


Abstract
Recent studies suggest that a novel positive allosteric modulator (PAM) of the metabotropic glutamate receptor (mGluRs), mGluR5, termed 4-nitro-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (VU-29), potentiates mGluR5 responses by actions at a site that is overlapping with the binding site of 2-methyl-6-(phenylethynyl)pyridine (MPEP), a previously identified negative allosteric modulator of this receptor. It is interesting that a structurally distinct PAM, N-{4-Chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide (CPPHA), does not to bind to the MPEP site. We now report that CPPHA potentiates mGluR5 responses by a mechanism that is distinct from that of VU-29. VU-29- and CPPHA-induced potentiation of mGluR5 responses are blocked by a neutral ligand at the MPEP allosteric site termed 5-methyl-2-(phenylethynyl)pyridine (5MPEP). However, increasing concentrations of 5MPEP induce parallel rightward shifts in the VU-29 concentration-response curve, whereas 5MPEP inhibits CPPHA potentiation in a noncompetitive manner. Consistent with this, a mutation (A809V/mGluR5) that reduces binding of ligands to the MPEP site eliminates the effect of VU-29 but has no effect on the response to CPPHA. On the other hand, a mutation (F585I/mGluRs) that eliminates the effect of CPPHA does not alter the response to VU-29. CPPHA is also a PAM at mGluR1. It is interesting that the corresponding mutation of F585I/mGluR5 in mGluR1 (F599I/mGluR1) eliminates CPPHA's effect without altering the potentiation of a known PAM of mGluR1, (S)-2-(4-fluorophenyl)-1-(toluene-4-sulfonyl)pyrrolidine (Ro 67-7476). Likewise, another mutation (V757L/mGluR1) that abolishes potentiation of Ro 67-7476 has no effect on CPPHA. Finally, CPPHA does not displace binding of a radioligand for the mGluR1 allosteric antagonist characterized previously. Together, these data suggest that CPPHA acts at a novel allosteric site on both mGluR1 and -5 to potentiate responses to activation of these receptors.
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system. In addition to eliciting fast excitatory synaptic responses, glutamate has neuromodulatory effects by activation of G protein-coupled receptors (GPCRs) termed metabotropic glutamate receptors (mGluRs). There are eight subtypes of mGluRs divided into three groups. Group I mGluRs (mGluR1 and mGluR5) couple to Gαq/11 and activate phospholipase C. Group II (mGluR2 and mGluR3) and group III (mGluR4, mGluR6, mGluR7, and mGluR8) mGluRs couple to the inhibition of adenylyl cyclase and other effectors through Gαi/o (Conn and Pin, 1997; Schoepp et al., 1999; Coutinho and Knopfel, 2002). Group I mGluRs are distributed widely in multiple brain regions. Positive allosteric modulators (PAMs) of group I mGluRs have emerged recently as useful tools to study their physiological roles and provide a promising approach for developing novel therapeutic agents for the treatment of psychiatric and cognitive disorders (Anwyl, 1999; Marino et al., 2001; Marino and Conn, 2002; O'Brien et al., 2003, 2004; Campbell et al., 2004; Moghaddam, 2004; Kinney et al., 2005).

Structures of allosteric modulators of mGluR5. CPPHA, DFB, CDPPB, and VU-29 are positive allosteric modulators of mGluR5, whereas MPEP is a negative allosteric modulator. 5MPEP is a positional isomer of MPEP that is a neutral ligand at the MPEP allosteric site.
Three distinct families of mGluR5 PAMs have been developed, including 3,3′-difluorobenzaldazine (DFB), 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB) and its analog VU-29, and N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide (CPPHA) (O'Brien et al., 2003, 2004; Kinney et al., 2005; Chen et al., 2007). Figure 1 shows the structures of each of these mGluR5 PAMS and other allosteric site ligands used in the present studies. The mGluR5 PAMs do not activate the receptor directly but potentiate its response to glutamate. Unlike allosteric modulators of other GPCRs, mGluR5 PAMs do not affect binding of ligands to the orthosteric glutamate binding site (Christopoulos and Kenakin, 2002; O'Brien et al., 2003, 2004; Kinney et al., 2005). DFB and CDPPB displace binding of a radioligand to a well-characterized binding site for mGluR5 negative allosteric modulators (NAMs), such as MPEP. Furthermore, we have provided evidence that the CDPPB family of PAMs (including VU-29) potentiates mGluR5 responses by actions at this site (Chen et al., 2007). It is interesting that a third mGluR5 PAM, CPPHA, does not bind to the MPEP site at concentrations several orders of magnitude higher than its potency as a PAM (O'Brien et al., 2004), suggesting that CPPHA acts by a different mechanism than do CDPPB, VU-29, and DFB. However, PAMs and orthosteric agonists activate mGluR5 in a cooperative manner so that relatively low occupancy at the MPEP site can induce robust potentiation of mGluR5 responses (de Paulis et al., 2006; Chen et al., 2007). Thus, CPPHA could potentiate mGluR5 by actions at this site but have low affinity and high cooperativity so that that low binding is not detectable at concentrations that have been used in radioligand binding studies. We now report a series of studies that suggest that CPPHA potentiates responses to activation of both mGluR5 and mGluR1 by actions at a site that is clearly distinct from the allosteric sites identified previously on either of these receptor subtypes. These data suggest that multiple distinct allosteric sites exist on mGluRs that can serve as targets for PAMs that induce qualitatively similar effects on receptor function.
Materials and Methods
Mutagenesis and Transient Transfection. HEK 293A cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 1 mM l-glutamine, and 1× antibiotic-antimycotic (all from Invitrogen, Carlsbad, CA). Cells were collected and plated in clear-bottomed 96-well plates (Costar; Corning Life Sciences, Acton, MA) pretreated with poly(d-lysine) (Sigma, St. Louis, MO) in normal growth medium with a density of 40,000 cells/well overnight before transfection. Cells were transiently transfected with wild-type and mutant forms of rat mGluR5a/mGluR1 cDNA using the pRK5 vector (Clontech, Mountain View, CA). Point mutations were generated using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). Construction of the N-terminal truncated mutant of mGluR5 (mGluR5HD) was performed as reported by Goudet et al. (2004). Construction of the N-terminal truncated mutant of mGluR1 (mGluR1HD) was performed similarly by inserting between the Mlu-I and Xba-I sites of pRK5-NHA the mGluR1a cDNA coding for the HD and a shortened C-terminal tail between the residues P582 and I875C, obtained by polymerase chain reaction. All mutations were verified by sequencing. The transfection plasmid was prepared using Sigma Mini Prep kit (Sigma-Aldrich, St. Louis, MO). Cells were transfected with Lipofectamine (Invitrogen) for 6 h according to the manufacturer's instructions (80 ng of DNA and 0.2 μl of Lipofectamine per well) before switching to normal growth medium. Rat GLAST pCDNA3.1 (20 ng/well) was coexpressed with mGluR5 pRK5 to reduce extracellular glutamate concentration for the test of VU-29. Glutamate/glutamine-free medium (glutamine-free DMEM plus 10% dialyzed fetal bovine serum; Invitrogen) was applied to substitute growth medium at least 4 h before performing functional assays. Cell culture, transfection, and starvation were performed at 37°C in an atmosphere of 95% air plus 5% carbon dioxide. Transfected cells were tested approximately 48 h after transfection. Rat mGluR2 and human mGluR4 were coexpressed with Gqi5, which enables coupling to the calcium mobilization as reported by Galici et al. (2006).
Secondary Rat Astrocytes Culture. Secondary rat cortical astrocytes were prepared as described previously (Peavy et al., 2001; Zhang et al., 2005; Chen et al., 2007). Astrocytes were plated into poly(d-lysine)-coated 96-well plates with a density of 30,000 cells/well on day 0 in DMEM containing 10% FBS, 1 mM l-glutamine (Invitrogen), and 1× antibiotic-antimycotic (Invitrogen) overnight. Then, G-5 supplement (Invitrogen), which contains epidermal growth factor (10 ng/ml), basic fibroblast growth factor (5 ng/ml), insulin (5 μg/ml), and other factors, was added to the growth medium on day 1 and switched to glutamine-free DMEM with 10% dialyzed FBS on day 3. Calcium mobilization assay was performed on day 4. Cell culture and starving were performed at 37°C with 5% carbon dioxide.

A, 5MPEP blocked CPPHA-induced potentiation of mGluR5-mediated intracellular calcium flux in the secondary cultured rat cortical astrocytes. Pretreatment with 10 μM 5MPEP did not affect the response to glutamate (EC20 value, approximately 300 nM). CPPHA (3 μM) enhanced the response. 5MPEP blocked the potentiation significantly (*, P < 0.01, Student's t test). B, 5MPEP blocks the effects of VU-29 and CPPHA in a concentration-dependent manner in the secondary cultured rat cortical astrocytes via calcium mobilization assay. Data were normalized to the maximum potentiation of each potentiator determined by 3 μM CPPHA or 60 nM VU-29, respectively, shown as Control A. Control B shows the EC20 value response of glutamate without any allosteric potentiator. The potency of 5MPEP on VU-29 was 0.745 ± 0.2 μM, whereas it was 2.27 ± 0.72 μM for CPPHA (Student's t test, P < 0.05). Concentration-response curves were generated from three independent experiments, each performed in triplicate. Error bars represent S.E.M. of three separate experiments.

5MPEP induces parallel rightward shifts in the VU-29 CRC but has noncompetitive actions on the response to CPPHA effects on calcium mobilization in rat astrocytes. A, concentration-response curves of VU-29 were shifted to the right in a concentration-dependent manner by 5MPEP at concentrations up to 10 μM. For the Schild regression, X is the dose ratio, which is calculated by the EC50 value of VU-29 CRC in the presence of 5MPEP divided by the EC50 value of VU-29 CRC in the absence of 5MPEP. The fitted equation is log(X - 1) = 0.97 × log[5MPEP] - 7.009; r2 = 0.9921. B, 5MPEP blocks the CRC of CPPHA in the noncompetitive manner. Increasing concentrations of 5MPEP induced progressive decreases in the maximum response to CPPHA. Data were obtained from four separate experiments, each performed in triplicate. Error bars represent S.E.M.
Calcium Fluorescence Measurement. Cells were loaded with calcium-sensitive dye according to the manufacturer's instructions (Calcium 3 kit; Molecular Devices, Sunnyvale, CA) after incubation in glutamate/glutamine-free medium (DMEM and 10% dialyzed fetal bovine serum) for 5 h. Compound A (1 ml) from Calcium 3 kit was dissolved in 20 ml of 1× Hanks' balanced salt solution (HBSS; Invitrogen) containing 2.5 mM probenecid (Sigma), adjusted to pH 7.4. Cells were loaded for 50 min at 37°C with 5% carbon dioxide. Dye was then carefully removed, and cells were washed with HBSS containing probenecid. Cells were maintained in the same buffer at room temperature for the following assay. For calcium fluorescence measurement of rat cortical astrocytes, allosteric modulators were added 5 min before the addition of agonist manually. For transient transfected cells, allosteric modulators were added 1 min before the addition of agonist using Flexstation II (Molecular Devices) at a speed of 52 μl/s. Calcium flux was measured at 25°C. All of the peaks of the calcium response were normalized to the maximum response to a saturated dose of glutamate (10 μM). The submaximal concentration (EC20) of glutamate was determined for every separate experiment, allowing for a response varying from 10 to 30% of the maximum peak.
Inositol Phosphate Measurement. Inositol phosphate (IP) accumulation experiments were performed in 96-well microplates as already described by Goudet et al. (2004). In brief, after transfection of the truncated mGluR1 or mGluR5, HEK 293 cells were incubated overnight with [3H]myoinositol (16 Ci/mmol; GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). The following day, cell medium was washed, and ambient glutamate was degraded by incubation in the presence of glutamate pyruvate transaminase. Cells were stimulated by the indicated compounds for 30 min, and then the medium was removed and cells incubated with ice-cold 0.1 M formic acid, which induced cell lysis. Then [3H]IP produced after receptor stimulation was recovered by ion exchange chromatography using a Dowex resin (Bio-Rad Laboratories, Hercules, CA). IP samples were then eluted by a highly concentrated formate solution (4 M), collected in a 96-well sample plate, and mixed with liquid scintillator (PerkinElmer Life and Analytical Sciences, Waltham, MA). The radioactivity remaining in the membranes is used to normalize the IP produced. Membranes were solubilized with a solution of NaCl (0.1 M) containing 10% of Triton X-100 (Sigma), collected, and mixed with liquid scintillator in a 96-well sample plate. Radioactivity was counted using a Wallac 1450 Microbeta scintillation and luminescence counter (PerkinElmer Life and Analytical Sciences). Results are expressed as the ratio between IP and the total radioactivity corresponding to IP plus membrane. All points are realized in triplicate.

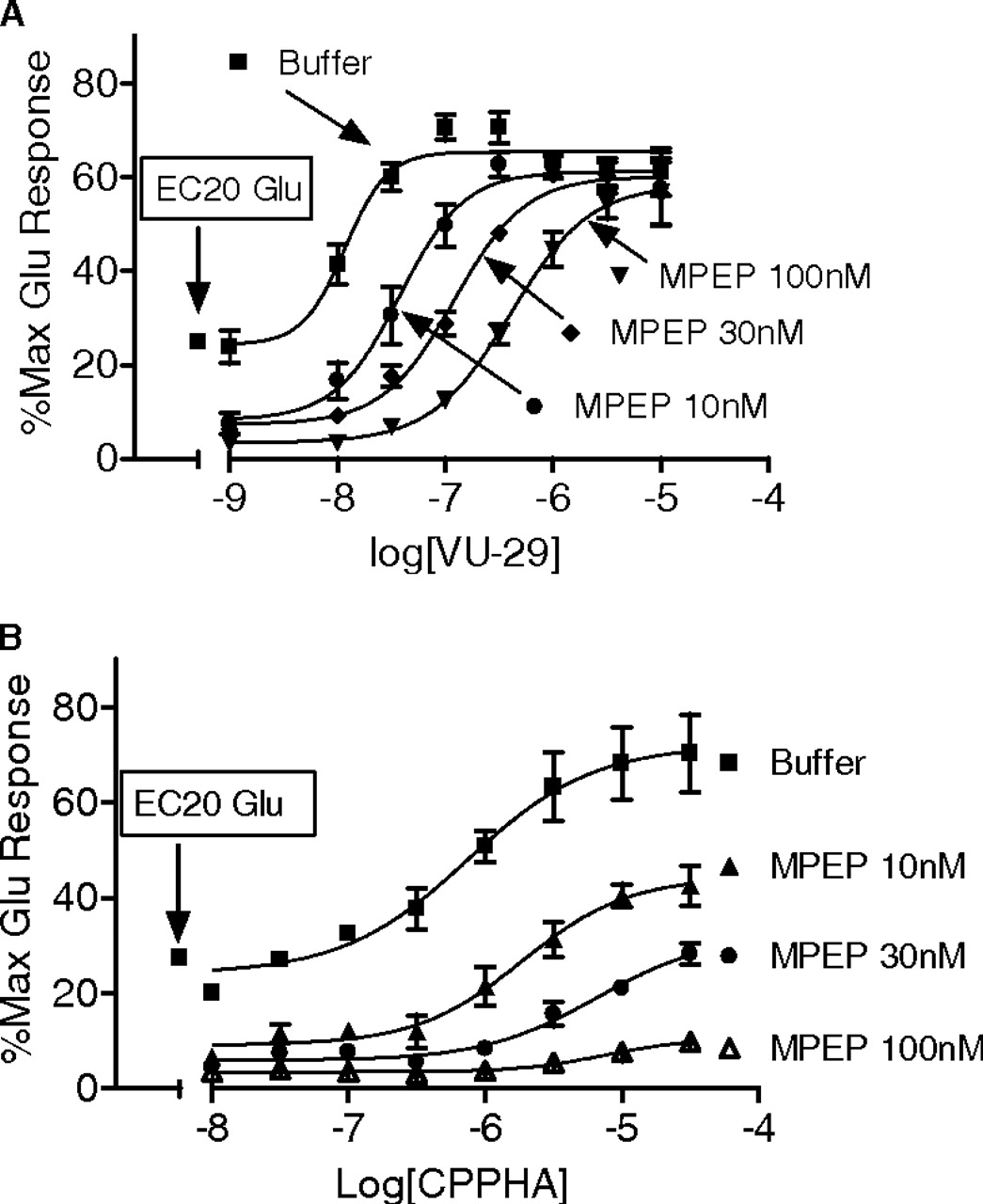
MPEP differentially shifts the concentration-response curves of VU-29 and CPPHA using the calcium mobilization assay of cultured rat cortical astrocytes. A, MPEP induced a parallel rightward shift in the CRC of VU-29 with no decrease of its maximum potentiation, suggesting competitive blockade. B, MPEP decreased the maximum effect of CPPHA, suggesting a noncompetitive inhibitory action. Data were obtained from three separate experiments, each performed in triplicate. Error bars represent S.E.M.

Selectivity of CPPHA for different mGluR subtypes in HEK 293 cells. A and B, 10 μM CPPHA shifted the glutamate CRC for mGluR5 and mGluR1. C, 3 or 10 μM CPPHA did not affect the CRC of mGluR2 significantly. D, 10 μM CPPHA slightly antagonized the response of mGluR4. Data were obtained from three separate experiments, each performed in triplicate. Error bars represent S.E.M.
[3H]R214127 Radioligand Binding Assays. Membranes preparation from BHK cells stably expressing rat mGluR1a and [3H]R214127 competition binding assay was performed by following the protocol described in Hemstapat et al. (2006). Compounds were dissolved in dimethyl sulfoxide (DMSO) as stocks kept in -20°C. The final DMSO concentration was 0.12% in the assay.
Compound Preparation.Figure 1 shows the structures of each of the allosteric modulators used in these studies. Compounds were dissolved in dimethyl sulfoxide (Sigma) and stored at -80°C. Stock solutions were dissolved in 1× HBSS containing 0.1% bovine serum albumin (Sigma) on the day of experiment. Final DMSO concentration was 0.12 to 0.15% for all of the assays.
Results
5MPEP Blocked Responses to CPPHA and VU-29 with Different Potencies. We used secondary cultured rat cortical astrocytes for functional assays of mGluR5 activity (calcium mobilization assay). As described previously, the calcium fluorescence response at an EC20 concentration of glutamate (approximately 300 nM) was potentiated by 3 μM CPPHA (Fig. 2A; Student's t test, P < 0.01). The neutral MPEP site ligand 5MPEP (10 μM) (Rodriguez et al., 2005) had no effect on the EC20 glutamate response. However, 5MPEP blocked potentiation of this response by 3 μM CPPHA (Fig. 2A; Student's t test, P < 0.01) in a manner similar to that shown previously for VU-29 (Chen et al., 2007). Comparable submaximal concentrations of CPPHA and VU-29 were chosen (3 μM CPPHA and 60 nM VU-29) to generate 5MPEP concentration-response curves. The potency of 5MPEP at blocking the response to VU-29 was 0.75 ± 0.2 μM, whereas its potency at blocking the response to CPPHA was 2.27 ± 0.7 μM (Fig. 2B). These values were significantly different (Student's t test, P < 0.05). Furthermore, the slope of the inhibition curve for blockade of the response to CPPHA seemed to be more shallow than that for blockade of the response to VU-29.
5MPEP and MPEP Had Actions Consistent with Noncompetitive Blockade of the Response to CPPHA. To determine whether 5MPEP blocks responses to VU-29 and CPPHA by similar mechanisms, we determined the effects of increasing concentrations of 5MPEP on the concentration-response relationships of VU-29 and CPPHA-induced potentiation of mGluR5 responses (Fig. 3). 5MPEP induced progressive parallel rightward shifts of the VU-29 CRC when added at concentrations up to 10 μM. Thus, VU-29 was capable of overcoming inhibition by 5MPEP across this concentration range. However, we found previously that VU-29 fully potentiates mGluR5 at concentrations that only partially occupy the MPEP site. This suggests that there is a positive cooperativity between orthosteric agonists and the allosteric potentiators and that mGluR5 present in these cells can fully activate calcium mobilization when the receptor is partially occupied by the positive allosteric modulators (Chen et al., 2007). Thus, the finding that VU-29 was capable of overcoming inhibition by 5MPEP across this concentration range could be explained by the cooperativity between VU-29 and glutamate in activating mGluR5 (Chen et al., 2007). Because this leads to full potentiation with partial occupancy of the allosteric site by VU-29, it could have an effect that is directly analogous to the impact of spare receptors when performing Schild analysis with orthosteric site ligands. However, our previous studies provide strong support for the hypothesis that VU-29 has its potentiator effects by actions at the MPEP site (Chen et al., 2007). Furthermore, the current Schild analysis reveals a linear regression line (r2 = 0.9921) with a slope of approximately 1 (0.97). The x-intercept was 100 nM, which is consistent with the Ki value of 5MPEP determined in previous competition binding studies (Rodriguez et al., 2005). Taken together, these data are consistent with a competitive interaction between 5MPEP and VU-29. In contrast, increasing concentrations of 5MPEP induced progressive decreases in the maximum potentiation of CPPHA (Fig. 3B), suggesting that 5MPEP blocks the response to CPPHA by a noncompetitive mechanism of action.

The potencies of CPPHA on mGluR1 and mGluR5 are slightly different using calcium mobilization assay on transfected HEK cells. We preincubated cells with multiple concentrations of CPPHA for 5 min before adding EC20 concentrations of glutamate to elicit calcium mobilization. CPPHA had a potency of 239 ± 27 nM on rmGluR5 (A) and a potency of 3455 ± 517 nM on rmGluR1d (B). Data were obtained from three separate experiments performed in triplicate. Error bars represent S.E.M.

Activity of CPPHA and VU-29 on N-terminal truncated mutant forms of mGluR1 and mGluR5. A, CPPHA and VU-29 directly activate the truncation mutant containing the heptahelical domain of mGluR5 (mGlu5HD). B, CPPHA activates the mutant containing the heptahelical domain of mGluR1 (mGlu1HD), whereas VU-29 is without effect on mGlu1HD. N-terminal truncated mGluR1 and mGluR5 were transiently expressed in the HEK 293A cells. Their activity was assessed by measuring inositol phosphate production induced by receptor activation. Data displayed are mean ± S.E.M. of triplicate determinations from a representative experiment. At least four separate experiments were performed. Error bars represent S.E.M.
We next performed similar experiments with MPEP. Because MPEP is an mGluR5 NAM instead of neutral modulator, this compound decreased the baseline response of the concentration-response curves of both VU-29 and CPPHA. Consistent with 5MPEP, MPEP induced concentration-dependent parallel rightward shifts in the VU-29 CRC but downward shifts in CRC for CPPHA. This is consistent with the effects of 5MPEP and the hypothesis that ligands acting at the MPEP site do not interact with CPPHA in a competitive manner (Fig. 4, A and B).
CPPHA Had PAM Activity at Both mGluR1 and mGluR5. We next evaluated the selectivity of CPPHA as a potentiator of mGluR5 relative to mGluR1 and representative mGluR subtypes belonging to group II (mGluR2) and group III (mGluR4). CPPHA (10 μM) potentiated responses of both mGluR5 and mGluR1d to glutamate but had no effect on responses to mGluR2 (Fig. 5, A-C). Consistent with the original report, CPPHA induced slight antagonism of the mGluR4-mediated response (Fig. 5D). CPPHA also potentiated rmGluR1a- and 1b-mediated calcium mobilization (data not shown).

Sequence alignments of mGluR1, -5 and -2 transmembrane domains. Boldface type indicates the amino acids that have been mutated.
CRC analysis revealed that CPPHA had a potency of 239 ± 27 nM on rmGluR5 and a potency of 3460 ± 517 nM on rmGluR1d (Fig. 6, A and B). Thus, whereas somewhat selective for mGluR5, CPPHA has clear effects at both group I mGluR subtypes. This 10-fold selectivity probably accounts for the failure to see an effect of a single lower concentration of CPPHA on mGluR1 in previous studies (O'Brien et al., 2004).
CPPHA Acted in the 7TM Domain of Group I mGluRs at a Site Distinct from That of VU-29. The studies above suggest that CPPHA and VU-29 probably act at distinct sites. Because VU-29 and other allosteric modulators identified previously of mGluR5 act at a common site that is shared with MPEP, this raises the question of what domains of the receptor are most important for the action of CPPHA. The MPEP site and sites for other previously identified allosteric modulators of mGluRs are in the 7TM domain of the receptor. However, it is conceivable that an allosteric modulator could act in the extracellular or intracellular domains. The finding that CPPHA is an allosteric potentiator of mGluR1d suggests that CPPHA is unlikely to act in the intracellular C-terminal domain of the receptor. mGluR1d is a C-terminal truncated receptor that lacks the large intracellular C-terminal domain. We reported previously that other allosteric potentiators of mGluRs act as agonists of N-terminal truncation mutants that lack the large extracellular domain and consist primarily of the heptahelical domain (HD) (Goudet et al., 2004; Chen et al., 2007). Thus, we determined the effect of CPPHA on the N-terminal truncated mutant of mGluR5 described previously (mGluR5HD). If CPPHA acts in the N-terminal domain of the receptor, it should not have activity at this N-terminal truncation mutant. It is interesting that CPPHA induced a robust activation of the truncated mutant, as assessed by activation of phosphoinositide hydrolysis. This was qualitatively similar to the effect of VU-29 (Fig. 7A). We also determined the activity of CPPHA and VU-29 on the N-terminal truncation mutant of mGluR1 (mGluR1HD). CP-PHA directly and dose-dependently activates the truncation mutant, whereas VU-29 is without effect (Fig. 7B). These data suggest that, like other allosteric modulators, CPPHA is likely to act in the heptahelical domain of mGluR1 and mGluR5.
We took advantage of the ability of CPPHA to potentiate responses at mGluR1 and mGluR5 but not mGluR2 to guide the selection of mutations that may provide insight into specific amino acids that may be required for CPPHA action. Thus, we aligned the 7TM domain of these receptors and searched for residues that were identical between mGluR1 and mGluR5 but in which there was a different amino acid in mGluR2. We selected 20 amino acids in the transmembrane domain that were identical between mGluR1 and -5 but were divergent in mGluR2. Mutants of mGluR5 were constructed in which each of these 20 amino acids was singly mutated to the corresponding amino acids in mGluR2. Sequence homology and all of the mutants studied are shown in Fig. 8. All mutants were tested for responses to 1 μM CPPHA in transiently transfected HEK cells using the calcium mobilization assay. Among the mutants, only A582P/F585I/mGluR5 and F585I/mGluR5 in the first transmembrane domain eliminated the potentiation of mGluR5 by 1 μM CPPHA (Fig. 9B). CPPHA induced a significant potentiation of responses for all other mutants (data not shown) and wild-type receptor (Fig. 9A). In contrast, the potentiation of 200 nM VU-29 remained for F585I/mGluR5 compared with wild type (Fig. 9, A and B).

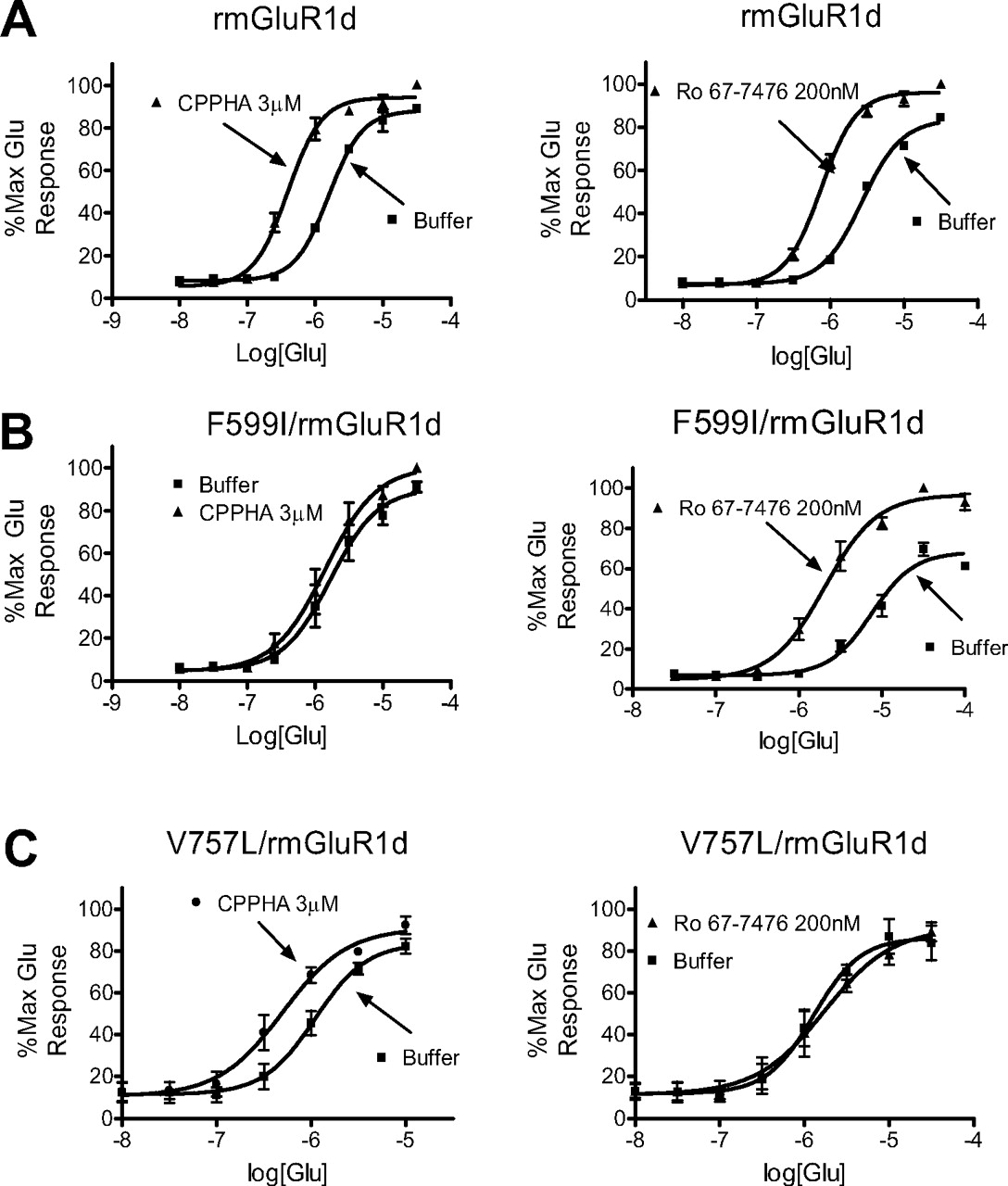
A,1 μM CPPHA and 200 nM VU-29 induced leftward shifts of the glutamate CRC for wild-type rmGluR5 in transiently transfected HEK 293 cells as measured by the calcium mobilization assay. B, single point mutation F585I completely abolished CPPHA potentiation without changing VU-29 potentiation. C, A809V eliminated the VU-29 potentiation, but CPPHA potentiation remained intact. Data shown represent mean ± S.E.M. of three separate experiments, each performed in triplicate.
We reported previously that potentiation of VU-29 was abolished by the mutation A809V, which also eliminated MPEP site binding (Pagano et al., 2000; Malherbe et al., 2003; Chen et al., 2007). As shown in Fig. 9C, A809V eliminates the effect of VU-29 without affecting the potentiation by CPPHA.
To further verify the impact of this mutation on CPPHA responses, we determined the effect of mutation of the homologous amino acid in mGluR1. In transiently transfected HEK cells, both 3 μM CPPHA and 200 nM Ro 67-7476 potentiate wild-type mGluR1-mediated calcium mobilization (Fig. 10A). However, F599I/mGluR1, the homologous mutation of F585I/mGluR5, eliminated the potentiation of mGluR1 responses by CPPHA but not a mGluR1 PAM characterized previously, Ro 67-7476 (Knoflach et al., 2001; Hemstapat et al., 2006) (Fig. 10B). Furthermore, another mutation, V757L/mGluR1, abolished the potentiation of mGluR1 by Ro 67-7476 as reported previously (Knoflach et al., 2001; Hemstapat et al., 2006) but did not abolish the potentiation of CPPHA (Fig. 10C).
CPPHA Did Not Compete for Binding of the Allosteric Antagonist R214127 Site on mGluR1. The allosteric mGluR1 antagonist radioligand [3H]R214127 binds to mGluR1 at a site that is believed to be homologous with the MPEP site on mGluR5 (Pagano et al., 2000; Lavreysen et al., 2003). If CPPHA acts at a site that is conserved between mGluR1 and mGluR5 but is distinct from the MPEP and R214127 sites on these receptors, CPPHA should not compete for binding to the R214127 site. Thus, we determined the effect of CPPHA on specific binding of 2.5 nM [3H]R214127 to mGluR1. Binding of [3H]R214127 to membranes from cells expressing mGluR1 was reduced in a concentration-dependent manner by unlabeled R214127 with an IC50 value of 3.4 nM (Fig. 11). Under the same conditions, CPPHA did not affect the [3H]R214127 binding up to 32 μM (Fig. 11).

A, 3 μM CPPHA and 200 nM Ro 67-7476 potentiated the CRC of glutamate to the left for wild-type rmGluR1d in transiently transfected HEK cells via calcium mobilization assay. B, single point mutation F599I completely abolished CPPHA potentiation without changing Ro 67-7476 potentiation. C, V757L eliminated the Ro 67-7476 potentiation, but CPPHA potentiation remained intact. Data are shown as mean ± S.E.M. of three separate experiments.
Discussion
The data reported provide strong support for the hypothesis that CPPHA acts at a novel allosteric site on mGluR5 and mGluR1 to potentiate activation of the receptor by orthosteric agonists. Our data suggest that the action of CPPHA is similar to actions of other known allosteric modulators in that it is mediated by actions in the 7TM domain, but that the specific site differs from previously defined sites for allosteric potentiators or allosteric antagonists on these receptors. Most of the mGluR5 allosteric antagonists are derived from the same chemical scaffold and are believed to act on a shared site represented by the MPEP binding site (Gasparini et al., 1999; Varney et al., 1999). There are three families of PAMs for mGluR5 that are represented by DFB, CDPPB, and CPPHA (O'Brien et al., 2003, 2004; Kinney et al., 2005). VU-29 belongs to the CDPPB chemical class. The recent finding that DFB and CDPPB/VU-29 act through binding to an overlapping site of MPEP (Mühlemann et al., 2006; Chen et al., 2007) suggests that the MPEP site is a common site shared by multiple allosteric modulators. Likewise, multiple allosteric ligands at mGluR1 seem to interact with a common site that is homologous with the MPEP site on mGluR5. Thus, all known mGluR1 NAMs that have been examined interact with the allosteric site labeled by R214127 (Litschig et al., 1999; Pagano et al., 2000). Furthermore, some mGluR1 PAMs characterized previously have been postulated to share a homologous binding pocket with MPEP in mGluR5 based on critical amino acids implicated in a detailed mutagenesis study (Pagano et al., 2000; Knoflach et al., 2001; Hemstapat et al., 2006). However, competition binding assays revealed that two other PAMs, Ro 67-7476 and VU-71, do not displace the binding of radiolabeled R214127 (Hemstapat et al., 2006). Thus, it is unlikely that these mGluR1 PAMs act at this previously characterized site for mGluR1 NAMs.

CPPHA does not bind to the R214127 NAM site on mGluR1. R214127 induced a concentration-dependent decrease in binding to of 2.5 nM [3H]R214127 to a membrane preparation from an mGluR1 stable cell line with a IC50 value of 3.4 nM. CPPHA did not affect the binding at concentrations up to 32 μM. Data are shown as mean ± S.E.M. of three separate experiments, each performed in triplicate.
Although the current studies suggest that CPPHA does not act by direct interactions with the MPEP site, our studies also provide important data suggesting that the ligands at the MPEP site can regulate effects of ligands at the CPPHA site indirectly. This is based on the finding that the neutral MPEP site ligand, 5MPEP, noncompetitively inhibits CPPHA action. It is consistent with our previous observation that CPPHA potentiation is blocked by another neutral allosteric modulator, 3,3′-dichlorobenzaldazine (Chen et al., 2004). This may imply that ligands at the MPEP site and the CPPHA site can allosterically regulate one another. However, if CPPHA allosterically regulates MPEP site ligand affinity and vice versa, radioligand binding studies should reveal a noncompetitive effect of CPPHA on binding of ligands to the MPEP site. At present, the relationship between these allosteric sites is not understood.
The finding that mGluR5 PAMS can be identified that act on distinct allosteric sites raises the interesting question of whether PAMs that act at these sites will have identical effects on mGluR5 function. In the simplest view, these compounds would induce similar increases in activity of mGluR5 regardless of the signaling pathway or cell population involved. However, a growing body of evidence suggests that different traditional orthosteric agonists can differentially activate different signaling pathways of a single GPCR, a phenomenon referred to as agonist receptor trafficking (Berg et al., 1998; Brink et al., 2000; Gazi et al., 2003). Based on this, it is possible that allosteric potentiators of mGluRs could differentially regulate coupling of these receptors to different signaling pathways. Furthermore, it is conceivable that PAMs that act at different sites could differentially regulate coupling of mGluR5 to different pathways. mGluR5 can couple to multiple signaling pathways and physiological responses. For instance, we reported that mGluR5 in cortical astrocytes activates phosphoinositide hydrolysis and phosphorylation of extracellular signal-regulated kinase (ERK2) by completely independent mechanisms (Peavy et al., 2001, 2002). Furthermore, we and a number of other investigators have found that activation of mGluR5 can have a wide variety of effects on different neuronal populations, including cell depolarization, modulation of different potassium currents, potentiation of N-methyl-d-aspartate receptor currents, and a variety of other responses (Valenti et al., 2002). It is likely that these responses are mediated by different signaling mechanisms and could be differentially regulated. It is interesting that we recently reported that that the MPEP sites PAM, DFB, and CPPHA have subtly different effects on mGluR5-mediated responses in astrocytes (Zhang et al., 2005). Both PAMs potentiate mGluR5-mediated increases in intracellular calcium with similar profiles. This is consistent with the similarities of effects of CPPHA and CDPPB on calcium mobilization shown in the present article. It is also interesting to note that DFB and CDPPB have different effects on coupling of mGluR5 to ERK1/2 phosphorylation in these same cells (Zhang et al., 2005). DFB induces a leftward shift of the agonist concentration-response curve for ERK1/2 phosphorylation similar to that seen with calcium mobilization. However, CPPHA had a small agonist effect when added alone and potentiated the effect of low concentrations of agonist but inhibited the effect of high concentrations of agonist on the ERK1/2 phosphorylation response. When considered in light of the present findings, this raises the possibility that ligands at these two allosteric sites may differentially regulate coupling of mGluR5 to these two signaling pathways. This is also interesting in light of previous studies showing that mGluR5 PAMs that act at the MPEP site (i.e., CDPPB) have behavioral effects in animal models that can predict antipsychotic-like efficacy. It is not known whether compounds that act at the CPPHA site will have this effect. In future studies, it will be important to systematically compare effects of mGluR5 PAMs that act at these different allosteric sites in native systems and in vivo.
Footnotes
-
This work was supported by grants from the National Institute of Mental Health, National Institute of Neurological Disorders and Stroke, the Stanley Foundation, and the National Alliance for Research on Schizophrenia and Affective Disorders. Vanderbilt is a center within the National Institutes of Health-supported Molecular Libraries Screening Centers Network.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; CDPPB, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide; CPPHA, N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide; CRC, concentration-response curve; DFB, 3,3′-difluorobenzaldazine; DMEM, Dulbecco's modified Eagle's medium; HEK, human embryonic kidney; HBSS, Hanks' balanced salt solution; IP, inositol phosphate; mGluR, metabotropic glutamate receptor; MPEP, 2-methyl-6-(phenylethynyl)pyridine; NAM, negative allosteric modulator; PAM, positive allosteric modulator; Ro 67-7476, (S)-2-(4-fluorophenyl)-1-(toluene-4-sulfonyl)pyrrolidine; R214127, 1-(3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-2-phenyl-1-ethanone; VU-29, 4-nitro-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide; 5MPEP, 5-methyl-2-(phenylethynyl)pyridine; 7TM, seven transmembrane; FBS, fetal bovine serum; IP, inositol phosphate; DMSO, dimethyl sulfoxide; HD, heptahelical domain; VU-71, 4-nitro-N-(1,4-diphenyl-1H-pyrazol-5-yl)benzamide; ERK, extracellular signal-regulated kinase.
- Received July 17, 2007.
- Accepted December 4, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}