


Abstract
CXC chemokine receptor CXCR3 and/or its main three ligands CXCL9, CXCL10, and CXCL11 are highly upregulated in a variety of diseases. As such, considerable efforts have been made to develop small-molecule receptor CXCR3 antagonists, yielding distinct chemical classes of antagonists blocking binding and/or function of CXCR3 chemokines. Although it is suggested that these compounds bind in an allosteric fashion, thus far no evidence has been provided regarding the molecular details of their interaction with CXCR3. Using site-directed mutagenesis complemented with in silico homology modeling, we report the binding modes of two high-affinity CXCR3 antagonists of distinct chemotypes: VUF11211 [(S)-5-chloro-6-(4-(1-(4-chlorobenzyl)piperidin-4-yl)-3-ethylpiperazin-1-yl)-N-ethylnicotinamide] (piperazinyl-piperidine) with a rigid elongated structure containing two basic groups and NBI-74330 [(R)-N-(1-(3-(4-ethoxyphenyl)-4-oxo-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)ethyl)-2-(4-fluoro-3-(trifluoromethyl)phenyl)-N-(pyridin-3-ylmethyl)acetamide] (8-azaquinazolinone) without any basic group. Here we show that NBI-74330 is anchored in the transmembrane minor pocket lined by helices 2 (W2.60, D2.63), 3 (F3.32), and 7 (S7.39, Y7.43), whereas VUF11211 extends from the minor pocket into the major pocket of the transmembrane domains, located between residues in helices 1 (Y1.39), 2 (W2.60), 3 (F3.32), 4 (D4.60), 6 (Y6.51), and 7 (S7.39, Y7.43). Mutation of these residues did not affect CXCL11 binding significantly, confirming the allosteric nature of the interaction of these small molecules with CXCR3. Moreover, the model derived from our in silico–guided studies fits well with the already published structure–activity relationship data on these ligands. Altogether, in this study, we show overlapping, yet different binding sites for two high-affinity CXCR3 antagonists, which offer new opportunities for the structure-based design of allosteric modulators for CXCR3.
Introduction
The chemokine system is intimately involved in leukocyte homeostasis and in directing immune cells to sites of infection or inflammation (Viola and Luster, 2008). Inappropriate expression of chemokine ligands or their respective G protein–coupled receptors (GPCRs) can result in disproportionate infiltration of specific immune cells into (inflamed) tissues or confer chemokine sensitivity to cells normally nonresponsive to chemokines (O’Hayre et al., 2008). Ultimately, this leads to development of autoimmune diseases, chronic inflammation, or tumor growth and metastasis (Koelink et al., 2012).
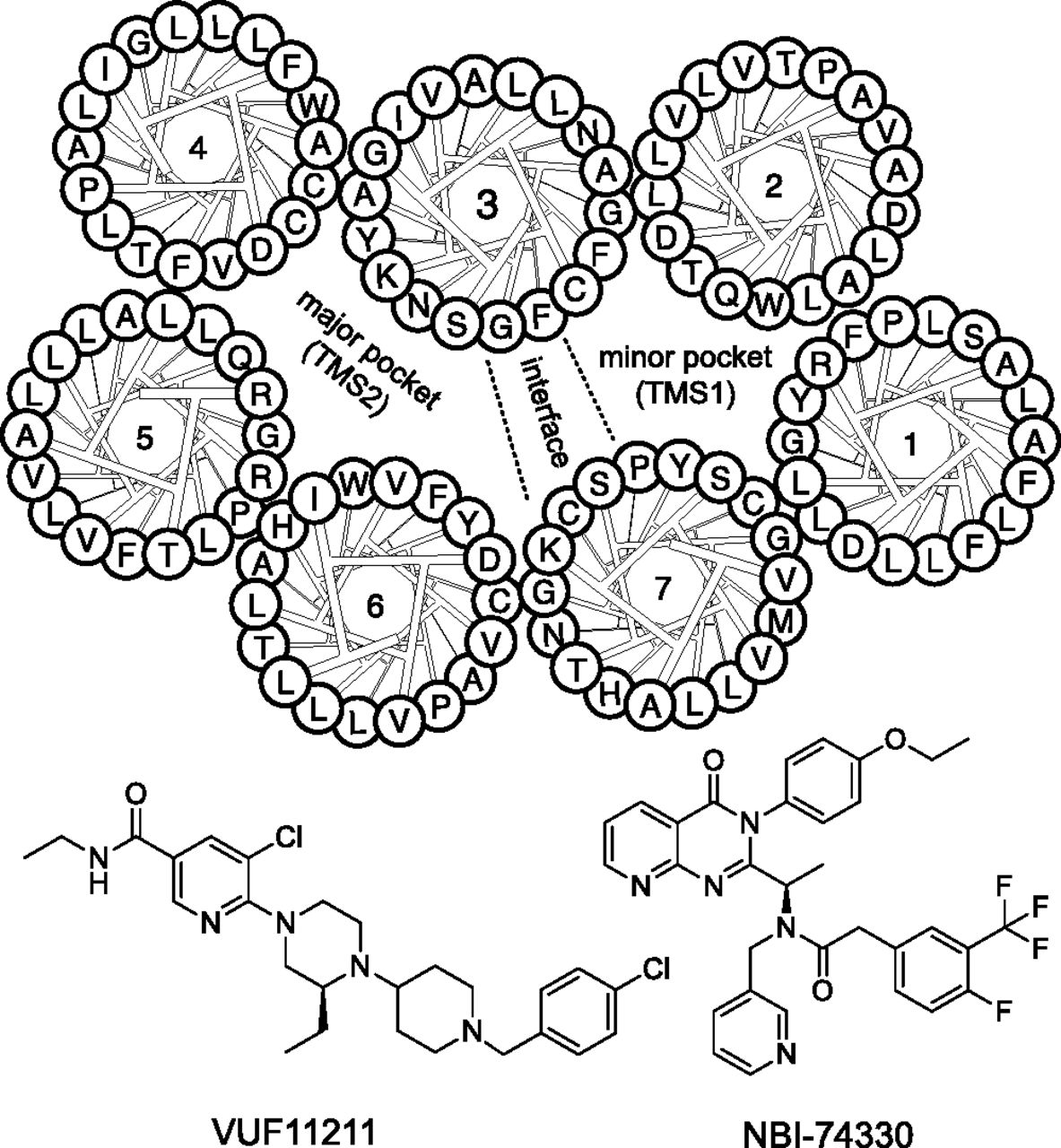
Chemokines are the endogenous peptide ligands of chemokine receptors with molecular masses of approximately 10 kDa. Chemokines are 10- to 50-fold larger than the average small molecule that binds these receptors. Nonetheless, many of these small ligands are able to inhibit chemokine-induced responses and/or the binding of these chemokines with nanomolar potencies (Scholten et al., 2012a). Intuitively, such size differences would suggest allosteric noncompetitive mechanisms of action for small-molecule antagonists acting via distinct binding sites. In general, the interaction of chemokines with their receptors can be described by a two-step model, in which a chemokine first binds to the N terminus of its respective GPCR. Subsequently, the N terminus of the chemokine is positioned such that it interacts with the extracellular loops (ELs) and transmembrane (TM) domains of the GPCR. The lack of structural data on chemokine-GPCR binding hinders molecular understanding on how small molecules act at this subclass of family A GPCRs. Fortunately, structural information on chemokine receptors has started to emerge with the recent publication of crystal structures of CXC and CC chemokine receptors (CXCR and CCR, respectively) CXCR4 and CCR5 (Wu et al., 2010; Tan et al., 2013), opening up new possibilities for structure-based drug design on the chemokine receptor family (Scholten et al., 2012a; Kooistra et al., 2013). In general, three pockets are distinguished in GPCRs, two in the TM domains, including the minor pocket or TM site 1 (TMS1) lined by TM helices 1 and 2, and the major pocket (TM site 2 or TMS2) delimited by helices 4, 5, and 6 (Fig. 1). Residues in helices 3 and 7 constitute the interface between both pockets, pointing either to one or the other pocket. Furthermore, a third pocket, lining the intracellular surface of the GPCR, was recently suggested as a binding site for certain CXCR2 ligands (Nicholls et al., 2008; Salchow et al., 2010). In CXCR4, the small molecule IT1t (1,3-dicyclohexyl-2-(3-methyl-6,6-dimethyl-5,6-dihydroimidazo[1,2-b]thiazole)-2-thiopseudourea) binds in TMS1, whereas the CVX-15 peptide (Arg-Arg-Nal-Cys-Tyr-Gln-Lys-dPro-Pro-Tyr-Arg-Cit-Cys-Arg-Gly-dPro) interacts only with residues in TMS2, showing that ligands for the same receptor can bind to different pockets in the TM domains of chemokine receptors (Wu et al., 2010).

Chemical structures of CXCR3 antagonists used in this study and a helical wheel representation of the transmembrane domains of CXCR3 with TMS1 and TMS2 generally involved in binding of small ligands to chemokine receptors. Residues from TM3 and TM7 constitute the interface between both pockets (also see Fig. 2).
The CXC chemokine receptor CXCR3 is a key regulator of T cell responses and has been linked to several diseases. Overexpression of the CXCR3 receptor and/or its ligands (CXCL9, CXCL10, CXCL11) is often observed in rheumatoid arthritis and transplant rejection (Lacotte et al., 2009). In the past decade, many efforts have focused on the discovery of small molecule CXCR3 antagonists, leading to the disclosure of ligands with a multitude of different chemotypes (Wijtmans et al., 2008, 2010). Initially, 8-azaquinazolinone compounds from Amgen [AMG487; (R)-N-(1-(3-(4-ethoxyphenyl)-4-oxo-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)ethyl)-N-(pyridin-3-ylmethyl)-2-(4-(trifluoromethoxy)phenyl)acetamide] and Neurocrine Biosciences [NBI-74330; (R)-N-(1-(3-(4-ethoxyphenyl)-4-oxo-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)ethyl)-2-(4-fluoro-3-(trifluoromethyl)phenyl)-N-(pyridin-3-ylmethyl)acetamide] were shown to bind CXCR3 with nanomolar affinities (Heise et al., 2005; Johnson et al., 2007; Verzijl et al., 2008), and are effective in animal models of disease (Walser et al., 2006; van Wanrooij et al., 2008). AMG487 was even assessed in clinical trials for treatment of psoriasis, but was discontinued after a phase IIa trial (Tonn et al., 2009). Moreover, a piperazinyl-piperidine compound class with high-affinity CXCR3 ligands has been disclosed by Schering-Plough (now Merck Sharp & Dohme, Whitehouse Station, NJ) (McGuinness et al., 2006, 2009; Shao et al., 2011). This compound series is effective in rodent models of CXCR3-associated disease, including transplant rejection and rheumatoid arthritis (Jenh et al., 2012).
Despite the interest in small-molecule antagonists for CXCR3, little information is available about their interaction with CXCR3 at the molecular level as to whether they bind to TMS1, TMS2, or both. In this study, we aimed to elucidate the binding mode of two high-affinity CXCR3 ligands from the 8-azaquinazolinone and the piperazinyl-piperidine class using site-directed mutagenesis complemented with in silico modeling of CXCR3. We show that NBI-74330, from the azaquinazolinone class from Amgen (Heise et al., 2005; Johnson et al., 2007), mainly binds to TMS1, whereas a chlorobenzyl derivative of the piperazinyl-piperidine class disclosed by Merck (McGuinness et al., 2006), with one of the highest CXCR3 affinities [named VUF11211; (S)-5-chloro-6-(4-(1-(4-chlorobenzyl)piperidin-4-yl)-3-ethylpiperazin-1-yl)-N-ethylnicotinamide] (Shao et al., 2011), binds to both TMS1 and TMS2 (Fig. 1).
Materials and Methods
Dulbecco’s modified Eagle’s medium and trypsin were purchased from PAA Laboratories GmbH (Pasching, Austria), penicillin and streptomycin were obtained from Lonza (Verviers, Belgium), fetal bovine serum was purchased from Integro B.V. (Dieren, The Netherlands), and [125I]CXCL11 (±1000 Ci/mmol) was obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). Unlabeled chemokines were purchased from PeproTech (Rocky Hill, NJ). Unless stated otherwise, all other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
CXCR3 Ligands.
VUF11211 [compound 18i in the study by Shao et al. (2011)] was synthesized in enantiopure form in our group according to the general synthetic procedures patented by Merck (McGuinness et al., 2006). Details of the synthetic procedures are provided in the Supplemental Methods. The synthesis of NBI-74330 was previously described (Storelli et al., 2007).
DNA Constructs and Site-Directed Mutagenesis.
The DNA coding for human CXCR3 was a gift from Dr. B. Moser (Cardiff University School of Medicine, Cardiff, UK) and was inserted in the expression vector pcDEF3. Mutants were generated by using polymerase chain reaction (PCR) primers containing one or more mismatches in the center of the primer, flanked by 15–20 bp. At first, two individual PCRs were performed simultaneously to amplify the first part of the receptor until the desired mutation, and the second part of the receptor from the mutation until the BGH polyA sequence. The forward primer used to generate the first part (pcDEF3-FW; 5′-gggtggagactgaagttaggcc-3′) recognizes part of the EF1α promoter, whereas the reverse primer for the second part (pcDEF3-RV; 5′ggaaggcacgggggaggggc-3′) targets part of the BGH polyA sequence of the vector. The reverse primer for the first part and the forward primer for the second are reverse complementary and recognize CXCR3 around the desired mutation as mentioned above. See Supplemental Table 1 for the sequences of these mutation-specific primers. Subsequently, a second PCR with primers pcDEF3-FW and pcDEF3-RV was performed to fuse both receptor DNA fragments, making use of the overlapping sequence in both individual parts as internal primer. Finally, the resulting products were digested using BamHI and XbaI restriction enzymes and ligated into pcDEF3. Sequences were confirmed using Sequencing (Macrogen, Amsterdam, The Netherlands).
In Silico CXCR3 Model Construction.
A three-dimensional model for the CXCR3 receptor was constructed with Molecular Operating Environment version 2011.10 (Chemical Computing Group, Inc., Montreal, ON, Canada) based on the structure of CXCR4 cocrystallized with the small molecule IT1t (Protein Data Bank ID 3ODU) (Wu et al., 2010). The primary sequence of CXCR3 (GenBank accession no. P49682) was aligned to that of the CXCR4 crystal structure. N-terminal residues 1–41 were omitted from the model due to a lack of crystallographic data. An initial model was constructed for the CXCR3−VUF11211 complex as a basis for the binding model for NBI-74330. VUF11211 was docked into the model using GOLD v4 (Verdonk et al., 2003). Subsequently, protein–ligand interactions were optimized in Molecular Operating Environment by energy minimization, during which all heavy atoms were tethered with a 10.0 kcal/mol restraint, similar to the protocol our group recently described (de Kruijf et al., 2011). However, since the CXCR4−IT1t template contains lipids protruding into protein between TM5 and TM6 and the C terminus blocking the extracellular opening (Roumen et al., 2012), the binding pocket of a CXCR3 model is spacious and VUF11211 cannot form an interaction with W2686.48. To explain all site-directed mutagenesis data, the CXCR3 model was optimized within this region by moving TM6 by 2 Å closer to TM3 and TM5, followed by the same energy minimization protocol (de Kruijf et al., 2011). This resulted in a TM arrangement comparable to that of the aminergic receptors (Shimamura et al., 2011). Using the finalized CXCR3 model, NBI-74330 was docked into the protein pocket with GOLD resulting in a pose that is in accordance with the site-directed mutagenesis data from this study, and optimized by energy minimization.
Residue numbering throughout the manuscript is displayed as absolute sequence numbers with the Ballesteros–Weinstein notation in superscript (e.g., W1092.60). If residues are compared between different receptors, only the Ballesteros–Weinstein notation is used (e.g., W2.60) (Ballesteros and Weinstein, 1995).
Cell Culture and Transfection.
Human embryonic kidney (HEK) 293T cells were grown at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, penicillin, and streptomycin. Cells were transfected with 2.5 μg pcDEF3-CXCR3 wild-type (WT) or pcDEF3 containing the mutant CXCR3 DNA, and 2.5 μg pcDEF3 per 2 million cells, using linear polyethylenimine with a molecular mass of 25 kDa (Polysciences, Warrington, PA) as previously described (Verzijl et al., 2008).
Enzyme-Linked Immunosorbent Expression Assay.
The day after transfection, cells were trypsinized, resuspended into fresh culture medium, and plated in poly(l-lysine)–coated 48-well assay plates. The enzyme-linked immunosorbent assay (ELISA) procedure was performed as reported earlier (Scholten et al., 2012b). Briefly, 48 hours after transfection, the cells were fixed with 4% formaldehyde solution, permeabilized by 0.5% Nonidet P40, and stained with anti-CXCR3 antibody mAb160 (R&D Systems, Minneapolis, MN), and subsequently with goat anti-mouse horseradish peroxidase–conjugated antibody (BioRad Laboratories, Hercules, CA). O-phenylenediamine was used as a substrate for the enzyme coupled to the secondary antibody. The resulting color was detected using a Powerwave X340 absorbance plate reader (BioTek, Bad Friedrichshall, Germany).
Membrane Preparation.
Membrane preparation was performed as previously described (Verzijl et al., 2008). In brief, cell membrane fractions from HEK293T cells, transiently transfected with WT or mutant CXCR3, were prepared by washing with ice-cold phosphate-buffered saline. Subsequently, the cells were collected in tubes and centrifuged at 1500g for 10 minutes. The pellet was resuspended in ice-cold membrane buffer (15 mM Tris, pH 7.5, 1 mM EGTA, 0.3 mM EDTA, and 2 mM MgCl2), and homogenized using a Teflon-glass homogenizer and rotor. The membranes were subjected to two freeze-thaw cycles using liquid nitrogen, and centrifuged at 40,000g for 25 minutes. The pellet was resuspended in Tris-sucrose buffer (20 mM Tris, 250 mM sucrose, pH 7.4) and aliquots were stored at −80°C.
Radioligand Binding Assays.
For [125I]CXCL11 binding, between 2 and 10 μg of membranes were used per well depending on the mutant, in 96-well clear plates (Greiner Bio One, Alphen a/d Rijn, The Netherlands). For displacement binding experiments, membranes were incubated in binding buffer [50 mM HEPES, pH 7.4, 1 mM CaCl2, 5 mM MgCl2, 100 mM NaCl, and 0.5% (w/v) bovine serum albumin fraction V] with approximately 70 pM [125I]CXCL11 and a concentration range of cold CXCL11, VUF11211, or NBI-74330 for 2 hours at room temperature. Next, the membranes were harvested by filtration through Unifilter 96-well GF/C plates (PerkinElmer) presoaked with 0.5% polyethylenimine, using ice-cold wash buffer (binding buffer supplemented with 0.5 M NaCl). Bound radioactivity was determined with a MicroBeta scintillation counter (PerkinElmer).
Data Analysis.
Prism software (version 5.0d; GraphPad Software, Inc., La Jolla, CA) was used to plot and analyze the data.
Results
To dissect the binding modes of the two selected CXCR3 antagonists, we considered the homology between different chemokine receptors, focusing on residues that were previously shown to be involved in the binding of small molecule antagonists to a variety of CC and CXC chemokine receptors. An alignment of transmembrane binding pocket residues from CC and CXC chemokine receptors is shown in Fig. 2, with amino acid residues highlighted that affect ligand binding when mutated (de Mendonça et al., 2005; Watson et al., 2005; Vaidehi et al., 2006; Jensen et al., 2007; Kondru et al., 2008; Wong et al., 2008; Wu et al., 2010). In the next sections, we will focus on specific features of the different binding pockets.

Alignment of residues in the binding pockets of chemokine receptors. Residue numbers are in Ballesteros–Weinstein notation. Amino acids residing in the minor pocket (TMS1), major pocket (TMS2) or interface are indicated in blue, orange, and gray, respectively. The Ballesteros–Weinstein residue numbers (Ballesteros and Weinstein, 1995) are used to enumerate residues in TM helices, whereas the numbering scheme proposed by de Graaf et al. (2008) is used to enumerate the conserved cysteine residue (45.50) and the residue downstream from this cysteine residue (45.51) in EL2. Residues are colored per receptor when mutation of that particular residue is reported to affect ligand affinity or antagonism. For CCR5 and CXCR4, interactions with small molecules suggested by mutagenesis and computational modeling studies are shown in the first row, whereas the second row highlights residues that make contact with ligands in their respective crystal structures: maraviroc for CCR5 and IT1t and CVX-15 for CXCR4. Interacting residues from these crystal structures are indicated with bold black borders. Residues specific for IT1t in the CXCR4 cocrystal structure are indicated as dark blue squares with white text, specific interactions with peptide CVX-15 are shown with orange or dark gray squares with white text, whereas residues that interact with both IT1t and CVX-15 are indicated with bold black text. Data are obtained from primary literature, reviewed in two recent reviews (Roumen et al., 2012; Scholten et al., 2012a). Note that no data are included on ligands like metal chelators requiring modification of receptors to bind at all.
Negatively Charged Ligand Anchors.
In general, small-molecule ligands for chemokine receptors are characterized by a positively charged quaternary ammonium and aromatic groups around it (Wijtmans et al., 2008). This positive charge often contributes significantly to the affinity of the ligand, as observed for the biaryl- and piperazinyl-piperidine class of CXCR3 ligands (Shao et al., 2011; Wijtmans et al., 2011). Calculations with Marvin tools (version 5.2.0; ChemAxon Kft, Budapest, Hungary) suggest that protonation of VUF11211 at pH 7.4 occurs either at the piperidine nitrogen (45%) or the trialkyl-nitrogen of the piperazine (43%), leaving 13% of the compound nonprotonated.
As can be deduced from Fig. 2, a negatively charged glutamic acid at position 7.39 is highly conserved in the chemokine receptor family. This residue is often found to be the ionic anchor for a positive charge in chemokine receptor ligands such as CCR1, CCR5, CXCR4, and viral chemokine receptor US28 (Casarosa et al., 2003; Vaidehi et al., 2006; Kondru et al., 2008; Wong et al., 2008). Yet CXCR3 does not possess a negatively charged residue at this position and we therefore considered other potential anchors that could accommodate the positive charge in VUF11211. The chemokine receptor alignment in Fig. 2 and the detailed information from the recently solved CXCR4 cocrystal structures indicate that other negatively charged residues can also be involved in anchoring chemokine receptor ligands. Aspartates D2.63, D4.60, and D6.58 are involved in binding of small ligands to CXCR4 (Wong et al., 2008; Wu et al., 2010), whereas D2.63 has been shown to be involved in the binding of small molecule CXCR3 agonists (Nedjai et al., 2012). To identify the negatively charged residues in CXCR3 that act as partners for ionic interaction with the positively charged VUF11211, we started this study by mutating 10 negatively charged aspartate or glutamate residues (Table 1): seven residing in the TM domains, one in the N terminus close to TM1, and two in EL2. All of these residues were mutated to asparagine, thereby preventing any ionic interaction.
Effects of mutation of negatively charged residues in CXCR3 on protein expression, CXCL11 binding, and affinities of small-molecule CXCR3 antagonists VUF11211 and NBI-74330
Overview of both total receptor expression levels determined using whole cell–based ELISA, and affinity data for the three compounds. The latter was generated by performing [125I]CXCL11 radioligand displacement binding studies on membranes prepared from HEK293T cells transiently expressing CXCR3 WT or mutants. Values are presented as the mean ± S.E.M. from at least three individual experiments. The Ballesteros–Weinstein residue numbers are indicated in superscript for residues in TM helices, whereas the numbering scheme proposed by de Graaf and colleagues is used to enumerate residues in EL2 (Ballesteros and Weinstein, 1995; de Graaf et al., 2008).
HEK293T cells were transiently transfected with cDNA coding for the CXCR3 WT or mutant receptors. Next, protein expression levels were determined for each mutant using a whole cell–based ELISA (Table 1). All of the mutants were expressed between 68% and 121% of the level of WT CXCR3. Membranes were prepared from the transfected cells (see Materials and Methods) and used for competition binding experiments using [125I]CXCL11 as the radioligand (Table 1). The affinity of CXCL11 for all mutants was determined to investigate the effect of the introduced mutations on CXCL11 binding. The affinity of CXCL11 was hardly affected (max only ±2-fold) by any of these mutations as shown in Fig. 3A and Table 1, with D2826.62N as the exception. Although this mutant protein could be detected by whole cell–based ELISA (68% of the level of WT CXCR3), no [125I]CXCL11 binding to this mutant receptor could be detected, impeding further studies. Next, the ability of VUF11211 and NBI-74330 to displace [125I]CXCL11 from CXCR3 WT and mutants was determined. Only one mutant, D1864.60N, resulted in a 10-fold decrease in affinity for VUF11211 (pIC50 from 7.8 to 6.8) (Fig. 3B; Table 1), in which the other aspartic and glutamic acid to asparagine mutations hardly affected the potency of VUF11211 to displace [125I]CXCL11 from the mutant CXCR3 proteins (Table 1). Surprisingly, mutation of aspartic acid D1122.63 reduced potency of NBI-74330 to displace [125I]CXCL11 binding by more than 10-fold compared with WT (pIC50 7.2 to 6.1) (Fig. 3C; Table 1) despite the absence of a positive charge in the compound.
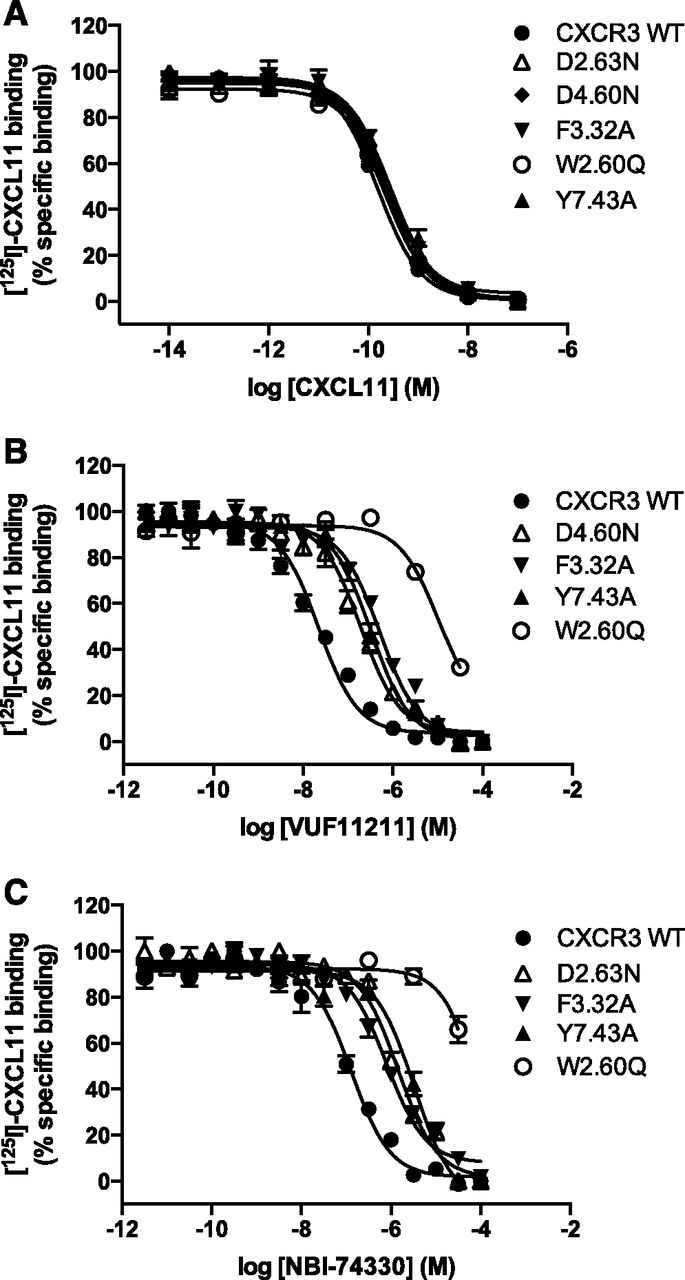
[125I]CXCL11 binding to membranes prepared from HEK293T cells transiently transfected with WT or selected CXCR3 receptor mutants. (A) [125I]CXCL11 homologous binding on WT (●), D1122.63N (▵), D1864.60N (♦), F1313.32A (▾), W1092.60Q (○), and Y3087.43A (▴). (B) [125I]CXCL11 displacement by VUF11211 from CXCR3 WT (●), D1864.60N (▵), F1313.32A (▾), Y3087.43A (▴), and W2.60Q (○) by VUF11211 ligand. (C) [125I]CXCL11 displacement by NBI-74330 from CXCR3 WT (●), D1122.63N (▵), F1313.32A (▾), Y3087.43A (▴), and W1092.60Q (○) by NBI-74330 ligand. Graphs represent grouped data from three or more experiments.
Aromatic Cages in CXCR3.
Next to ionic interactions, many chemokine receptor ligands engage in interactions with hydrophobic aromatic amino acids present in the TM binding cavities of this receptor class. Figure 2 shows that many ligands appear to interact with such residues, including the conserved Y1.39 and W2.60 residues in TMS1, Y/F3.32 and F3.36 in the interface, and W6.48 and Y/F6.51 in TMS2 (Roumen et al., 2012; Scholten et al., 2012a). These conserved residues form the so-called “aromatic cages” within the transmembrane region in which hydrophobic and aromatic moieties of chemokine receptor antagonists can be positioned (Surgand et al., 2006). Examples are BX-471 [(R)-1-(5-chloro-2-(2-(4-(4-fluorobenzyl)-2-methylpiperazin-1-yl)-2-oxoethoxy)phenyl)urea] and UCB-35625 (1,4-trans-1-(1-cycloocten-1-ylmethyl)-4-[[(2,7-dichloro-9H-xanthen-9-yl)carbonyl]amino]-1-ethylpiperidinium iodide) binding to CCR1 (de Mendonça et al., 2005; Vaidehi et al., 2006), maraviroc (4,4-difluoro-N-((S)-3-((1R,3R,5S)-3-(3-isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl)-1-phenylpropyl)cyclohexanecarboxamide) and aplaviroc (4-(4-{[(3R)-1-butyl-3-[(R)-cyclohexylhydroxymethyl]-2,5-dioxo-1,4,9-triazaspiro[5.5]undecan-9-yl]methyl}phenoxy)benzoic acid) to CCR5 (Watson et al., 2005; Kondru et al., 2008), LMD-009 (8-[3-(2-methoxyphenoxy)benzyl]-1-phenethyl-1,3,8-triaza-spiro[4.5]decan-4-one) to CCR8 (Jensen et al., 2007), and AMD3100 (1,1′-[1,4-phenylenebis(methylene)]bis-1,4,8,11-tetraazacyclotetradecane octahydrochloride) to CXCR4 (Wong et al., 2008). To study the involvement of aromatic stacking interactions in the binding of VUF11211 and NBI-74330, the aromatic phenylalanine F1313.32 and F1353.36 and tyrosine residues Y601.39, Y2716.51, and Y3087.43 were mutated to alanine. All mutants were expressed at levels ranging from 41% to 106% of WT CXCR3 and had no appreciable effect on the affinity of [125I]CXCL11 (Table 2). The mutation of Y2716.51 to alanine resulted in a 7- and 10-fold reduction in affinity for NBI-74330 and VUF11211, respectively. In addition, both small ligands displayed lower affinity (8- to 30-fold) at F1313.32A and Y3087.43A mutants (Fig. 3; Table 2). F1313.32 was also mutated to a histidine residue (which is present at position 3.32 in CXCR5; see Fig. 2) to investigate the possibility of aromatic interactions between the ligands and a different type of aromatic ring. This mutant was comparable to WT with respect to [125I]CXCL11 binding affinity, and only resulted in very minor decreases in affinity for VUF11211 and NBI-74330.
Effects of mutation of aromatic and polar residues in CXCR3 on protein expression, CXCL11 binding, and affinities of small molecule CXCR3 antagonists VUF11211 and NBI-74330
Overview of both total receptor expression determined using whole cell–based ELISA, and affinity data for the three compounds. pKd and pIC50 values were obtained by performing [125I]CXCL11 radioligand displacement binding studies on membranes prepared from HEK293T cells transiently expressing CXCR3 WT or mutants. Values are presented as the mean ± S.E.M. from at least three individual experiments.
In chemokine receptors, W1092.60 is a highly conserved tryptophan residue (Fig. 2) close to the TxP motif (Govaerts et al., 2001), indicating that it is important for chemokine receptor stability and function, and W2686.48 has been hypothesized as important for receptor function for numerous GPCRs (Elling et al., 2006; Schwartz et al., 2006). Several chemokine receptors feature a glutamine residue at these positions that is similar in size to a tryptophan, yet lacks aromaticity (Fig. 2). Therefore, W1092.60 and W2686.48 were selected for mutation to a glutamine. These mutant proteins were expressed at 81 and 60% compared with WT levels, respectively. Moreover, affinity of [125I]CXCL11 was unaltered at these mutants. The W2686.48Q mutation lowered the binding affinity of only VUF11211 (6-fold; pIC50 from 7.8 to 7.0; Table 2). Yet the W1092.60 residue appears to be very important for the binding of both NBI-74330 as well as VUF11211, because its mutation to glutamine led to 500-fold and 630-fold decreases in affinity for NBI-74330 and VUF11211, respectively (Fig. 3; Table 2).
Hydrogen Bonding.
Next to aromatic stacking, opportunities also exist for the ligands to engage in hydrogen bonding with residues in the TM domains. As such, the serine (S3017.36 and S3047.39) and tyrosine residues (Y601.39 and Y3087.43) were mutated to nonhydroxylated amino acids (alanine and phenylalanine, respectively) to investigate involvement of potential hydrogen bonding with VUF11211 and NBI-74330. All mutants showed expressions similar to WT levels (Table 2). None of these mutations had a significant effect on potency of both NBI-74330 and VUF11211 to displace [125I]CXCL11, whereas the affinity of CXCL11 decreased 5-fold for only the S3047.39A mutant (Table 2).
The lack of significant effects on binding of NBI-74330 or VUF11211 by testing these individual mutations might be the result of possible hydrogen bonding networks between CXCR3 and the ligands. As a consequence, other residues in the vicinity might compensate for the loss of a single hydrogen bond interaction. Therefore, a triple mutant of residues in close proximity to each other in our CXCR3 homology model, namely Y601.39F/S3047.39A/Y3087.43F, was constructed. The triple mutation had no effect on [125I]CXCL11 binding, but caused a 10-fold decrease in affinity of VUF11211, suggestive of a hydrogen bonding network for this CXCR3 antagonist. No significant decrease in affinity was observed for NBI-74330 (Table 2).
Additional CXCR3 Mutations.
For several residues, additional mutants were constructed to investigate specific interactions. G1283.29 is a variable residue among chemokine receptors indicating a location potentially important for selectivity between chemokine receptors. In addition, CXCR4 contains a histidine residue at this position, important for interaction with both IT1t and CVX-15 ligands (Wu et al., 2010). Moreover, introduction of a histidine at this position (G1283.29H) in CXCR3 anchored small metal chelators to the receptor (Rosenkilde et al., 2007).
Because histidine is greater in size than glycine, the residue at this position was mutated to a histidine (G1283.29H) in analogy with CXCR4, to investigate the allowed space around this residue. This mutant was well expressed in HEK293T cells (80% of WT). Interestingly, radioligand displacement potencies of CXCL11, NBI74330, and VUF11211 were all affected by this mutation, by 5-, 100-, or 800-fold, respectively (Table 2), indicating that the larger histidine likely introduces a steric clash within the binding pocket of CXCR3 affecting the binding of all of the ligands under investigation.
Finally, residue S3047.39 was given special attention due to its analogous residue E7.39, which is relatively conserved in the chemokine receptor family and is often involved in binding of small molecules (Fig. 2). Moreover, in CXCR4, the E7.39 interacts with the small-molecule antagonist IT1t compound in CXCR4 cocrystallized with this compound. In CXCR3, the S3047.39 was mutated to a glutamic acid residue to investigate the influence of a larger polar residue in the center of the protein cavity and a leucine mutation (S3047.39L) was also included to determine whether this influence was due to the polarity or due to the size of the residue. Both mutants were expressed to a similar extent as WT, and also bind CXCL11 with similar affinity. The S3047.39L mutation resulted in decreased potency to displace [125I]CXCL11 by 8- or 20-fold for NBI-74330 and VUF11211, respectively (Table 2). The presence of the glutamic acid at the same position was tolerated for VUF11211 but not for NBI-74330, for which a loss in affinity of 80-fold was observed.
Discussion
To rationalize the effects of CXCR3 mutations on CXCL11, NBI-74330, and VUF11211 binding (Fig. 4), a CXCR3 homology model was constructed based on CXCR4 cocrystallized with ligand IT1t in the TMS1 and CVX-15 in TMS2 (Surgand et al., 2006; Wu et al., 2010). The obtained site-directed mutagenesis data were used to guide the docking of NBI-74330 and VUF11211 into the homology model of CXCR3, and the models were fine-tuned by literature structure–activity relationship (SAR) data on both chemical series (Fig. 4).

(A and B) Helical wheel diagrams are shown for a top view of the TM domains of CXCR3 with effects of mutations highlighted for VUF11211 (A) and NBI-74330 (B). Residues that show a 10-fold or more decrease in affinity upon mutation are indicated in red, and mutations that result in a decrease in affinity between 5- and 10-fold are indicated in orange. Residues that give a significant decrease (10-fold or more) in affinity when mutated together are shown in blue. Other residues that were mutated but did not give a significant change in affinity are colored gray. (C and D) Two-dimensional interaction plots are shown for VUF11211 (C) and NBI-74330 (D). Side chains of proposed interacting residues are shown in gray. Suggested receptor-ligand interactions are depicted as dashed lines. Polar and hydrogen bonding interactions are shown as blue dashed lines, whereas aromatic interactions are shown as gray dashed lines. The ligands are shown in green (VUF11211) and magenta (NBI-74330). (E and F) Homology models of CXCR3 with the two ligands VUF11211 (E) and NBI-74330 (F). The ligands are shown in green (VUF11211) and magenta (NBI-74330), and the TM helices are shown in yellow. Side chains of proposed interacting residues are shown in gray. Hydrogen bonds/polar interactions are shown as dashed blue lines. For reasons of clarity, aromatic interactions are not shown in these panels (see C and D). Although VUF11211 is predicted to have a charge at the piperidine nitrogen (45%) or the trialkyl-nitrogen of the piperazine (43%), our data together with literature SAR, highly suggests that the piperidine nitrogen engages in binding the D1864.60 residue.
Proposed Binding Mode of VUF11211.
Binding of VUF11211 to CXCR3 is affected by several mutations introduced in the TM domains, whereas binding of the endogenous agonist CXCL11 is largely unchanged. In addition, displacement of a radiolabeled variant of VUF11211 by CXCL11 was incomplete (unpublished data). Moreover, a closely related compound (SCH-546738; (S)-3-amino-6-chloro-5-(4-(1-(4-chlorobenzyl)piperidin-4-yl)-3-ethylpiperazin-1-yl)pyrazine-2-carboxamide) exhibited noncompetitive antagonistic behavior on CXCL11-mediated receptor activity (Jenh et al., 2012). Overall, these findings indicate that VUF11211 affects the binding of CXCL11 to CXCR3 in an allosteric fashion.
Our CXCR3 mutation data and homology modeling studies indicate that VUF11211 interacts with residues in both TMS1 and TMS2 (Fig. 4, A, C, and E). We hypothesize that D1864.60 serves as the anchor for the positive charge at the piperidine nitrogen of VUF11211, since a 10-fold drop in affinity was observed at the D1864.60N mutant (Fig. 4A; Table 1). This hypothesis is supported by SAR studies that highlight the importance of the basicity of the piperidine ring (Fig. 4A) (Shao et al., 2011). Rigidification of the benzyl moiety either by ring closure or intramolecular hydrogen bonding, at the cost of basicity, could maintain ligand affinity, indicating the importance of directionality for the chlorobenzyl moiety (Kim et al., 2011; Shao et al., 2011). In the proposed binding mode, the chlorobenzyl moiety resides in a small space between TM4 and TM5 (Fig. 4, A, C, and E). The importance of the S-ethyl moiety on the piperazine core was proven by various substitutions that showed a preference for a small apolar group over larger or polar moieties (McGuinness et al., 2009; Shao et al., 2011). This implies that rotation of the pyridine and the piperidine rings with respect to the piperazine ring are restricted and that there is limited space around the ethyl moiety. In our model, the piperazine moiety is close to TM6 and the finding that W2686.48Q and Y2716.51A mutants decrease affinity for VUF11211 7- and 10-fold, respectively, emphasizes the size and shape limitation of the pocket by TM6. The loss of affinity at mutants W1092.60Q (>600-fold, aromatic interaction), G1283.29H (±800-fold, space), and F1313.32A (20-fold, aromatic interaction) indicate a specific fit of the pyridyl moiety in the binding pocket of CXCR3 (Fig. 4, A, C, and E). Finally, modification of the amide moiety by removal or repositioning of the nitrogen atom indicated a role for hydrogen bonding of the nitrogen atom (±10-fold difference in affinity) (McGuinness et al., 2009; Shao et al., 2011). Removal of the hydroxyl moiety from the residues Y601.39, S3047.39, or Y3087.43 showed that none of them specifically formed an interaction with the amide moiety of VUF11211. Since the triple mutation (Y601.39F/S3047.39A/Y3087.43F) showed a 10-fold reduction in affinity, we suggest that at least these three residues that are in close proximity to VUF11211 might form a hydrogen bonding network within the CXCR3 binding pocket (Fig. 4, A and C). Apparently, these amino acid residues are able to compensate for the removal of the hydrogen bonding capabilities of a nearby residue. The involvement of hydrogen bonding is also indicated by mutation of S3047.39 to glutamic acid or leucine. The results obtained with the S3047.39E mutant show that a hydrogen bond acceptor moiety (i.e., the alcohol oxygen in S3407.39 or carboxylate oxygen atoms in E3047.39) is required, which is proposed to interact with the amide moiety. However, the introduction of a large hydrophobic group with S3047.39L reduces the affinity of VUF11211 by 20-fold.
Binding Hypothesis of NBI-74330.
Multiple mutations affected the binding of NBI-74330, whereas CXCL11 affinity remained unchanged. Furthermore, NBI-74330 exhibited noncompetitive insurmountable antagonism in various functional assays, including phospholipase C activation (Heise et al., 2005; Verzijl et al., 2008). In addition, CXCL11 could not completely displace the radiolabeled closely related NBI-74330 analog RAMX3 (Bernat et al., 2012). Altogether, these pharmacological data combined with our in silico–guided mutagenesis studies suggest an allosteric mode of action for NBI-74330.
Since NBI-74330 does not possess highly basic moieties and hence lacks H-bond donor atoms, the reduction of affinity observed at the D1122.63N mutant is remarkable (Fig. 3C; Table 1). The quinazolinone nitrogen atoms and associated positive partial charge on the 7-position of the ring are important for CXCR3 antagonism (Johnson et al., 2007; Storelli et al., 2007; Liu et al., 2009). As such, we propose that an aromatic −CH group of the 8-azaquinazolinone moiety of NBI-74330 forms a weak hydrogen bond to D1122.63, like N-heteroaromatic −CH groups in pyridine (Balevicius et al., 2007) and quinolin-8-ol (Zhang et al., 2013) rings (Fig. 4, D and F). The ligand SAR and CXCR3 mutagenesis data, however, do not exclude indirect water-mediated H-bond interactions between the pyridine nitrogen and the carboxylate group of D1122.63, like the water-mediated H-bond network between the tetrazole moiety of maraviroc and the hydroxyl group of Y1.39 in the CCR5 crystal structure (Fig. 5C) (Tan et al., 2013). In the proposed binding mode, the quinazolinone ring stacks with W1092.60, which is corroborated by the site-directed mutagenesis data (500-fold reduction in NBI-74330 affinity at the W1092.60Q mutant). The ethoxyphenyl moiety is located close to TM3, in line with the 100-fold reduction of NBI-74330 affinity at G1283.29H mutant, likely due to a steric clash in the mutant receptor (Fig. 4, D and F).
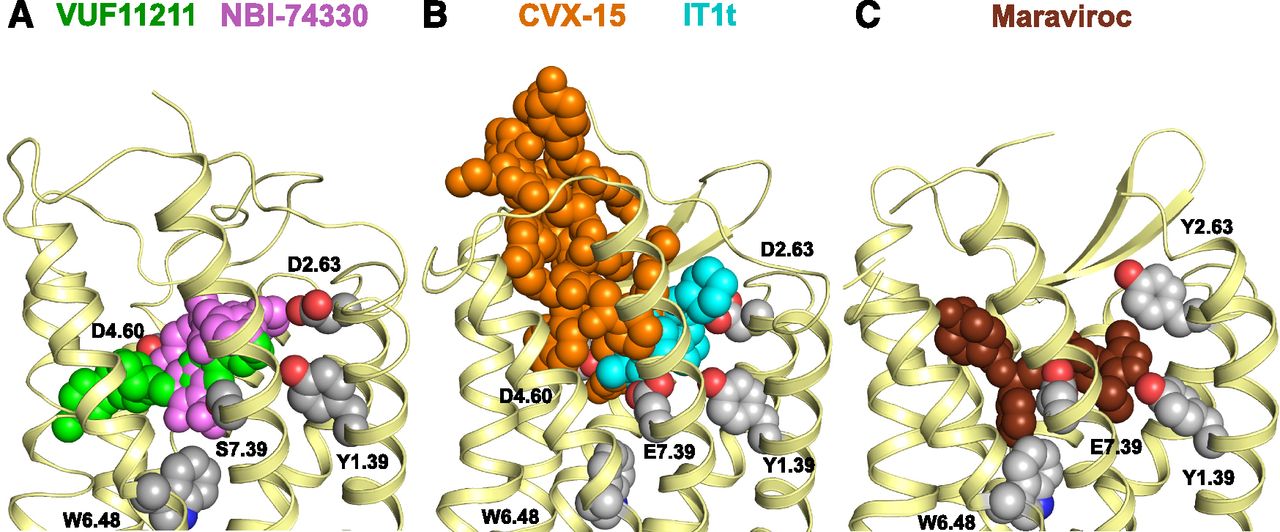
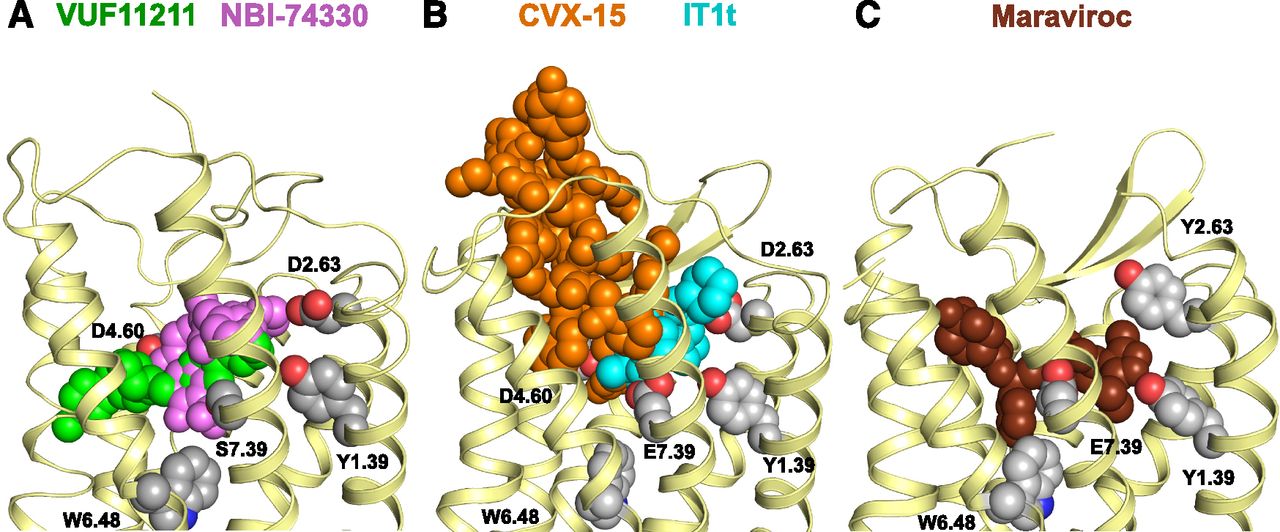
(A) Side view of the homology model of CXCR3 with the binding modes for VUF11211 bridging both subpockets (TMS1 and TMS2; green) and NBI-74330 mainly in TMS1 (magenta). (B) Side view of the CXCR4 receptor bound to the small ligand IT1t in TMS1 (cyan) and the peptide CVX-15 in TMS2 (orange), obtained by X-ray crystallography. (C) Side view of the CCR5 receptor bound to the small ligand maraviroc in both TMS1 and TMS2, obtained by X-ray crystallography.
The electron-withdrawing character of the trifluoromethyl moiety is also an important determinant for ligand affinity (>100-fold better over unsubstituted benzyl) (Storelli et al., 2005, 2007). Polar aromatic interactions between CXCR3 and NBI-74330 are identified by F1313.32A, Y2716.51A, and Y3087.43A, reducing affinity by 10-, 6-, and 30-fold, respectively (Fig. 4, B, D, and F; Table 2). Substitution of the serine at position 3047.39 by a glutamic acid showed a large reduction in affinity (80-fold), which is mainly caused by the increased hydrophobicity (20-fold reduction in the case of S3047.39L).
CXCR3 Antagonist Binding Pockets.
Different mutations in the TM region impacted NBI-74330 or VUF11211 binding, yet did not significantly affect CXCL11 affinity (Fig. 3), suggesting that CXCL11 does not bind to the TM domains (Xanthou et al., 2003; Trotta et al., 2009) and that these small molecules are allosteric CXCR3 ligands. However, chemokines are considerably larger, and most of their interaction energy comes from binding to the N terminus and ELs of the receptor. A small part of the chemokine (N terminus) is thought to interact with the receptor TM bundle for receptor activation. As such, potential overlap in interacting residues of the CXCR3 N terminus and small molecules in TMS1/2 cannot be ruled out at this point.
Interestingly, and similar to CXCR2 and CXCR4, these ligands bind differentially in the TM pockets within CXCR3. In our homology model of CXCR3, NBI-74330 binds mainly to TMS1 residues (Fig. 5A), as also observed for the antagonist IT1t cocrystallized with the CXCR4 receptor (Wu et al., 2010) (cyan in Fig. 5B versus magenta in Fig. 5A). NBI-74330 seems to span TMS2 to a small extent, in which only the Y2716.51A affected its binding affinity (6-fold). The TMS1 binding pocket has also been identified for ligands in other chemokine receptors such as UCB-35625 in CCR1 (Y1.39, Y3.32, and E7.39) (de Mendonça et al., 2005), LMD-009 in CCR8 (Y1.39, Q2.60, S3.29, Y3.32, and E7.39) (Jensen et al., 2007), and IT1t in CXCR4 (D2.63 and E7.39) (Wu et al., 2010). On the other hand, VUF11211, with its elongated shape, is anchored in TMS1 and traverses TMS2 to a much larger degree than NBI-74330, interacting with residues including W1092.60, F1313.32, D1864.60, and Y2716.51 (Fig. 5A). Interestingly, a recent X-ray crystallography study shows that maraviroc also binds in both pockets in CCR5 (e.g., Y1.39, W2.60, Y3.32, W6.48, Y6.51, I5.42, E7.39) (Tan et al., 2013) (Fig. 5C), whereas the CVX-15 peptide exclusively binds to TMS2 in a CXCR4 crystal structure (Fig. 5B) (Wu et al., 2010). The relatively large CVX-15 peptide stretches out to the ELs (Fig. 5B), whereas VUF11211 seems to bind in a more horizontal fashion (Fig. 5A). Ligands for other chemokine receptors also stretch both binding pockets such as BX-471 in CCR1 (Y1.39, Y3.33, I6.55, Y7.43) (Vaidehi et al., 2006) and AMD3100 in CXCR4 (Y1.39, W2.60, Y3.32, D4.60, D6.58, E7.39) (Wong et al., 2008; Wu et al., 2010). In conclusion, we report for the first time the molecular details of the binding of two high-affinity CXCR3 antagonists of distinct chemotypes. Fundamental knowledge on ligand interactions with the CXCR3 receptor may fuel the structure-based design and optimization of CXCR3 ligands in the future.
Acknowledgments
The authors thank Elwin Janssen for technical assistance in the determination of optical purities.
D.J.S., M.W., I.J.P.d.E., M.J.S., C.d.G., and R.L. participate in the European Cooperation in Science and Technology Action CM1207 [GPCR-Ligand Interactions, Structures, and Transmembrane Signalling: A European Research Network (GLISTEN)].
Authorship Contributions
Participated in research design: Scholten, Roumen, Wijtmans, Verkade-Vreeker, Custers, Lai, de Hooge, Canals, de Esch, Smit, de Graaf, Leurs.
Conducted experiments: Scholten, Roumen, Wijtmans, Verkade-Vreeker, Custers, Lai, de Hooge, Canals, de Graaf.
Contributed new reagents or analytic tools: Wijtmans, Custers, Lai, de Esch, de Graaf.
Performed data analysis: Scholten, Roumen, Wijtmans, Verkade-Vreeker, Custers, de Hooge, Canals, Smit, de Graaf, Leurs.
Wrote or contributed to the writing of the manuscript: Scholten, Roumen, Wijtmans, Smit, de Graaf, Leurs.
Footnotes
- Received August 4, 2013.
- Accepted October 30, 2013.
↵1 Current affiliation: Drug Discovery Biology and Department of Pharmacology, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, Victoria, Australia.
D.J.S. and L.R. contributed equally to this work.
This work was supported by The Netherlands Organization for Scientific Research [Grant 700.59.408 to C.d.G.] and was performed within the framework of the Dutch Top Institute Pharma GPCR Forum [Project D1-105].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AMD3100
- 1,1′-[1,4-phenylenebis(methylene)]bis-1,4,8,11-tetraazacyclotetradecane octahydrochloride
- AMG487
- (R)-N-(1-(3-(4-ethoxyphenyl)-4-oxo-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)ethyl)-N-(pyridin-3-ylmethyl)-2-(4-(trifluoromethoxy)phenyl)acetamide
- aplaviroc
- 4-(4-{[(3R)-1-butyl-3-[(R)-cyclohexylhydroxymethyl]-2,5-dioxo-1,4,9-triazaspiro[5.5]undecan-9-yl]methyl}phenoxy)benzoic acid; BX-471, (R)-1-(5-chloro-2-(2-(4-(4-fluorobenzyl)-2-methylpiperazin-1-yl)-2-oxoethoxy)phenyl)urea
- CCR
- CC chemokine receptor
- CVX-15
- Arg-Arg-Nal-Cys-Tyr-Gln-Lys-dPro-Pro-Tyr-Arg-Cit-Cys-Arg-Gly-dPro
- CXCL
- CXC chemokine ligand
- CXCR
- CXC chemokine receptor
- EL
- extracellular loop
- ELISA
- enzyme-linked immunosorbent assay
- GPCR
- G protein–coupled receptor
- HEK
- human embryonic kidney
- IT1t
- 1,3-dicyclohexyl-2-(3-methyl-6,6-dimethyl-5,6-dihydroimidazo[1,2-b]thiazole)-2-thiopseudourea
- LMD-009
- 8-[3-(2-methoxyphenoxy)benzyl]-1-phenethyl-1,3,8-triaza-spiro[4.5]decan-4-one
- maraviroc
- 4,4-difluoro-N-((S)-3-((1R,3R,5S)-3-(3-isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl)-1-phenylpropyl)cyclohexanecarboxamide
- NBI-74330
- (R)-N-(1-(3-(4-ethoxyphenyl)-4-oxo-3,4-dihydropyrido[2,3-d]pyrimidin-2-yl)ethyl)-2-(4-fluoro-3-(trifluoromethyl)phenyl)-N-(pyridin-3-ylmethyl)acetamide
- PCR
- polymerase chain reaction
- SAR
- structure–activity relationship
- SCH-546738
- (S)-3-amino-6-chloro-5-(4-(1-(4-chlorobenzyl)piperidin-4-yl)-3-ethylpiperazin-1-yl)pyrazine-2-carboxamide
- TM
- transmembrane
- TMS
- transmembrane site
- UCB-35625
- 1,4-trans-1-(1-cycloocten-1-ylmethyl)-4-[[(2,7-dichloro-9H-xanthen-9-yl)carbonyl]amino]-1-ethylpiperidinium iodide
- VUF11211
- (S)-5-chloro-6-(4-(1-(4-chlorobenzyl)piperidin-4-yl)-3-ethylpiperazin-1-yl)-N-ethylnicotinamide
- WT
- wild-type
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}