


Abstract
Activators of M1 muscarinic acetylcholine receptors (mAChRs) may provide novel treatments for schizophrenia and Alzheimer's disease. Unfortunately, the development of M1-active compounds has resulted in nonselective activation of the highly related M2 to M5 mAChR subtypes, which results in dose-limiting side effects. Using a functional screening approach, we identified several novel ligands that potentiated agonist activation of M1 with low micromolar potencies and induced 5-fold or greater leftward shifts of the acetylcholine (ACh) concentration-response curve. These ligands did not compete for binding at the ACh binding site, indicating that they modulate receptor activity by binding to allosteric sites. The two most selective compounds, cyclopentyl 1,6-dimethyl-4-(6-nitrobenzo[d][1,3]-dioxol-5-yl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (VU0090157) and (E)-2-(4-ethoxyphenylamino)-N′-((2-hydroxynaphthalen-1-yl)methylene)acetohydrazide (VU0029767), induced progressive shifts in ACh affinity at M1 that were consistent with their effects in a functional assay, suggesting that the mechanism for enhancement of M1 activity by these compounds is by increasing agonist affinity. These compounds were strikingly different, however, in their ability to potentiate responses at a mutant M1 receptor with decreased affinity for ACh and in their ability to affect responses of the allosteric M1 agonist, 1-[1′-(2-tolyl)-1,4′-bipiperidin-4-yl]-1,3-dihydro-2H-benzimidazol-2-one. Furthermore, these two compounds were distinct in their abilities to potentiate M1-mediated activation of phosphoinositide hydrolysis and phospholipase D. The discovery of multiple structurally distinct positive allosteric modulators of M1 is an exciting advance in establishing the potential of allosteric modulators for selective activation of this receptor. These data also suggest that structurally diverse M1 potentiators may act by distinct mechanisms and differentially regulate receptor coupling to downstream signaling pathways.
The psychotic and cognitive symptoms of neuropsychiatric disorders such as schizophrenia and Alzheimer's disease (AD) remain serious unmet medical challenges. Patients with schizophrenia exhibit a constellation of symptoms that include positive, negative, and cognitive symptom clusters. Although current antipsychotic agents are effective in reducing positive symptoms such as hallucinations and delusions in most patients, negative symptoms such as anhedonia and blunted affect, as well as deficits in cognitive function, are not effectively treated with current medications (Vohora, 2007). In addition to the unmet medical needs of schizophrenia, the devastating cognitive and neuropsychiatric symptoms characteristic of AD present urgent needs for new therapeutic interventions (Saddichha and Pandey, 2008).
A large number of animal and clinical studies suggest that activation of muscarinic acetylcholine receptors (mAChRs) may provide a novel strategy for the treatment of multiple symptoms of both schizophrenia and AD (Langmead et al., 2008). The mAChRs are G protein-coupled receptors (GPCRs) with five subtypes termed M1 through M5 that respond to the endogenous neurotransmitter acetylcholine (ACh); M1, M3, and M5 couple to Gq, whereas M2 and M4 couple via Gi/o to downstream signaling pathways and associated effector systems (Wess, 1996; Langmead et al., 2008). It is interesting that mAChR agonists and acetylcholinesterase inhibitors have established efficacy in improving cognitive performance in patients with AD (Davis et al., 1992; Bodick et al., 1997; Rogers et al., 1998; Raskind et al., 1999). In addition, the mAChR agonist xanomeline has robust efficacy in reducing delusions, hallucinations, and other psychotic symptoms in patients with AD (Bodick et al., 1997) and schizophrenia (Shekhar et al., 2008). mAChR agonists also reduce pathological processing of amyloid precursor protein, raising the exciting possibility that these agents may also have disease modifying effects in patients with AD (Fisher, 2008; Jones et al., 2008).
Despite these advances, current mAChR agonists have dose-limiting adverse effects that preclude their use as therapeutic agents. The most prominent adverse effects of mAChR agonists (bradycardia, gastrointestinal distress, salivation, and sweating) are mediated by peripheral M2 and M3 mAChRs (Okamoto et al., 2002; Bymaster et al., 2003; Wess et al., 2007). In contrast, M1 is probably important for the effects of mAChR activation on cognition and sensory processing (Conn et al., 2008b; Fisher, 2008; Jones et al., 2008; Langmead et al., 2008). Unfortunately, efforts to develop selective orthosteric agonists for the M1 receptor have been unsuccessful, probably because the ACh binding site is highly conserved, making it difficult to develop agonists for M1 that are devoid of activity at the other mAChR subtypes. In addition, there are a number of theoretical problems associated with the use of direct acting agonists of M1, including rapid desensitization that could lead to tolerance and induction of M1-mediated generalized seizures (Hamilton et al., 1997; Bymaster et al., 2003; Wess et al., 2007).
An alternative approach that has been highly successful for ligand-gated ion channels is the development of positive allosteric modulators (PAMs) or allosteric potentiators of receptor function. Allosteric potentiators do not directly activate the receptor but bind to an allosteric site removed from the orthosteric neurotransmitter binding site and potentiate the effects of the endogenous agonist. The classic example of this approach is the use of benzodiazepines to potentiate GABA-A receptor function and provide safe and effective drugs for the treatment of insomnia and anxiety disorders without the potentially lethal effects of direct-acting GABA-A receptor agonists (Mohler et al., 2002). We and others have now expanded this approach to GPCRs (Conn et al., 2008a), including the discovery of highly selective allosteric potentiators of M4 (Brady et al., 2008; Chan et al., 2008; Shirey et al., 2008). These findings raise the possibility that allosteric potentiators of M1 may provide a viable approach to selectively activate M1. Brucine, an analog of strychnine, is a weak allosteric potentiator of M1 (Lazareno et al., 1998; Birdsall et al., 1999), providing proof-of-concept that M1 PAMs can enhance receptor activity. However, brucine exhibits low potency as an M1 PAM, induces only a slight increase in the potency of ACh, and is not selective for M1. If identifying M1 PAMs offers a viable approach to the development of novel activators of this receptor, it will be critical to discover compounds that belong to structurally diverse chemical scaffolds that possess robust M1 PAM activity. We now report the discovery of multiple structurally distinct M1 PAMs. These compounds induce significant increases in ACh potency and affinity and include compounds that are selective for M1. Detailed molecular pharmacological studies suggest that representative compounds are mechanistically distinct and can differentially regulate signal transduction downstream of M1 activation.
Materials and Methods
Constructs, Cell Lines, and Maintenance. Rat M1 cDNA, provided by T.I. Bonner (National Institutes of Health, Bethesda, MD), was used to generate the orthosteric-site mutant rat M1Y381A using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). All tissue culture reagents were purchased from Invitrogen Corporation (Carlsbad, CA) unless otherwise noted. Cell lines used for these studies stably expressed rat M1, human M2, human M3, rat M4, or human M5 in the Chinese hamster ovary (CHO-K1) parental background. CHO-K1 cells expressing M1, M3, or M5 receptors were cultured in Ham's F-12 medium with 10% FBS, 20 mM HEPES, and 50 μg/ml G418. Cells expressing the M2 receptor along with the chimeric G protein Gqi5 were cultured in the same media with 500 μg/ml hygromycin. M4/Gqi5 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS, 20 mM HEPES, 1 mM sodium pyruvate, 1× nonessential amino acids, 2 mM l-glutamine, 400 μg/ml G418 (Mediatech, Inc., Herndon, VA), and 500 μg/ml hygromycin. Receptor densities for these cells lines were determined as described in Shirey et al. (2008) and were the following: M1, 509 fmol/mg; M2-Gqi5, 505 fmol/mg; M3, 2313 fmol/mg; M4-Gqi5, 437 fmol/mg; and M5, 968 fmol/mg. Cells expressing the rat M1Y381A orthosteric-site mutant receptor were cultured in the same medium, except without hygromycin. Cells expressing the human metabotropic glutamate receptor 4 (mGluR4) receptor with Gqi5 were cultured in DMEM with 10% dialyzed FBS, 20 mM HEPES, 1 mM sodium pyruvate, 20 μg/ml proline, 400 μg/ml G418, and 5 nM methotrexate (Calbiochem/EMD Chemicals, San Diego, CA).
Primary High-Throughput Screening. Rat M1/CHO cells (10,000 cells/20 μl/well) were plated in black-walled, clear-bottomed, tissue culture-treated, 384-well plates (Greiner Bio-One, Monroe, NC) in Ham's F-12 medium with 10% FBS and 20 mM HEPES. The cells were grown overnight at 37°C in the presence of 5% CO2. The next day, the medium was removed using a VSpin (Velocity 11; Agilent Technologies, Santa Clara, CA) fitted with a modified bucket allowing the 384-well plate to be mounted inverted over a catch basin and spun at 80g for 10 s with 40% acceleration and deceleration.
The medium was replaced, using a Combi (Thermo Fisher Scientific, Waltham, MA), with 20 μl of 2 μM Fluo-4/acetoxymethyl ester (Invitrogen) for 45 min at 37°C. Fluo-4/acetoxymethyl ester was prepared as a 2.3 mM stock in DMSO, mixed in a 1:1 ratio with 10% (w/v) Pluronic acid F-127, and diluted in assay buffer (Hanks' balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid; Sigma-Aldrich, St. Louis, MO). Dye solution was removed using the VSpin and replaced with 20 μl of assay buffer using a Combi. Test compounds were transferred to daughter plates using an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA) and then diluted into assay buffer, using a Combi, to generate a 20 μM stock. Ca2+ flux was measured using the Functional Drug Screening System 6000 (FDSS6000; Hamamatsu, Tokyo, Japan). Baseline readings were taken (10 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm), and then test compounds (20 μl/well) were added using the FDSS's integrated pipettor. For the primary screen, cells were incubated with test compounds (final concentration, 10 μM) for 3.2 min, and then an EC20 concentration of carbachol was applied. The overall assay protocol was automated using the instruments noted above integrated with a robotic arm (F3; Thermo Fisher Scientific) under the control of a Polara scheduler (Thermo Fisher Scientific). All data were recorded to instrument local drives and later migrated to a network drive. FDSS data were analyzed using a custom analysis application and were associated with unique compound identifiers based on liquid handler transfer logs and plate barcode readings captured by the Echo and by Polara. “Hits” were selected by comparing the amplitude of the responses at the time of EC20 addition plus and minus test compounds. Wells with responses that differed from vehicle wells by 3 S.D. or greater were selected as hits for further study.
Concentration-Response Experiments and Counter-Screening. Compounds were serially diluted 1:3 into 10-point concentration-response curves in 100% DMSO and were transferred to daughter plates using the Echo. Compounds were then diluted to a 2× final concentration using assay buffer; the final DMSO concentration in these experiments was 0.3%. Test compounds were applied, followed 3 min later by an EC20 concentration of agonist. For human mGluR4/Gqi5/CHO counter-screening, cells were plated in DMEM, 10% dialyzed fetal bovine serum, 20 mM HEPES, and 1 mM sodium pyruvate at 30,000 cells/well, and the agonist glutamate was used rather than carbachol. Concentration-response experiments were performed in triplicate for each compound on M1 and in a single experiment for mGluR4. Compound activity at M1 was then confirmed using ACh as the agonist. Similar calcium mobilization assays were used to confirm activity of the compounds highlighted here at M1 in the presence of ACh and to test for concentration-dependent potentiation or antagonism of the other muscarinic subtypes as described in Shirey et al. (2008). Curves were fitted using a four-parameter logistical equation in Microsoft Excel (Redmond, WA) with the XLfit add-in (IDBS, Bridgewater, NJ). Subsequent confirmations of concentration-response parameters were performed in triplicate or quadruplicate on multiple days using independent serial dilutions of source compounds. Data from each day were fitted using a four-parameter logistical equation in GraphPad Prism to generate mean ± S.E.M. values (GraphPad Software, Inc., San Diego, CA).
Competition Binding. Membranes were prepared from M1-expressing CHO cells as described previously (Shirey et al., 2008). Binding reactions contained 0.1 nM l-[N-methyl-3H]scopolamine ([3H]NMS), 20 μg of membranes, and compound or vehicle (0.3% DMSO, final, to define total binding) or 1 μM atropine (to define nonspecific binding) in a total volume of 500 μl. The KD value of [3H]NMS was determined empirically to be 0.21 nM. Compounds were serially diluted in DMSO and then diluted in assay buffer (100 mM NaCl, 10 mM MgCl2, and 20 mM HEPES, pH 7.4) to give a final DMSO concentration of 0.3% in the binding reaction. Binding reactions were incubated for 2 h at room temperature on a Lab-Line Titer plate shaker (Lab-Line Instruments, Melrose Park, IL) at setting 7 (∼750 rpm). Reactions were stopped, and membranes were collected onto 96-well Barex microplates with GF/B filter (1 μm pore size) using a Brandel harvester and washed three times with ice-cold harvesting buffer (50 mM Tris-HCl and 0.9% NaCl, pH 7.4). Filter plates were dried overnight and counted in a TopCount scintillation counter (PerkinElmer Life and Analytical Sciences, Waltham, MA). For acetylcholine affinity experiments, the KI value of an acetylcholine competition curve was determined in the absence and presence of fixed concentrations (3-30 μM, final) of test compound. Actual [3H]NMS concentrations were back-calculated after counting aliquots of 5× [3H]NMS added to the reaction. Radioligand depletion was routinely kept to approximately 5% or less.
PI Hydrolysis. M1-expressing CHO cells were plated at 1.25 × 105 cells/well in the presence of 0.5 μCi myo-[3H]inositol in 24-well tissue culture plates and allowed to incubate overnight at 37°C, 5% CO2. Krebs buffer (108 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3, 10 mM glucose, and 20 mM LiCl) was prepared the morning of the assay and was allowed to equilibrate to 95:5 O2/CO2 at 37°C for at least 1 h to obtain a pH of 7.4. After aspirating inositol-loading media, 0.5 ml of 2× test compound or vehicle (30 μM final concentration, 0.3% DMSO) and then 0.5 ml of 2× acetylcholine was added to M1 cells, followed by a 1-h incubation at 37°C. Reactions were stopped, and inositol phosphates were extracted for 40 min at room temperature by adding 10 mM formic acid to each well. Inositol phosphates were separated on anion exchange columns containing AG 1-X8 resin (100-200 mesh, formate form; Bio-Rad, Hercules, CA). Columns were washed twice with H2O before loading formic acid extracts. Columns were then washed once with H2O, once with 5 mM myo-inositol (in H2O), and once with H2O before elution with 200 mM NH4HCO2/0.1 M H2CO2; counts were measured using a scintillation counter (LS 6500; Beckman Coulter, Fullerton, CA).
Activation of Phospholipase D. Phospholipase D activity in rat M1 CHO cells was assessed using a mass spectrometry-based assay as described previously (Brown et al., 2007). In brief, CHO cells expressing M1 muscarinic receptors were plated at 5 × 105 cells/well in 6-well tissue culture plates in growth media (Ham's F-12 medium with 10% FBS and 20 mM HEPES) in a 37°C humidified atmosphere with 5% CO2. After 45 h (∼80-90% confluence), growth media were removed, and cells were incubated in serum-free DMEM for 2 h. The cells were challenged with either vehicle or the indicated concentrations of ACh and/or potentiators with and without 0.3% 1-BuOH-d10. Cells were incubated in the indicated conditions for 30 min at 37°C. After incubation, treatment media was removed, cells were washed with ice-cold 1 × PBS, and 800 μl of ice-cold 0.1 N HCl/MeOH (1:1), and 400 μl of CHCl3 was added immediately. Next, 10 μl of 10 μg/ml 1,2-dipalmitoyl-sn-glycero-3-phosphomethanol [32:0 phosphatidylmethanol (PtdMeOH)] was added as an internal standard. The samples were vortexed (1 min) and centrifuged (5 min, 18,000g). The lower phase was then isolated and evaporated, and extracted phospholipids were dissolved in 80 μl of CH3OH/CHCl3 (9:1). Before analysis, 1 μl of NH4OH was added to each sample to promote protonation of lipid species. Mass spectral analysis was performed on a Finnigan TSQ Quantum triple quadrupole mass spectrometer (Thermo Fisher Scientific) equipped with a Harvard Apparatus (Holliston, MA) syringe pump and an electrospray source. Samples were analyzed at an infusion rate of 10 μl/min. The lipid samples were analyzed in negative ionization mode over the range of m/z 350 to 1200. The phospholipase D (PLD) activity was calculated as the ratio of the peak intensity of the produced PtdBuOH species to that of the internal standard (32:0 PtdMeOH, m/z 661 in negative mode mass spectrometry). In particular, 32:1 PtdBuOH-d9 at m/z 710, 34:1 PtdBuOH-d9 at m/z 738, and 36:2 PtdBuOH-d9 at m/z 764 were produced during stimulation, and these species were used to calculate the enzyme activity. The PLD activity (PtdBuOH/PtdMeOH) of agonist treatment alone was normalized to a value of 1, and the ratio of PLD activity of the samples treated with agonist and potentiators to that of the agonist alone was expressed as the relative ratio.
Compounds. Carbamylcholine chloride (carbachol), acetylcholine chloride, and atropine were purchased from Sigma-Aldrich. [3H]-NMS was purchased from GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK). 1-BuOH-d10 was purchased from Acros Organics (Fairlawn, NJ). PtdMeOH (32:0) and diacylglycerol (24:0) were purchased from Avanti Polar Lipids (Alabaster, AL).
The Vanderbilt High-Throughput Screening Center compound collection was obtained from ChemBridge Corporation (San Diego, CA) and ChemDiv, Inc. (San Diego, CA) and stored in barcoded, Griener 384-well, flat-bottomed, polypropylene plates. The plates were thermally sealed with peelable seals using a PlateLoc (Velocity 11). Groups of 10 plates were vacuum-packed in thermally sealed freezer bags (FoodSaver, Jarden Corporation, Cleveland, OH) and stored frozen at -80°C. Primary hits identified in the screen were reordered from ChemBridge or ChemDiv as 10 mM DMSO stocks; these orders were accompanied by NMR spectra to confirm compound identity, and three of the compounds were resynthesized in house (described below).
Synthesis. All NMR spectra were recorded on a Bruker 400 MHz instrument (Bruker BioSpin Ltd., Milton, ON, Canada). 1H chemical shifts are reported in δ values in parts per million down-field from DMSO as the internal standard in DMSO. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, coupling constant (in hertz). 13C chemical shifts are reported in δ values in parts per million with the DMSO carbon peak set to 39.5 ppm. Low-resolution mass spectra were obtained on an Agilent 1200 series 6130 mass spectrometer with electrospray ionization (Agilent Technologies). High-resolution mass spectra were recorded on a Waters Q-TOF API-US (Waters Corporation, Milford, MA). Analytical thinlayer chromatography was performed on Analtech silica gel GF 250-μm plates. Analytical high-performance liquid chromatography was performed on an Agilent 1200 series with UV detection at 214 and 254 nm along with evaporative light-scattering detection. Preparative purification was performed on an ISCO combi-flash companion (Teledyne ISCO, Lincoln, NE). Solvents for extraction, washing, and chromatography were of high-performance liquid chromatography grade. All reagents were purchased from Sigma-Aldrich and were used without purification. All polymer-supported reagents were purchased from Biotage, Inc. (Charlottesville, VA).
VU0029767. To a solution of 4-ethoxyaniline (10.0 g, 72.9 mmol) in EtOH (100 ml) was added ethyl 2-bromoacetate (14.61 g, 87.0 mmol) and sodium acetate (8.97 g, 109.0 mmol) and stirred for 48 h at room temperature. Once complete, the reaction was then partitioned into H2O and EtOAc, and the organics were washed with brine and dried over MgSO4 before concentration in vacuo. The crude product was purified by passage through a silica plug eluting with 5% EtOAc in hexanes to afford the intermediate product ethyl 2-(4-ethoxyphenylamino) acetate (11.7 g, 72%) as a dark brown oil. To a solution of the acetate intermediate (4.0 g, 17.93 mmol) in MeOH (50 ml) was added hydrazine (177.9 ml, 177.9 mmol) and stirred for 16 h at 65°C. Once complete, the reaction was then partitioned into H2O and EtOAc, and the organics were washed with brine and dried over MgSO4 before concentration in vacuo. The crude product was purified by column chromatography (silica gel) eluting with 5% MeOH in CH2Cl2 to afford the intermediate product 2-(4-ethoxyphenylamino)-acetohydrazide (2.5 g, 67%) as a white solid. To a solution of the hydrazide intermediate (2.0 g, 9.56 mmol) in dimethylformamide (100 ml) was added 2-hydroxy-1-napthaldehyde (1.64 g, 9.56 mmol) and stirred for 16 h at room temperature. Once complete, the reaction was concentrated in vacuo and the crude product was purified by filtration with diethyl ether wash (3 × 250 ml) to afford title compound (3.41 g, 98%) as a yellow solid; 1H NMR (400 MHz, DMSO-d6) δ 9.38 (s, 1H), 8.09 (d, J = 4.2 Hz, 1H), 7.80 (dd, J = 8.8, 4.4 Hz, 2H), 7.50 (t, J = 7.2 Hz, 1H), 7.31 (t, J = 7.2 Hz, 1H), 7.12 (d, J = 8.8 Hz, 1H), 6.67 (d, J = 8.8 Hz, 2H), 6.49 (d, J = 8.8 Hz, 2H), 5.69 (t, J = 6.0 Hz, 1H), 3.80 (q, J = 6.8 Hz, 2H), 3.73 (d, J = 5.6 Hz, 1H), 3.32 (s, 2H), and 1.18 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.2, 157.8, 150.6, 146.3, 142.7, 132.6, 131.6, 128.9, 127.8, 123.5, 120.7, 118.8, 115.4, 113.5, 113.3, 108.4, 63.3, 47.4, and 14.8; LC-MS (214 nm), 3.09 min (>98%); m/z 364.2 [M+H].
VU0119498. To a solution of isatin (250 mg, 1.70 mmol) in ACN (8 ml) was added potassium carbonate (470 mg, 3.40 mmol), potassium iodide (28.2 mg, 0.17 mmol), and 4-bromobenzyl bromide (632 mg, 2.55 mmol) and irradiated at 160°C for 10 min. The reaction was judged complete and partitioned into H2O and CH2Cl2 and passed through a phase-separation column. The organics were then concentrated in vacuo. The crude product was purified by filtration with diethyl ether wash (3 × 30 ml) to afford title compound (500 mg, 93%) as a bright orange solid; 1H NMR (400 MHz, DMSO-d6) δ7.58 (m, 2H), 7.53 (d, J = 8.4 Hz, 2H), 7.40 (d, J = 8.4 Hz, 2H), 7.12 (t, J = 7.6 Hz, 1H), 6.95 (d, J = 3.2 Hz, 1H), and 4.89 (s, 2H). 13C NMR (100 MHz, DMSO-d6): δ 182.9, 158.3, 150.1, 137.9, 135.0, 131.5, 129.7, 124.6, 124.5, 123.3, 120.6, 110.9, and 42.3; LC-MS (214 nm), 3.26 min (>98%); m/z 315.99.
VU0090157. To a solution of 6-nitrobenzo[d][1,3]dioxole-5-carbaldehyde (195 mg, 1.0 mmol) in MeOH (100 ml) was added 2-cyanoethyl 3-oxobutanoate (186 mg, 1.2 mmol), 1-methylurea (111 mg, 1.5 mmol), and p-TsOH (17 mg, 0.1 mmol) and refluxed for 4 h. Once complete, the reaction was filtered, and the crude product was purified by recrystallization with MeOH and H2O to afford the intermediate product 2-cyanoethyl 1,6-dimethyl-4-(6-nitrobenzo[d][1,3]-dioxol-5-yl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (283 mg, 78%) as a white crystalline solid. To a solution of the carboxylate intermediate (1.0 g, 2.57 mmol) in acetone/H2O (6:2 ml) was added 2 M NaOH (4 ml) at 0°C, and the solution was warmed to room temperature and stirred for 2 h. Once complete, the reaction was acidified with 2 N HCl and partitioned into H2O and CH2Cl2, and the organics were concentrated in vacuo. The crude product was purified by recrystallization with MeOH and H2O to afford the intermediate product 1,6-dimethyl-4-(6-nitrobenzo[d][1,3]dioxol-5-yl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylic acid (790 mg, 92%) as a white crystalline solid. To a solution of the acid intermediate (500 mg, 1.49 mmol) in dimethylformamide (12 ml) was added cyclopentanol (385 mg, 4.48 mmol), ethylene dichloride (428 mg, 2.24 mmol), and polystyrene-bound 4-dimethylaminopyridine (1.5 mmol/g, 5.96 mmol) and irradiated at 100°C for 20 min. Once complete, the reaction was partitioned into 1 N HCl in H2O and CH2Cl2, and the organics were concentrated in vacuo. The crude product was purified by recrystallization in EtOAc and hexanes to afford the title compound (200 mg, 33%) as a brown crystalline solid. 1H NMR (400 MHz, DMSO-d6): δ 7.60 (s 1H), 7.58 (d, J = 1.6 Hz, 1H), 6.95 (s, 1H), 6.23 (s, 1H), 6.20 (s, 1H), 5.81 (d, J = 1.4 Hz, 1H), 4.93 (m, 1H), 3.16 (s, 3H), 2.57 (s, 3H), 1.66 (m, 1H), 1.55 (m, 1H), 1.40 (m, 4H), 1.19 (m, 1H), and 1.09 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.5, 152.6, 152.3, 151.8, 147.0, 141.3, 135.3, 106.9, 104.8, 103.4, 100.4, 75.9, 48.5, 32.0, 29.6, 23.0, and 15.9; LC-MS (214 nm), 3.14 min (>98%); m/z 404.2.
Results
Novel M1 PAMs Were Identified via a High-Throughput Screening Approach. To search for novel PAMs of the M1 muscarinic receptor, we performed a high-throughput screen. For these studies, we used a CHO cell line expressing the rat M1 muscarinic receptor and measured intracellular calcium mobilization. The screening protocol monitored fluorescence in each well at baseline and changes in fluorescence after the addition at 4 s of either vehicle or test compound (10 μM final nominal concentration). After an approximately 3.2-min incubation period, a submaximally effective concentration (EC20) of the muscarinic agonist carbachol was added.
Raw data were normalized by dividing the fluorescence traces by the minimum fluorescence reading obtained 2 to 5 s before the EC20 agonist addition. This allowed us to retain information for compounds that were slightly fluorescent or that induced small baseline changes. Potential PAMs were defined as compounds that increased the EC20 carbachol response by at least 3 S.D. above the control EC20 response. Vehicle, EC20, and ECmax controls were included on each plate, and data were analyzed on a plate-by-plate basis. Plates were randomly spot-checked to ensure the quality of the data, validate the criterion for choosing PAMs, and prevent the loss of compounds with weak activity. The primary screen of M1 yielded 1634 hits that increased the carbachol EC20 response by at least 3 S.D. over control.
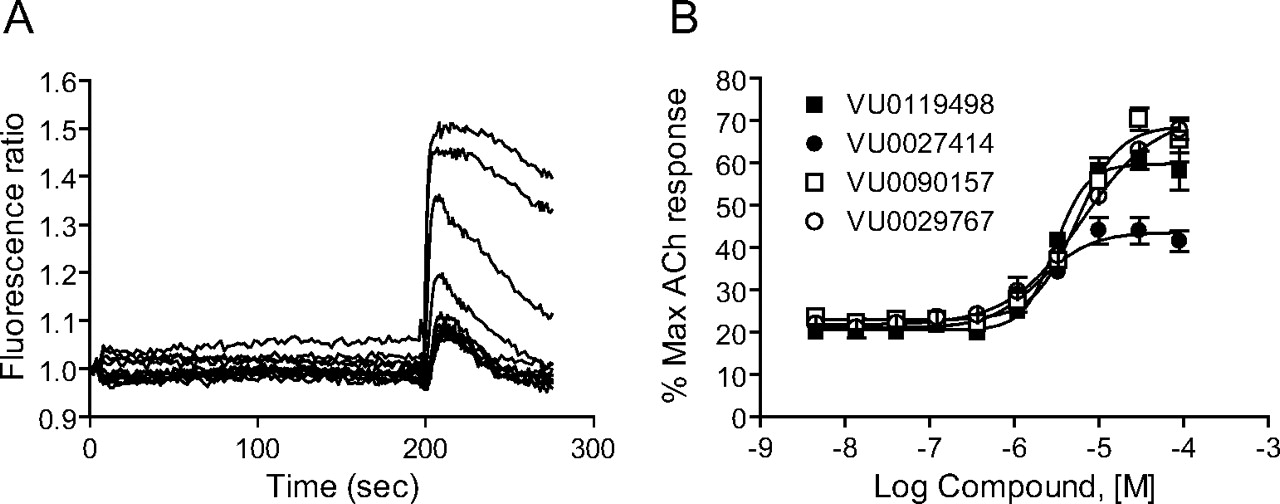
Initial retests focused on single-point (30 μM) screening of 961 compounds (with responses 3 S.D. above control) and concentration-response screening of 327 compounds that were deemed to have stronger potentiator activity (at least 5 S.D. above control) or mixed agonist/PAM activity. The remaining compounds (346) were unavailable for commercial reorder and have not yet been confirmed for activity. Of the 961 compounds in the weaker category, 34 compounds potentiated the acetylcholine response when tested at a 30 μM concentration. These 34 compounds, as well as the 327 compounds mentioned above, were formatted into 10-point concentration-response curves and tested for concentration-dependent ability to potentiate M1-mediated calcium responses. Compounds were also screened against a mGluR4/CHO line to determine whether compounds increased calcium levels by a nonspecific mechanism. One hundred five compounds were confirmed as potentiating the acetylcholine response in a concentration-dependent manner; 30 of these compounds potentiated (28 compounds) or antagonized (2 compounds) the mGluR4 response, indicating that approximately 71% of the compounds were selective for M1 over mGluR4. Four compounds that exhibited M1 PAM activity but were devoid of activity at mGluR4 were chosen for further characterization. The structures of these compounds are shown in Fig. 1. Figure 2A shows the effects of multiple concentrations of a representative M1 PAM hit, VU0090157, on the calcium fluorescence response to an EC20 concentration of the endogenous M1 agonist, ACh. Figure 2B shows the resulting concentration-response relationship for VU0090157 (white boxes) and the three additional PAMs shown in Fig. 1 (VU0119498, VU0027414, and VU0029767) for potentiation of an EC20 concentration of ACh. Each of these compounds exhibited potencies in the low micromolar range for potentiating M1-mediated calcium responses.

Structures of novel M1 PAMs VU0119498, VU0027414, VU0090157, and VU0029767.
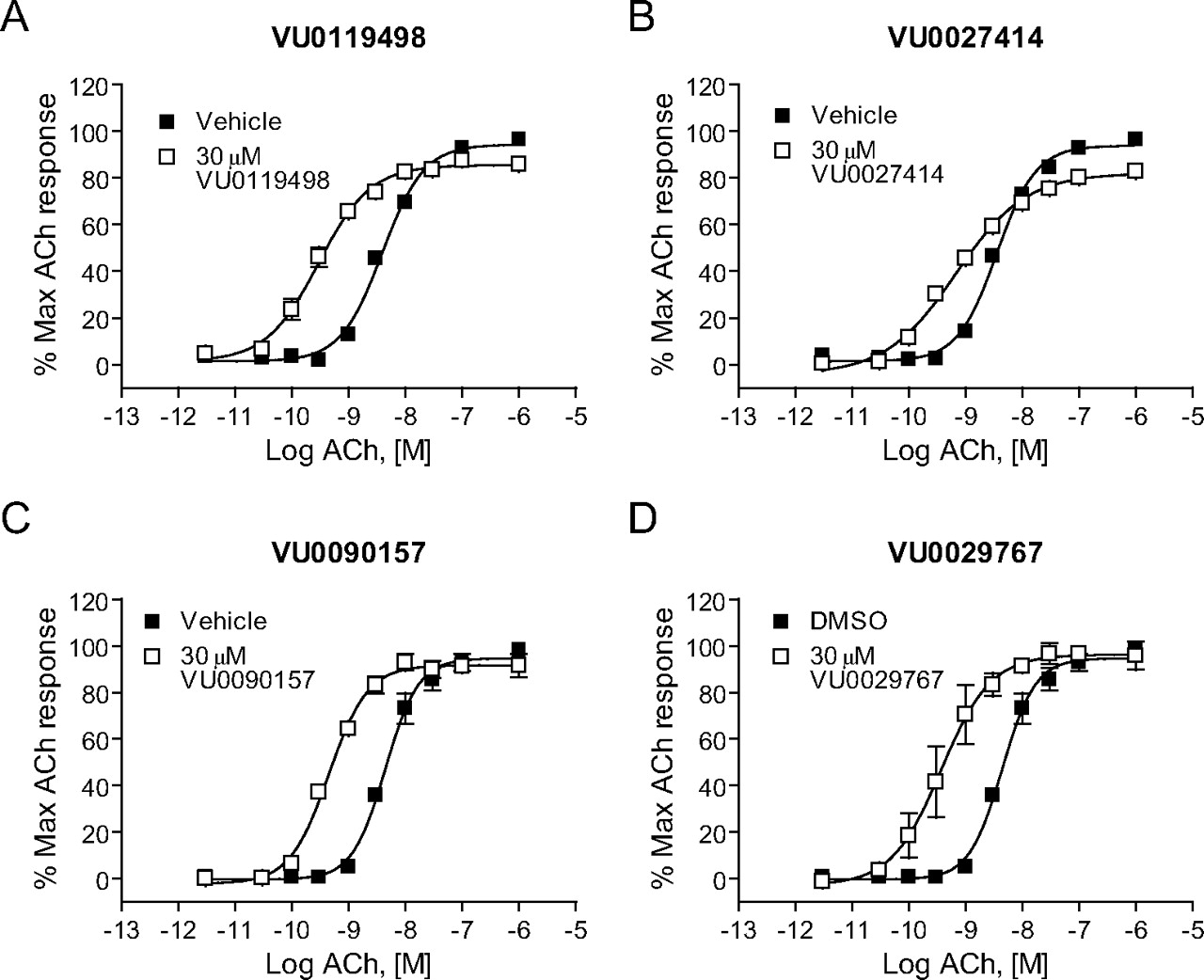
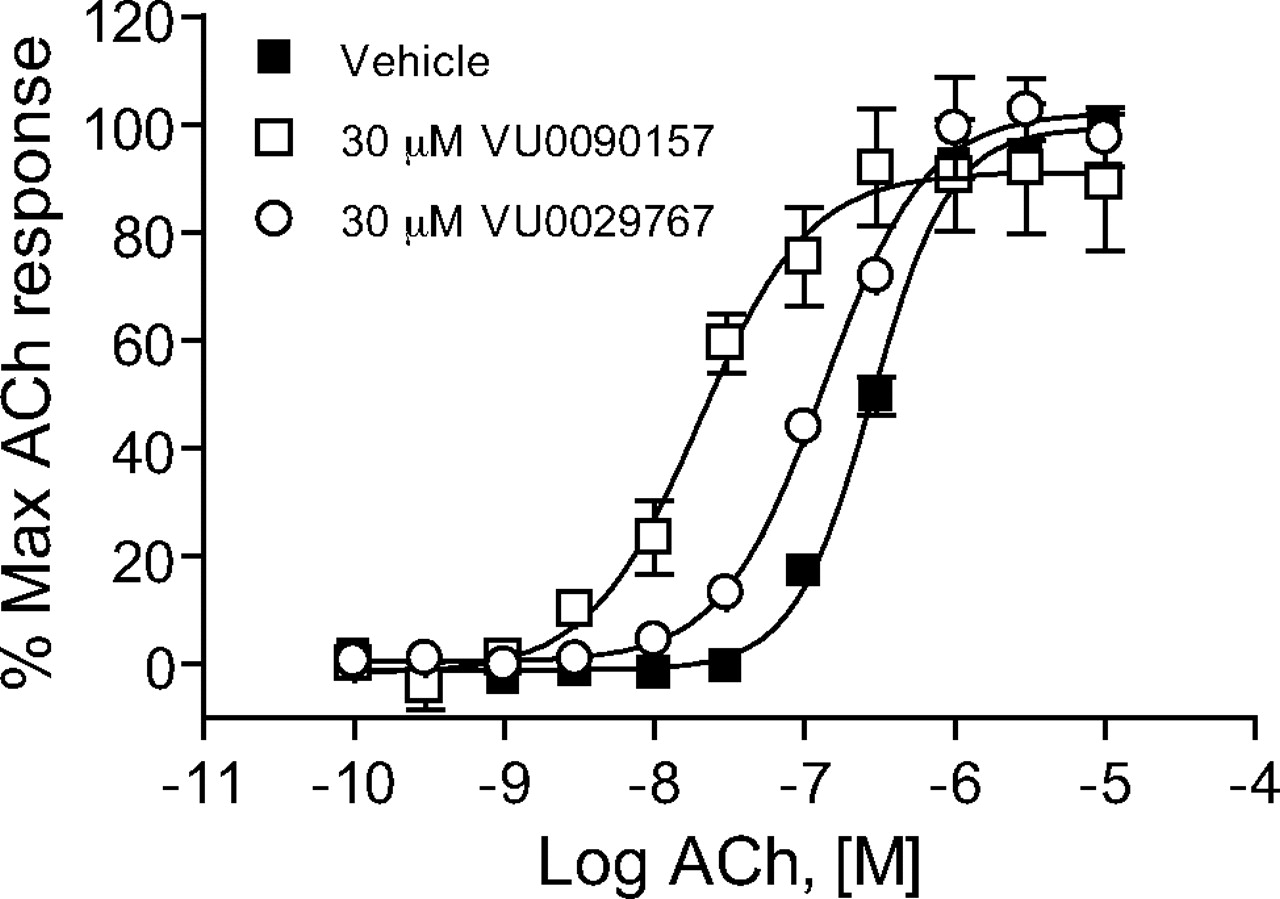
M1 PAMs Induce Robust Shifts in the ACh Concentration-Response Curves. The effects of the four representative M1 PAMs on potency and efficacy of ACh at activating M1 were determined by evaluating the concentration-response relationship for ACh in the absence and presence of each compound. Preincubation of M1/CHO cells with a 30 μM concentration of each M1 PAM before the addition of increasing concentrations of ACh revealed that each compound induced a robust leftward shift in the ACh concentration-response curve (Fig. 3). Shifts in ACh potency were most pronounced for VU0029767 (14.9 ± 5.9-fold, mean ± S.E.M.) and VU0119498 (14.0 ± 2.8-fold). VU0090157 also induced a robust shift in the ACh concentration-response relationship (10.1 ± 1.6-fold), whereas the increase in ACh potency induced by VU0027414 (5.3 ± 0.3-fold) was somewhat less pronounced than that observed for the other M1 PAMs. The relative efficacies of these compounds in shifting the ACh concentration-response curves corresponded with the maximal effects of these compounds on responses to an EC20 concentration of ACh, where VU0027414 induced a more modest effect than did the other M1 PAMs (Fig. 2B). The level of potentiation of M1 responses induced by these compounds is comparable with the efficacies that have been observed for some of the most robust allosteric potentiators of other GPCRs (Conn et al., 2008a) and represents a major advance relative to the 2-fold shift induced by brucine (Lazareno et al., 1998; Birdsall et al., 1999).
M1 PAMs Do Not Compete with an Orthosteric Antagonist for Binding at M1 Receptors. To determine whether these PAMs interact with M1 at the ACh (orthosteric) binding site, we performed competition binding studies with the orthosteric antagonist [3H]NMS (Fig. 4). Control experiments performed with the orthosteric antagonist atropine demonstrated a classic competitive interaction with [3H]NMS with a Ki value of 1.1 ± 0.09 nM (mean ± S.E.M.). In contrast, none of the four new M1 PAMs significantly competed with equilibrium binding of [3H]NMS at concentrations that maximally potentiated M1 responses to ACh. These data suggest that these compounds do not directly interact with the orthosteric ACh site on M1.
Novel M1 PAMs Exhibit Differential Selectivity Profiles for the Different mAChR Subtypes. It has been difficult to develop orthosteric mAChR ligands that are truly selective for individual mAChR subtypes. Allosteric sites may be less highly conserved, providing potential strategies for the development of more selective GPCR modulators. Examination of the effects of the four compounds presented here on each of the mAChR subtypes revealed clear distinctions in their selectivity profiles (Fig. 5). For example, VU0119498 potentiated ACh responses at each of the three Gq-coupled muscarinic receptors (M1, M3, and M5) but had no activity at the Gi/o-coupled mAChR subtypes M2 and M4 (Fig. 4A). VU0027414 potentiated M1 and M5 responses, antagonized M2 and M3, and had no effect at M4. It is interesting that VU0090157 and VU0029767 were more selective in potentiating responses to M1 relative to the other mAChR subtypes. VU0090157 weakly potentiated responses at M4 at the highest concentration but displayed significantly lower potency for M4 than for M1 and did not potentiate any of the other receptor subtypes. VU0029767 was the most selective M1 PAM and exhibited only a slight potentiation of the four other muscarinic subtypes at the highest concentrations tested. Based on these selectivity profiles and the finding that VU0090157 and VU0029767 were among the most robust and potent of the M1 PAMs, these two compounds were chosen for further studies.
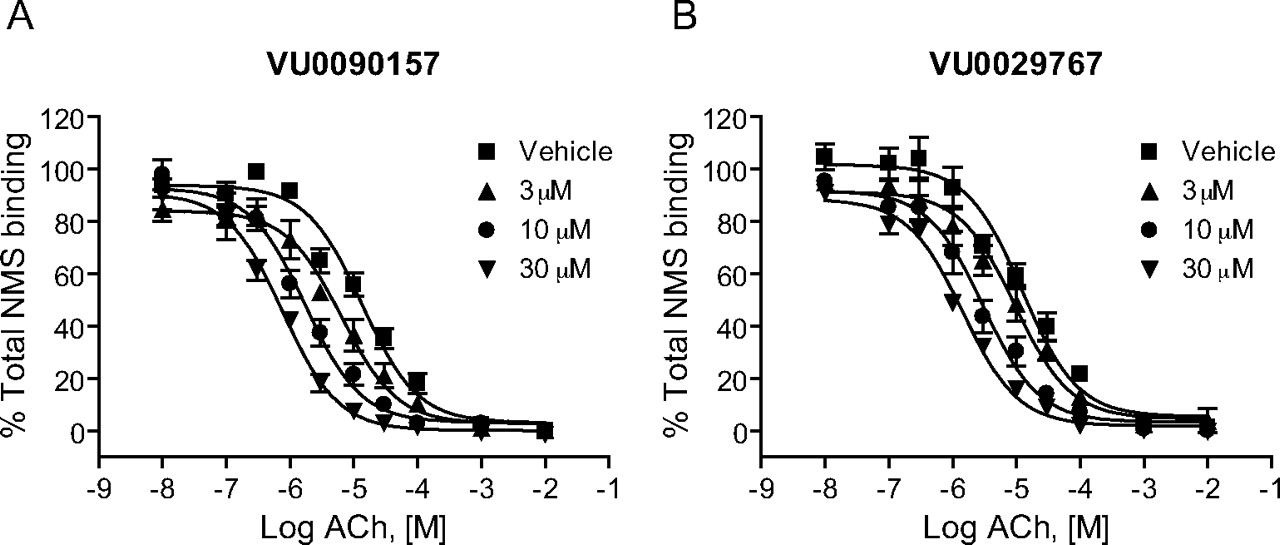
Novel M1 PAMs Increase Affinity of the Orthosteric Site for ACh. In theory, allosteric potentiators of GPCRs can act by increasing the affinity of the receptor for orthosteric agonists or by increasing efficiency of coupling of the receptor to G proteins (Conn et al., 2008a), and GPCR PAMs have been identified that act by a combination of these mechanisms (Shirey et al., 2008). To gain insight into the mechanisms of action of the novel M1 PAMs, we assessed the effects of these compounds on the affinity of ACh for the orthosteric site by performing competition binding studies (Fig. 6). ACh exhibited a competitive interaction with [3H]NMS with an estimated Ki value of approximately 9 μM. Increasing concentrations of each of the novel M1 PAMs progressively shifted the ACh competition curves to the left, suggesting that the compounds induced concentration-dependent increases in ACh affinity for the orthosteric site. The maximal affinity shifts of ACh measured in the presence of a 30 μM concentration of each compound were similar to the maximal shifts in ACh potency as measured in the functional calcium mobilization assays. For example, a maximally effective concentration of VU0090157 induced a 17.9 ± 3.5 (mean ± S.E.M.)-fold shift in ACh affinity, and VU0029767 induced an increase in ACh affinity of 8.8 ± 1.9-fold (mean ± S.E.M.). These results suggest that the increase in ACh affinity induced by these two M1 PAMs can account for the increase in ACh potency measured in the studies of ACh-induced calcium mobilization.

Novel M1 PAMs potentiated agonist responses in M1 CHO cells. A, fluorescence ratio trace generated by a potentiator identified via high-throughput screening, VU0090157, in the Ca2+ assay. Each point of the raw trace was divided by the fluorescence at time 0 to generate a ratio. Compound was added at 4 s, followed by an EC20 concentration of acetylcholine at 196 s. Traces reflect the increase in fluorescence obtained in the presence of increasing concentrations of compound. B, maximum fluorescence ratio values in the EC20 window (196-225 s) were plotted as a function of compound concentration to generate a concentration-response curve. Potencies of compounds were as follows (mean ± S.E.M.): VU0119498, 3.1 ± 0.2 μM; VU0027414, 2.1 ± 0.5 μM; VU0090157, 5.3 ± 0.7 μM; and VU0029767, 9.4 ± 4.4 μM. Data represent the mean ± S.E.M. of three independent experiments performed in quadruplicate.
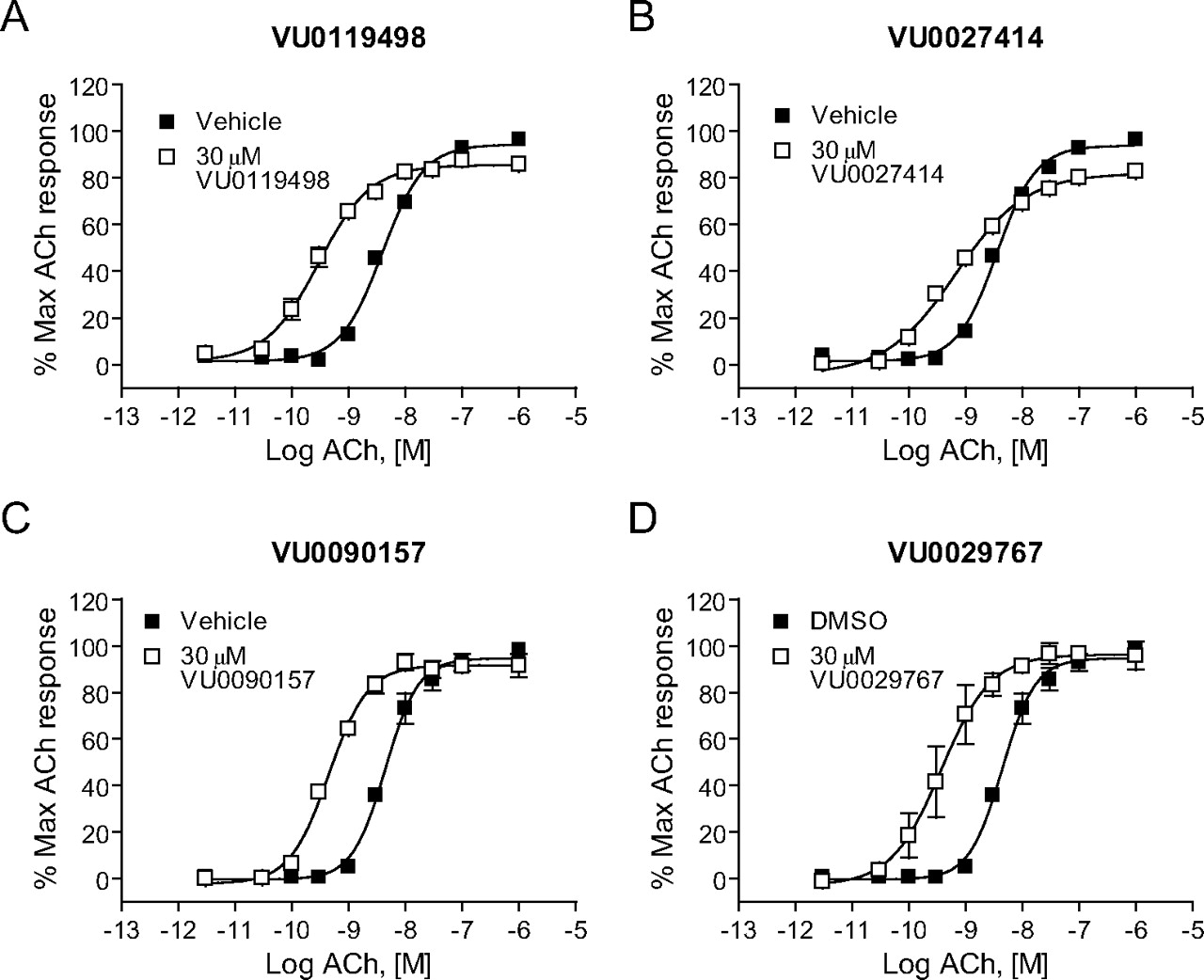
Novel M1 PAMs shifted the concentration-response curve for ACh at least 5-fold to the left. DMSO-matched vehicle or 30 μM VU0119498 (A), VU0027414 (B), VU0090157 (C), and VU0029767 (D) were applied to M1-CHO cells before the addition of increasing concentrations of ACh, and calcium mobilization was measured. Shifts of the ACh curve were the following (mean ± S.E.M.): VU0119498, 14.0 ± 2.8-fold (A); VU0027414, 5.3 ± 0.3-fold (B); VU0090157, 10.1 ± 1.6-fold (C); and VU0029767, 14.9 ± 5.9-fold (D). Data represent the mean ± S.E.M. of three independent experiments performed in triplicate.
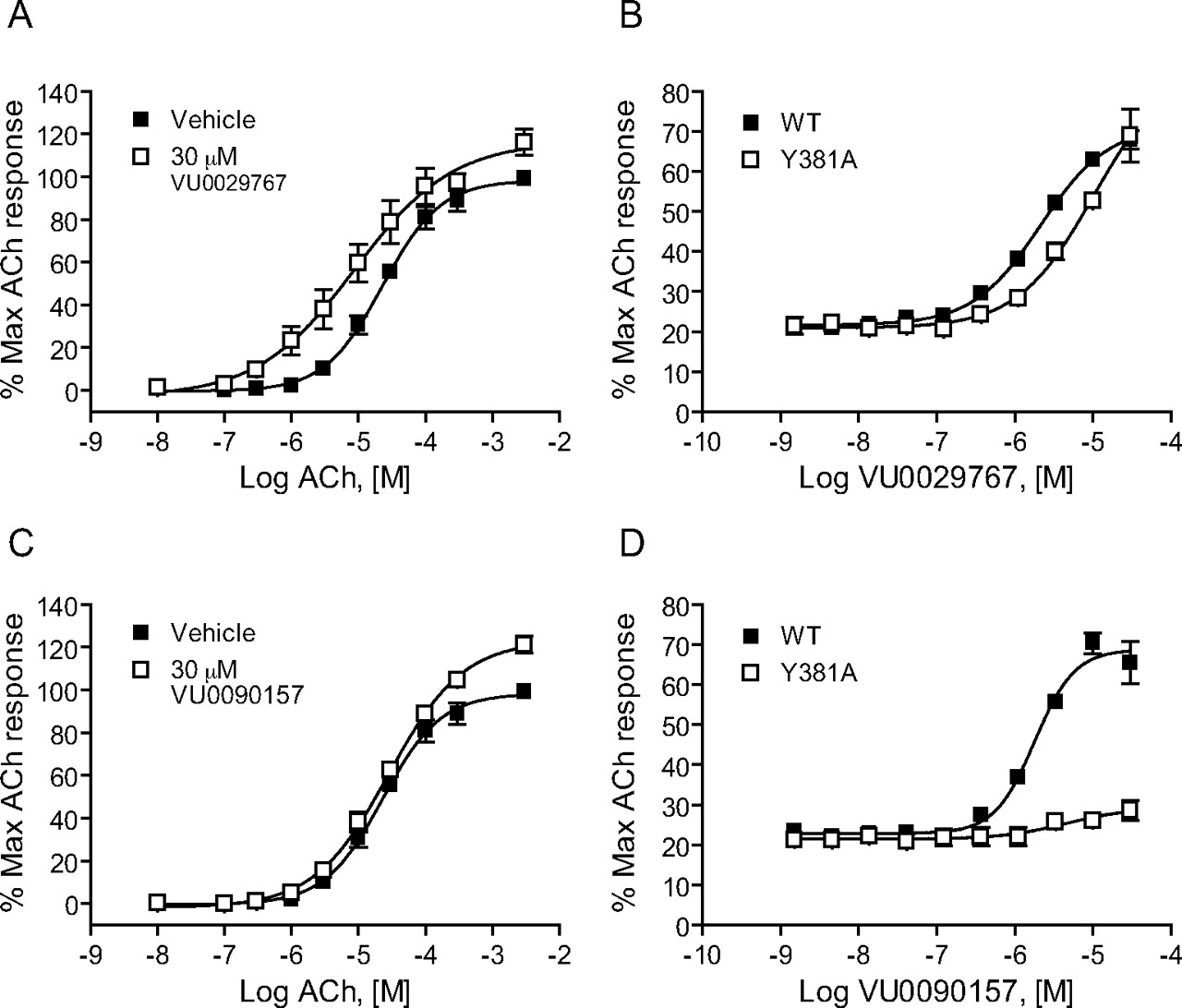
VU0029767 and VU0090157 Differentially Modulate a Form of M1 Bearing a Mutation in the Orthosteric Site. Previous studies aimed at mapping the orthosteric binding site on M1 suggest that a tyrosine residue at amino acid position 381 plays a critical role in ACh binding within the orthosteric binding pocket of this receptor. Mutation of this single tyrosine residue to alanine (M1Y381A) dramatically reduces the affinity and potency of ACh and other orthosteric ligands at M1 (Ward et al., 1999). Although the potency of ACh is greatly reduced by the mutation of this residue, high concentrations of ACh or other orthosteric ligands can activate M1Y381A and induce concentration-dependent activation of the mutant receptor (Fig. 7, A and C, ACh concentration range shifted roughly 1000-fold to the right for M1Y381A relative to WT M1). This mutant, therefore, serves as a valuable tool to begin probing the interaction of novel compounds with the M1 receptor. Because the new M1 PAMs are structurally unrelated, such a strategically located mutation of M1 may differentially affect their ability to increase the affinity of ACh for the orthosteric site and thus may provide insight into whether compounds act in a mechanistically distinct manner. VU0029767 induced a leftward shift in the ACh concentration-response curve at M1Y381A in a manner similar to that observed for the WT receptor (Fig. 7A). Furthermore, the potency of VU029767 at potentiating the response to an EC20 concentration of ACh was similar at M1Y381A and WT M1 receptors (Fig. 7B). It is interesting that VU0090157 did not increase the potency of ACh for M1Y381A activation (Fig. 7C) and did not significantly potentiate responses to an EC20 concentration of ACh at any concentration tested (Fig. 7D). This is in striking contrast to the ∼15-fold potentiation of ACh potency induced by VU0090157 at the WT receptor and suggests that VU029767 and VU0090157 may potentiate ACh responses at different sites and/or by stabilizing different conformations of the receptor.

Novel M1 PAMs exhibited minimal effects on [3H]NMS binding to the M1 receptor. Competition curves for PAMs VU0119498 (▪), VU0027414 (•), VU0090157 (□), and VU0029767 (○) were obtained in the presence of 0.1 nM [3H]NMS using membranes from M1-expressing CHO-K1 cells. The Ki value for atropine competition (▴) was determined to be 1.08 ± 0.09 nM. Data represent the mean ±S.E.M. of three independent experiments performed in triplicate.

Different muscarinic subtype-selectivity profiles were observed for various PAMs. Increasing concentrations of each PAM, VU0119498 (A), VU0027414 (B), VU0090157 (C), and VU0029767 (D), were added before the application of an appropriate EC20 concentration of ACh determined individually for each muscarinic receptor subtype, and calcium-generated fluorescence changes were measured. Average ACh EC20:M1 (▪, 1.4 nM), M2 (□, 13.2 nM), M3 (•, 0.12 nM), M4 (○, 23.7 nM), and M5 (▴, 0.20 nM). The M2 and M4 receptors were coexpressed with the chimeric G protein, Gqi5, to permit coupling of these Gi/o-coupled receptors to calcium mobilization. Data represent the mean ± S.E.M. of at least three independent experiments performed in quadruplicate.

Novel M1 PAMs altered the affinity of ACh for M1. Membranes from M1-expressing CHO-K1 cells were incubated with 0.1 nM [3H]NMS. ACh competition curves were generated in the absence and presence of increasing concentrations of VU0090157 and VU0029767. The Ki values of ACh in the presence of DMSO control were 9.7 ± 2.3 μM (VU0090157-matched experiments) and 8.7 ± 1.3 μM (VU0029767-matched experiments). VU0090157 (3, 10, and 30 μM) (A) shifted the ACh competition curve by 2.1 ± 0.2-, 7.9 ± 1.3-, and 17.9 ± 3.5-fold, respectively. VU0029767 (3, 10 and 30 μM) (B) shifted the ACh competition curve by 1.7 ± 0.8-, 4.9 ± 2.0-, and 8.8 ± 1.9-fold, respectively. Data represent the mean ± S.E.M. of three independent experiments performed in triplicate.

VU0029767 potentiated responses of a mutant M1 receptor with greatly reduced affinity for ACh, whereas VU0090157 did not. DMSO vehicle, 30 μM VU0029767 (A), or VU0090157 (C) was preapplied to M1Y381A cells before the addition of increasing concentrations of ACh. Compound concentration-response curves of VU0029767 (B) and VU0090157 (D) were generated in the presence of an EC20 concentration of acetylcholine (1.4 nM for WT and 7.5 μM for the M1Y381A receptor) in a calcium assay using cells expressing either the WT M1 receptor (▪) or the M1Y381A receptor (□). Data represent the mean ± S.E.M. of three independent experiments performed in quadruplicate.
VU029767 and VU0090157 Differentially Modulate Responses of M1 to a Recently Discovered Allosteric Agonist. We reported recently that a compound termed TBPB is a selective allosteric agonist of M1 that directly activates the M1 receptor (Jones et al., 2008). This is in contrast to the novel M1 PAMs reported here, which increase M1 activity by potentiating the effects of ACh. Combined pharmacological and mutational analyses suggest that TBPB activates M1 by a mechanism that is distinct from ACh and acts via an allosteric site or a site partially overlapping the ACh binding site. Because VU029767 and VU0090157 act by increasing the affinity of ACh for the orthosteric site, it is not clear whether these compounds would potentiate the agonist response to TBPB in a manner similar to that seen with ACh. Furthermore, if these compounds stabilize slightly different conformations of the receptor, they may have different effects on responses of M1 to TBPB. Consistent with previous results, TBPB elicited a concentration-dependent activation of M1, as measured in the calcium fluorescence assay (Fig. 8A). VU0029767 potentiated the response of TBPB as manifested by a leftward shift in the TBPB concentration-response curve and an increase in the maximal response to TBPB (Fig. 8A). VU0029767 also potentiated the response to an EC20 concentration of TBPB in a concentration-dependent manner, with a potency consistent with the potency of this compound for potentiating the response to ACh (Fig. 8B). In contrast to VU0029767, VU0090157 did not shift the TBPB concentration-response relationship (Fig. 8A) and had little effect on the response to an EC20 concentration of TBPB (Fig. 8B).

VU0029767 potentiated responses to the selective M1 agonist TBPB, whereas VU0090157 did not. A, a 30 μM concentration of VU0029767 shifted the concentration-response curve for TBPB 5.4 ± 1.3-fold to the left, whereas 30 μM VU0090157 did not induce responses significantly different from vehicle control (potency of TBPB plus DMSO, 210 ± 30 nM; TBPB plus 30 μM VU0090157, 145 ± 23 nM; 1.4 ± 0.1-fold shift). B, compound concentration-response curves of VU0090157 (▪) and VU0029767 (□) were generated in the presence of an EC20 concentration of TBPB (53 nM) in a calcium assay using cells expressing the wild-type M1 receptor. VU0029767 potentiated TBPB-mediated responses in a concentration-dependent manner with a potency and efficacy similar to its effects in potentiating ACh responses in the same cell line. In contrast, VU0090157 only weakly potentiated TBPB-mediated responses. Data represent the mean ± S.E.M. of three independent experiments performed in quadruplicate.
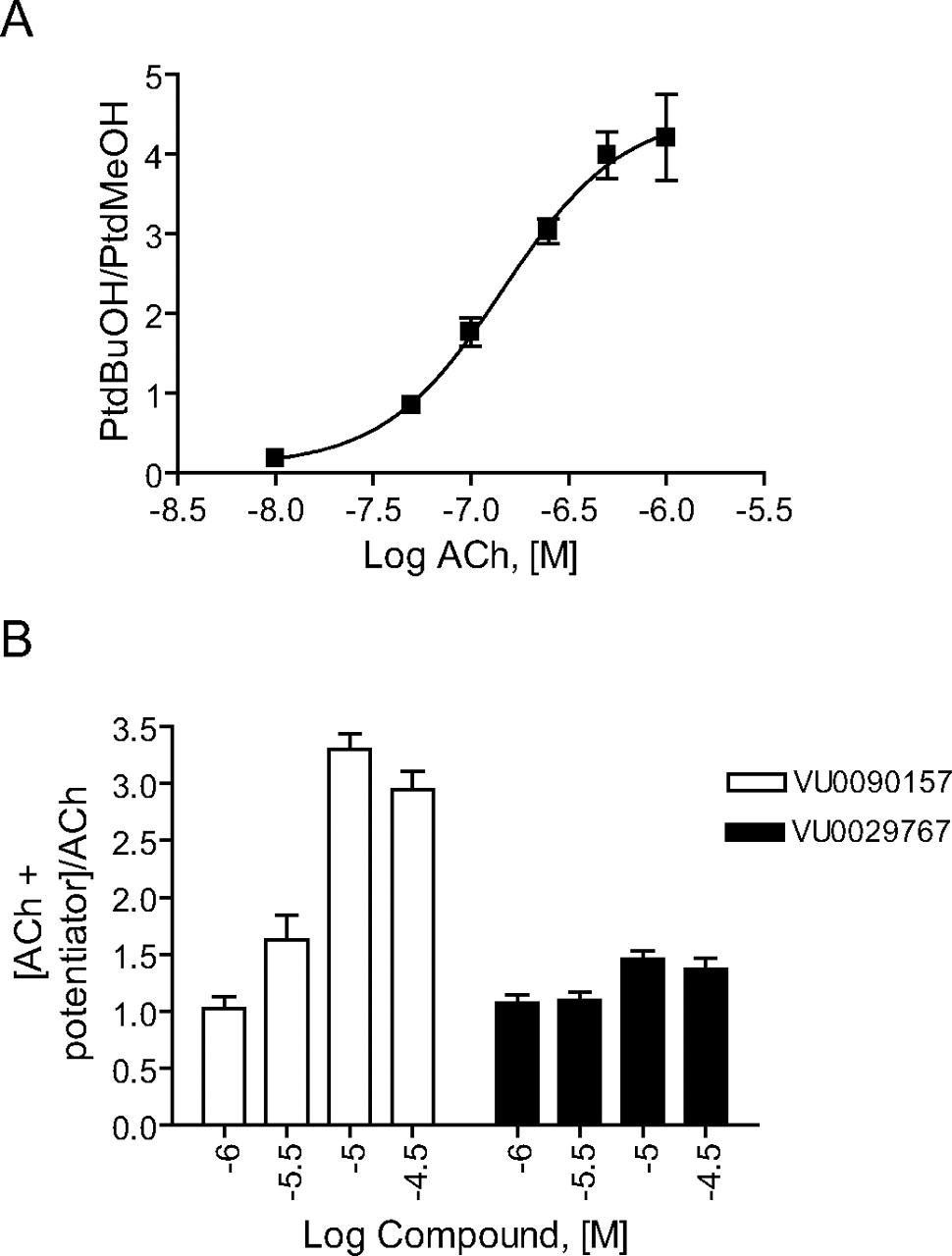
VU0090157 and VU0029767 Differentially Modulate Coupling of M1 to Activation of Phospholipases C and D. In recent years, it has become clear that traditional orthosteric agonists of GPCRs can differentially activate distinct signaling pathways of a single GPCR, a phenomenon referred to as agonist-directed trafficking or functional selectivity (Urban et al., 2007). This implies that different orthosteric agonists can stabilize unique conformations of the GPCR that favor coupling to distinct downstream signaling pathways. In the simplest case, the novel M1 PAMs VU029767 and VU0090157 will increase the activity of M1 regardless of the specific effector system being monitored. Because these compounds seem to act by increasing ACh affinity, one might expect this to be the case, and the M1 PAMs might simply increase the potency of ACh for all responses normally elicited by ACh action via M1. However, the finding that VU029767 and VU0090157 have differential effects on ACh activation of M1Y381A and on responses to TBPB suggests that these compounds do not lead to the stabilization of identical activity states of M1 when activated by ACh. As a result, it is possible that these M1 PAMs will not have uniform effects on coupling of M1 to all signaling pathways but might differentially alter the coupling of M1 to various downstream effector systems. To begin to address this, we determined the effects of VU029767 and VU0090157 on M1-mediated activation of phosphoinositide hydrolysis (a measure of coupling to phospholipase C) and activation of PLD. Consistent with previous reports, ACh stimulated phosphoinositide hydrolysis in M1-CHO cells in a concentration-dependent manner (Fig. 9). As in the calcium mobilization studies, VU0090157 and VU0029767 induced leftward shifts in the ACh concentration-response curves (Fig. 9). These studies confirmed that the two most selective PAMs were able to increase ACh potency in a second assay that is believed to require activation of the same signaling pathway as that involved in the calcium mobilization response. However, it is interesting to note that VU0090157 induced a shift in ACh potency that was almost identical with that observed in the calcium assay (15.1 ± 1.2-fold), whereas VU0029767 had considerably lower efficacy in potentiating ACh activation of phosphoinositide hydrolysis (2.1 ± 0.1-fold shift of ACh potency) than in potentiating ACh-induced calcium mobilization or enhancing ACh affinity.
We next determined the effects of VU0090157 and VU0029767 on ACh-induced increases in PLD activity as a measure of a fundamentally distinct signaling pathway activated by this receptor in M1-CHO cells (Rumenapp et al., 1996; Lopez De Jesus et al., 2006). ACh induced a robust, concentration-dependent increase in PLD activity, as assessed by measurement of agonist-induced formation of PtdBuOH, with a potency of approximately 150 nM (Fig. 10A). After establishing the concentration-response relationship for ACh, we selected an EC20 concentration of ACh and determined the effects of increasing concentrations of VU0090157 and VU0029767 (Fig. 10B). Consistent with its effects in the calcium mobilization and phosphoinositide hydrolysis assays, VU0090157 induced a robust potentiation of the response to ACh with a potency in the low micromolar range. In contrast, VU0029767 had only a weak positive effect on the ACh response (Fig. 10B). This experiment further differentiates the actions of these two novel M1 PAMs and suggests that these compounds may differentially regulate the coupling of M1 to various signaling pathways.
Discussion
Discovery of multiple chemical classes of novel allosteric potentiators of M1 builds on major advances that have been achieved in recent years in developing highly selective allosteric modulators of GPCRs (May et al., 2007; Bridges and Lindsley, 2008; Conn et al., 2008a). Unlike recently discovered allosteric agonists of M1, such as AC42 and TBPB (Spalding et al., 2002; Sur et al., 2003; Jones et al., 2008), the allosteric potentiators described here do not activate M1 directly but rather potentiate responses to the orthosteric agonist ACh. Altogether, these foundational studies suggest that the identification of both PAMs and allosteric agonists is a viable approach in the discovery of novel activators of M1 that may have higher selectivity than has been possible with traditional orthosteric M1 agonists.

M1 PAM compounds potentiated PI hydrolysis in M1-expressing cells. ACh concentration-response curves were generated in the absence and presence of 30 μM VU0090157 (A) and VU0029767 (B). VU0090157 and VU0029767 shifted the ACh concentration-response 15.1 ± 1.2- and 2.1 ± 0.1-fold to the left, respectively. Data represent the mean ± S.E.M. of three independent experiments performed in quadruplicate.

VU0090157 and VU0029767 differentially affected the ACh-mediated PLD activation in M1-CHO cells. A, acetylcholine induced a dose-dependent increase in PLD activity with a potency of 148 ± 39 nM. B, increasing concentrations of VU0090157 and VU0029767 were incubated with an EC20 concentration of ACh, and PLD activity was measured. Data represent mean ± S.E.M. and are representative of three independent experiments performed in triplicate.
The M1 PAMs described here, VU0029767 and VU0090157, were similar in multiple respects. Both induced similar increases in M1-mediated mobilization of intracellular calcium, and both compounds seemed to act by increasing ACh affinity for the orthosteric site. Furthermore, neither compound reduced equilibrium binding of an orthosteric antagonist radioligand, [3H]NMS, suggesting that they do not bind to the orthosteric site or regulate this orthosteric antagonist's affinity. Considering these similarities, it is especially interesting that these two M1 PAMs seem to act by subtly different mechanisms. For instance, although VU0029767 induced similar potentiation of ACh responses at WT M1 and at a form of M1 in which a single point mutation known to reduce ACh affinity was introduced into the orthosteric site, VU0090157 was inactive in potentiating ACh-mediated responses of the mutant receptor. Furthermore, VU029767 induced similar potentiation of responses to the orthosteric agonist ACh and the allosteric agonist TBPB, whereas VU0090157 had no effect on the activation of M1 by TBPB. Although the reasons for these differences are not yet clear, these data suggest that the two compounds may stabilize different conformational states of the receptor, thereby increasing ACh responses through different mechanisms. We previously identified multiple allosteric potentiators for mGluR5 and have shown that these mGluR5 PAMs interact with at least two distinct allosteric sites on this receptor and can differentially modulate mGluR5 responses (O'Brien et al., 2003, 2004; Zhang et al., 2005; Chen et al., 2007, 2008). Likewise, we have provided evidence for the existence of multiple allosteric sites on mGluR1 (Hemstapat et al., 2006), mGluR2 (Hemstapat et al., 2007), and mGluR4 (Niswender et al., 2008). It is possible that M1 is similar to the mGluRs in having multiple allosteric sites and that VU0090157 and VU029767 act at different sites to regulate receptor function. However, it is also possible that these compounds interact with a single allosteric region but have distinct activities. Full characterization of the sites at which these compounds act will require detailed mutagenesis studies and discovery of high-affinity allosteric modulators that can be developed as radioligands for the allosteric sites.
In recent years, concepts about GPCR function have dramatically changed from a view of these receptors as simple on/off switches to a view in which receptors are seen as complex signaling molecules in equilibrium between inactive and active states that couple to different pathways—an equilibrium that is differentially regulated by ligands (Liggett, 2002). Studies of multiple GPCRs show that members of each of the major subfamilies of GPCRs can exhibit multiple subsets of active conformations that link to different and independent signaling pathways. Furthermore, traditional orthosteric agonists can differentially activate distinct signaling pathways of a single GPCR, a phenomenon referred to as agonist receptor trafficking or functional selectivity (Urban et al., 2007). Because VU0090157 and VU0029767 seem to act by increasing affinity of ACh for M1, one might expect that these M1 PAMs would not alter the signaling profile of ACh action on M1 but would simply increase ACh potency regardless of the response measured. However, the finding that VU0090157 and VU0029767 have differential effects on the orthosteric site mutant form of M1 and on responses to TBPB suggests that these compounds may stabilize different active states of the receptor and could differentially modulate ACh actions on different signaling pathways. Consistent with this, VU0090157 and VU0029767 similarly potentiated ACh-induced calcium mobilization but differed in their impact on coupling of M1 to activation of phospholipase D and phosphoinositide hydrolysis. VU0090157 potentiated ACh-induced activation of PLD and PI hydrolysis in a manner similar to that observed with calcium mobilization assays. In contrast, VU0029767 had almost no effect on M1-mediated increases in PLD activity and a very small effect on ACh-induced PI hydrolysis. Although the precise mechanism by which M1 activates PLD in these cells has not been studied in detail, previous studies suggest that mAChR-induced activation of PLD (but not phospholipase C) involves coupling to Gα12 or small G proteins such as R-Ras (Rumenapp et al., 1996, 2001; Lopez De Jesus et al., 2006). It is possible, therefore, that VU0029767 does not stabilize productive coupling complexes with non-Gαq signaling partners. The differences between the calcium and PI hydrolysis results for VU0029767, however, are interesting because the classic pathway activating both of these pathways stems from Gαq activation of phospholipase Cβ. One potential explanation is the temporal differences in the assays: calcium assay responses are monitored kinetically, whereas PI hydrolysis is measured in an endpoint assay. It is possible that VU0029767 induces very rapid desensitization of the receptor, thereby preventing dramatic accumulation of phosphoinositides but allowing for a strong calcium response. Future studies will address desensitization kinetics of M1 responses induced in the presence of the various M1 PAMs.
Differential effects of VU0029767 are reminiscent of the distinct effects of allosteric modulators of other GPCRs on various signaling pathways that have been described recently. For instance, N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide, a selective mGluR5 PAM, induces robust potentiation of mGluR5-mediated mobilization of intracellular calcium in rat astrocytes but can inhibit coupling of mGluR5 to activation of extracellular signal-regulated kinase 1/2 phosphorylation at the highest concentrations of orthosteric agonist (Zhang et al., 2005). Likewise, an allosteric modulator of the chemoattractant-receptor-homologous molecule on T-helper 2 cells was described recently that is inactive with respect to coupling to G protein-linked pathways but is a potent antagonist of G protein-independent β-arrestin coupling to the same receptor (Mathiesen et al., 2005). The finding that VU0090157 and VU0029767 have differential effects on M1 coupling to various signaling pathways raises the possibility that these compounds could have fundamentally different abilities to modulate M1 activity in the central nervous system. The ability to selectively modulate certain signaling pathways associated with activation of M1 introduces an additional mode for fine-tuning intracellular signaling with allosteric modulators. It is conceivable that allosteric regulators could be developed to preferentially regulate a specific pathway to achieve a desired response. However, in the absence of a clear understanding of the signaling pathways involved in potential therapeutic effects of M1 activation, this also raises a potential problem of inadvertently optimizing M1 PAMs that do not affect coupling of the receptor to the most important downstream signaling events. These possibilities suggest that it is critical to focus continued studies on developing a better understanding of the signaling pathways involved in different responses to M1 activation and to carefully analyze the effects of different classes of allosteric ligands on these pathways in native systems.
In addition to the implications of the current studies for the molecular pharmacology of M1 and allosteric modulators of GPCRs, these findings represent a further advance in developing a framework for discovery of novel drug leads and, ultimately, therapeutic agents for treatment of AD and schizophrenia. The therapeutic activity of mAChR agonists for the treatment of Alzheimer's disease and schizophrenia is believed to be mediated by activation of the M1 or M4 subtypes. In contrast, activation of peripheral M2 and M3 leads to adverse effects and has resulted in the discontinuation of the development of compounds that are not selective among the mAChRs (Wess et al., 2007; Conn et al., 2008b; Langmead et al., 2008). A major advance in discovery of M1-selective activators came with the discovery of AC42 as an allosteric M1 agonist that fully activates the receptor in the absence of orthosteric agonist and is highly selective for M1 relative to M2-M5 mAChRs. Although providing valuable proof-of-concept at the molecular level, AC42 lacked the potency, solubility, and ancillary pharmacology needed for use as a tool to probe M1 function. More recently, we reported characterization of TBPB as another highly selective allosteric agonist for the M1 mAChR with no agonist activity at any of the other mAChR subtypes (Jones et al., 2008). TBPB has properties that make it highly useful for in vitro and in vivo studies in native systems and was found to potentiate N-methyl-d-aspartate receptor currents by activation of M1 in hippocampal pyramidal cells but did not alter responses believed to be mediated by M2 and M4 mAChRs in these neurons. Furthermore, TBPB was efficacious in multiple rodent models predictive of antipsychotic-like activity in rats. Finally, TBPB had effects on processing of the amyloid precursor protein toward the nonamyloidogenic pathway and decreased Aβ production in vitro in a manner similar to that shown previously for a less selective mAChR agonist (Caccamo et al., 2006). These studies, combined with the clinical studies outlined above, provide strong proof-of-concept suggesting that selective activation of M1 may represent a novel strategy for the treatment of symptoms associated with schizophrenia and AD.
It is conceivable that allosteric potentiators of M1 could provide the needed selectivity for this receptor and also afford further advantages relative to allosteric agonists. For instance, there are a number of problems that are often associated with the use of direct-acting agonists as drugs. These include adverse effects associated with excessive activation of the receptor, greater receptor desensitization than may occur with more indirect approaches, and loss of activity dependence of receptor activation. Potential adverse effects of M1 agonists are a particular concern because excessive activation of this receptor has been shown to lead to generalized seizures in animal models (Hamilton et al., 1997). Although the relative utilities of allosteric agonists versus allosteric potentiators of M1 are not yet known, the present findings suggest that it will be possible to develop selective allosteric potentiators of M1. In future studies, it will be critical to optimize these compounds for use in behavioral studies and to make a direct comparison between M1-selective agonists and allosteric potentiators with respect to efficacy and adverse effects.
Footnotes
-
This work was supported by the National Institutes of Health National Institute of Mental Health [Grants MH073676, MH082867]; The Alzheimer's Association [Grant IIRG-07-57131]; the Vanderbilt Institute of Chemical Biology [Integrative Training in Therapeutic Discovery Grant T90-DA22873]; and LIPID Metabolites And Pathways Strategy [Grant U54-GM69339].
-
J.E.M. and C.M.N. contributed equally to this work.
-
ABBREVIATIONS: AD, Alzheimer's disease; GPCR, G protein-coupled receptor; mGluR, metabotropic glutamate receptor; CHO, Chinese hamster ovary; DMSO, dimethyl sulfoxide; ACh, acetylcholine; TBPB, 1-[1′-(2-tolyl)-1,4′-bipiperidin-4-yl]-1,3-dihydro-2H-benzimidazol-2-one; PAM, positive allosteric modulator; PtdMeOH, 1,2-dipalmitoyl-sn-glycero-3-phosphomethanol, phosphatidylmethanol; VU0090157, cyclopentyl 1,6-dimethyl-4-(6-nitrobenzo[d][1,3]dioxol-5-yl)-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate; VU0029767, (E)-2-(4-ethoxyphenylamino)-N′-((2-hydroxynaphthalen-1-yl)methylene)acetohydrazide; VU0119498, 1-(4-bromobenzyl)indoline-2,3-dione; mAChR, muscarinic acetylcholine receptor; FBS, fetal bovine serum; DMEM, Dulbecco's modified Eagle's medium; FDSS, Functional Drug Screening System; PI, phosphatidylinositol; LC-MS, liquid chromatography-mass spectrometry; WT, wild type; PLD, phospholipase D; [3H]NMS, l-[N-methyl-3H]scopolamine.
- Received October 17, 2008.
- Accepted December 1, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}