated that pretreatment with SFN is required to block P450-mediated activation of aflatoxin B1 in human hepatocytes (Gross-Steinmeyer et al., 2005) and that SFN is not a direct inhibitor of CYP3A4 catalytic activity, even at concentrations as high as 50 μM (K. Gross-Steinmeyer and D. L. Eaton, unpublished observations). Therefore, the inhibition of RIF-induced MDZ clearance by SFN most likely reflects the inhibition of CYP3A4 gene expression rather than CYP3A4 enzyme activity, and quantitative differences in SFN effects most likely reflect a partial discordance in the time course of change in mRNA and protein synthesis. Although we did not determine the effects of cotreatment only (no pretreatment) of SFN on MDZ activity in human hepatocytes, we would expect qualitatively similar results, and that the additional 24-h pretreatment with SFN before RIF administration is not needed to block the effects of RIF on CYP3A4 catalytic activity. However, it is worth noting that 24-h pretreatment with 10 μM SFN, followed by 24-h cotreatment of 10 μM SFN with RIF, abolished the RIF-mediated increase in MDZ activity, but that 48 h of 10 μM SFN had a much more limited repressive effect on constitutively expressed CYP3A4 activity. In contrast, SFN at the 25 μM dose level blocked RIF-mediated induction and decreased constitutive activity substantially. Although the reason for this dose-dependent SFN effect is not known with certainty, it suggests the possibility of a more complex mechanism of SXR antagonism that simple competitive binding or a mechanism of constitutive CYP3A4 expression that involves more than SXR activation.
Structural Determinants of SXR Antagonism by SFN. There are numerous other naturally occurring isothiocyanates present in a variety of cruciferous vegetables. Because SFN is structurally distinct from previously described natural or synthetic ligands of SXR, we tested naturally derived phytochemicals that represent structural analogs of SFN to elucidate which part of SFN contributes to its antagonistic effect (Fig. 6). The results suggested that the isothiocyanate moiety is critical for antagonism of SXR, because the nitrile breakdown product of SFN (SFN-nitrile) had no inhibitory activity at any concentration. Moreover, the methylsulfoxide part of the molecule also plays a role in SXR antagonism. Replacing the methylsulfoxide moiety with a phenyl (phenethyl isothiocyanate) group resulted in a substantial loss of the inhibitory effect of SFN; only the highest dose of 25 μM had a statistically significant inhibitory effect. Interestingly, the oxidation state of the methylsulfide moiety seems to be important as well. A fully reduced sulfur (erucin) had much less inhibitory activity toward SXR function (inhibition only at the highest dose), and the fully oxidized sulfur, cheirolin, had similar inhibitory effects with SFN at high concentrations, whereas it was less potent at low concentration. Similar to cheirolin, shortening the carbon chain from n-butyl to n-propyl (iberin) had little effect, although linker length and rotational flexibility may be important for optimal interaction with SXR. Because all of these compounds are natural products found in varying concentrations in cruciferous vegetables, the results may have practical value in addition to helping to elucidate the structure-function relationship for SFN as an SXR antagonist.

SFN inhibits SXR-mediated CYP3A4 expression in human primary hepatocytes and LS180 cells. Human primary hepatocytes from two different donors (A and B) or LS180 intestinal epithelial cells (C) were pretreated 24 h with 10 or 25 μM SFN before addition of 10 μM RIF or RU486 for 24 h as indicated. D, human primary hepatocytes and LS180 cells were treated with 10 or 25 μM SFN for 48 h. Total RNA from each sample was isolated and the expression of indicated genes (A-D) was determined by quantitative real-time-PCR. Statistically significant expression compared with control group (-SFN) is marked with asterisks: *, P < 0.05; **, P < 0.01; and ***, P < 0.001 (n = 3; Student's t test).
Discussion
We have demonstrated that SFN, a naturally occurring dietary isothiocyanate, is an antagonist of SXR and inhibits SXR-mediated CYP3A4 gene expression, and, subsequently, catalytic activity. To our knowledge, SFN is the first effective and relatively nontoxic SXR antagonist ever reported. Previous investigators (Synold et al., 2001) identified the natural product ecteinasidin-743 [ET-743; trabectedin (Yondelis); a marine-derived compound from the ascidian Ecetinascidia turbinanta] as an effective antagonist to human SXR. However, this compound is highly cytotoxic and has additional biological effects, including binding to the minor groove of DNA, disorganization of microtubular networks, perturbation of cell cycle, and interference with DNA repair pathways (van Kesteren et al., 2003). Because of its cytotoxic properties, ET-743 is being developed as a cancer chemotherapeutic agent, with some success (Jimeno et al., 2004; Zelek et al., 2006). In contrast to ET-743, SFN has been promoted as a nontoxic, putative chemopreventive agent to reduce cancer risk from pro-oxidant carcinogens (Talalay et al., 1995; Lee and Surh, 2005). We have demonstrated previously that SFN provides substantial protection against aflatoxin B1-induced genotoxicity in human primary hepatocytes (Gross-Steinmeyer et al., 2004). It is well established that SFN and other isothiocyanates are effective inducers of phase II detoxification pathways in animal models and that this is thought to be the primary chemopreventive mechanism (Fahey and Talalay, 1999; Conaway et al., 2002; Gao and Talalay, 2004). Consistent with animal studies, gene expression profiling of human primary hepatocytes treated with SFN showed increased mRNA levels of several detoxification enzymes, including NQO1 and glutamate cysteine ligase (both glutamate cysteine ligase M and glutamate cysteine ligase C) (Fig, 4C; K. Gross-Steinmeyer, T. K. Bammler, and D. L. Eaton, unpublished data). Most chemical carcinogens require P450 enzyme-mediated metabolic activation before exerting their effects (Conaway et al., 2002). Here, we show that SFN inhibits SXR transactivation by directly binding to SXR and inhibiting coactivator recruitment. Thus, a reduction in SXR-mediated CYP3A4 expression may also contribute to its cancer chemopreventive effects. However, in circumstances in which CYP3A4 and/or MDR1, or other SXR-regulated genes, play a significant role in detoxification, SFN-mediated inhibition of basal expression could potentially enhance toxicity. It is interesting to note that the lack of effect of SFN on rodent PXR suggests that this mechanism would not contribute to chemoprevention in rodents.
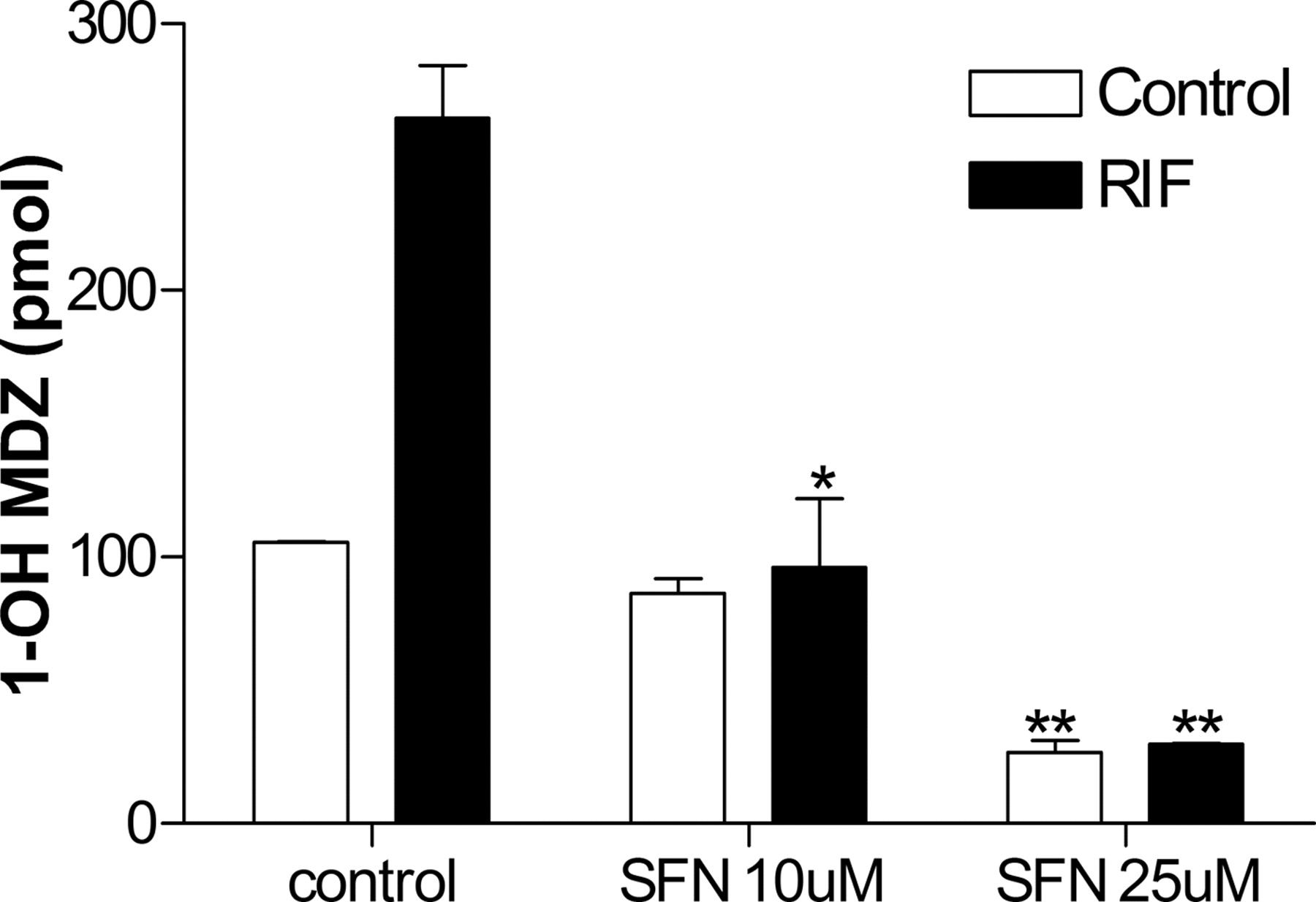
SFN inhibits CYP3A4-mediated MDZ clearance in human primary hepatocytes. Human primary hepatocytes were pretreated with 10 or 25 μM SFN for 24 h before addition of 10 μM RIF. After 24 h, cells were rinsed with phosphate-buffered saline and then incubated with 8 μM MDZ for 6 h. Supernatant media was collected and 1′OH-MDZ concentration was measured by liquid chromatography-MS. Data represent the mean ± S.E.M. of duplicate analyses of a single hepatocytes preparation.

Structural determinants of SXR antagonism by SFN. HepG2 cells were transiently transfected with full-length SXR together with a CYP3A4-luc reporter and CMX-β-galactosidase transfection control plasmid. After transfection, cells were treated with control medium or medium containing 10 μM RIF in the absence or presence of SFN analogs at the indicated concentrations for 24 h. Each bar represents the mean ± S.E.M. of three experiments. A one-way analysis of variance was performed for each treatment group. When statistical significance was found, each SFN dose was compared with the control using Dunnett's t test for multiple comparisons to a control. *, P < 0.05 and **, P < 0.01.
SXR is expressed at high levels in the liver and intestine where it acts as a xenobiotic sensor that regulates the expression of cytochrome P450 enzymes such as CYP3A4 and CYP2C8; conjugation enzymes such as UGT1A1; and ATP-binding cassette family transporters such as MDR1 and multidrug-resistance protein 2 (Synold et al., 2001). SXR is thus a master regulator of xenobiotic clearance, coordinately controlling steroid and xenobiotic metabolism and transport. Our study showed that SFN inhibits SXR function and the expression of its CYP3A4 target gene at low micromolar concentration. SFN is very abundant in broccoli and especially broccoli sprouts, with a reported concentration of approximately 10 μmol/g (Shapiro et al., 2001). In vitro data suggest that SFN is rapidly absorbed by cells, conjugated efficiently with glutathione and excreted mainly as the glutathione conjugate (Zhang and Callaway, 2002). However, the peak plasma concentration of unconjugated SFN in human subjects who ingest SFN in a “broccoli soup” can reach 4to5 μM (Gasper et al., 2005). Therefore, the concentration of SFN we used in our in vitro studies is potentially nutritionally relevant and certainly achievable in vivo via pharmacological treatments.
Although SFN is structurally unlike any previously identified class of SXR ligands, it can directly bind to SXR and strongly inhibit SXR coactivator recruitment (Figs. 2 and 3). Interestingly, compared with human SXR, SFN has little or no inhibitory effect on mouse or rat PXR at the same concentration (Fig. 1E). It is known that the induction of hepatic P450 enzymes, especially CYP3A, differs across vertebrate species, and interspecies difference in the pharmacology of SXR/PXR has been identified as the basis for much of this difference (Blumberg et al., 1998; LeCluyse, 2001). There are significant differences in the xenobiotic response between humans and rodents and these are completely explained by the structure and pharmacology of SXR and PXR. For example, the antibiotic rifampicin, the antidiabetic drug troglitazone and the cholesterol-reducing drug SR12813 were found to be effective activators of both human SXR and rabbit PXR, but they had little activity on mouse or rat PXR (Jones et al., 2000). In contrast, PCN is a more potent activator of rat and mouse PXR than of human SXR or rabbit PXR (Jones et al., 2000), but SFN has little effect on PCN-mediated activation of rat and/or mouse PXR (Fig. 1E). In addition, some highly chlorinated, nonplanar polychlorinated biphenyls have been identified as human SXR antagonists, but they act as rodent PXR agonists (Tabb et al., 2004). The crystal structure of the SXR LBD suggested which amino acid differences between SXR and PXR contribute to species differences in ligand activation of human SXR and mouse PXR and induction of CYP3A (Watkins et al., 2001). Further characterization of how SFN differentially interacts with human or rodent SXR/PXR ligand binding domains may explain the species-specific effects of SFN. From our experiments with SXR, it is clear that both ends of the molecule are critical for optimal SXR antagonism. This suggests bipolar anchoring of the relatively small molecule through hydrogen and possibly disulfide bonding with amino acid residues in the ligand binding domain.
Human CYP3A4 is expressed at high, but variable, levels in liver and small intestine. Large interindividual differences in hepatic and intestinal CYP3A4 activity contribute to difficulties in safe and effective dosing of narrow therapeutic index CYP3A4 substrates. Genetic differences in CYP3A4 or its regulatory genes have not explained much of this variability. Thus, interindividual differences in exposure to dietary or endogenous agents that modulate CYP3A4 transcription may contribute to functional CYP3A4 variability (Blumberg et al., 1998). Here, we show that SFN decreased constitutive CYP3A4 mRNA levels, in addition to attenuating RIF- and RU486-mediated CYP3A4 induction, in human intestinal cells and primary hepatocytes. These results suggest that dietary exposure to SFN and other dietary isothiocyanates from ingestion of cruciferous vegetables (especially broccoli or broccoli sprouts) could potentially contribute to the large interindividual variability in basal CYP3A4 expression. It is interesting to speculate on the mechanism by which SFN reduces constitutive expression of CYP3A4. One likely possibility is that SFN effectively competes with endogenous ligands of SXR that contribute to constitutive expression. However, it is not yet clear what endogenous ligands contribute to SXR-mediated regulation of constitutive expression of CYP3A4 in human liver or intestine. In addition, in the absence of an SXR agonist, SFN may enhance the interaction between SXR and corepressors. Further studies are necessary to determine whether inhibition of the effects of endogenous SXR ligand(s) via dietary isothiocyanates might contribute to variation in constitutive human CYP3A4 activity and/or other SXR-regulated genes.
Induction of CYP3A4 is a common cause of adverse drug-drug interactions. For example, it has been well documented that administration of RIF significantly induces CYP3A4 expression, thereby contributing to adverse drug interactions frequently associated with RIF treatment for tuberculosis. In a study of human volunteers, RIF caused a 95% decrease in the area under the curve of the plasma concentration-time curve of orally administered MDZ (Niemi et al., 2003). Oral midazolam, triazolam, simvastatin, verapamil, and most di-hydropyridine calcium channel antagonists are ineffective during RIF treatment. The plasma concentrations of the antimycotics itraconazole and ketoconazole and the HIV protease inhibitors indinavir, nelfinavir, and saquinavir are also greatly reduced by rifampicin, potentially resulting in reduced drug efficacy (Niemi et al., 2003). Indeed, the use of RIF with these HIV protease inhibitors is contraindicated to avoid treatment failures. Rifampicin can also cause acute transplant rejection in patients treated with immunosuppressive drugs, such as cyclosporin (Niemi et al., 2003). Although research on the causes of drug interactions has focused primarily on pharmaceutical agents, numerous examples exist where components of the diet modify P450 activity, particularly CYP3A4. For example, St. John's wort, a widely used herbal antidepressant, is able to interact with a variety of drugs. Hyperforin, the active constituent of St. John's wort, can induce drug metabolism through activation of SXR and induction of CYP3A4 expression (Moore et al., 2000). Our results indicate that SFN, a component of the human diet, is able to antagonize SXR activity and SXR-mediated CYP3A4 expression. Thus, pharmacological doses of SFN have the potential to reduce adverse drug responses that arise through the induction of CYP3A4 and other SXR target genes.
In summary, we have shown that SFN is a selective and effective antagonist of SXR function and drug-induced activation of SXR target genes, including CYP3A4. These findings suggest a complementary mechanism by which ingestion of the naturally occurring phytochemical may reduce the risk of certain cancers through a reduction in CYP3A4-mediated reactive metabolite formation. The data also suggest the potential use of SFN as an adjuvant to prevent CYP3A4 induction and accompanying adverse drug-drug interactions in patients receiving chronic therapy with SXR agonists.
Acknowledgments
We thank J. Calamia and J. C. Tay for technical support; J. Tracy for sample preparation; Dr. Barry M. Forman (City of Hope National Medical Center) for plasmids; and Dr. Stephen C. Strom (University of Pittsburgh, Pittsburgh, PA) for human primary hepatocytes.
Footnotes
-
This work was supported in part by National Institutes of Health Grant R01 GM60572 and Environmental Protection Agency Grant STAR R-83068601 to B.B., National Institutes of Health Grant R01 GM63666 (to K.E.T.), and National Institutes of Health Grant R01 ES05780 (to. D.L.E.); pilot Grant P30 CA15704 from the University of Washington/Fred Hutchinson Research Center Cancer Research Consortium; and the National Institute of Environmental Health Sciences Center for Ecogenetics and Environmental Health Grant P30 ES07033. Human hepatocytes were obtained through the Liver Tissue Procurement and Distribution System (University of Pittsburgh) funded by National Institutes of Health Contract N01-DK-92310. C.Z. is supported by a University of Washington School of Pharmacy Drug Metabolism, Transporter, and Pharmacogenomics Postdoctoral Fellowship funded by gifts from Abbott, Allergan, Amgen, Bristol-Myers-Squibb, Eli Lilly, Hoffman-La Roche, Johnson and Johnson, Merck, and Pfizer.
-
ABBREVIATIONS: SFN, sulforaphane; GST, glutathione S-transferase; NQO1, NAD(P)H:quinone oxidoreductase-1; UGT, UDP glucuronosyl-transferase; SXR, steroid and xenobiotic receptor; PXR, pregnane X receptor; h, human; RIF, rifampicin; MDR, multidrug resistance; HIV, human immunodeficiency virus; P450, cytochrome P450; MDZ, midazolam; PCN, pregnenolone 16α-carbonitrile; RU486, 17β-hydroxy-11β-[4-dimethylamino phenyl]-17α-[1-propynyl]estra-4,9-dien-3-one; Wy-14,643, pirinixic acid; 4-chloro-6-(2,3-xylidino)-2-pyrimidinyl)thioacetic acid; FBS, fetal bovine serum; DMEM, Dulbecco's modified Eagle's medium; luc, luciferase; SRC-1, steroid receptor coactivator-1; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate; PCR, polymerase chain reaction; MS, mass spectrometry; PPAR, peroxisome proliferator-activated receptor; LBD, ligand binding domain; TIF, transcriptional intermediary factor; ACTR, activator of thyroid and retinoic acid receptor; PBP, proliferator-activated receptor-binding protein; 1′OH-MDZ, 1′-hydoxymidazolam; SR12813, 3,5-di-tert-butyl-4-hydroxystyrene-β,β-diphosphonic acid tetraethyl ester.
-
↵1 Current affiliation: Laboratory of Biochemical Genetics and Metabolism, The Rockefeller University, New York, NY 10021.
- Received July 25, 2006.
- Accepted October 6, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}