


Abstract
Seven transmembrane receptors were originally named and characterized based on their ability to couple to heterotrimeric G proteins. The assortment of coupling partners for G protein–coupled receptors has subsequently expanded to include other effectors (most notably the βarrestins). This diversity of partners available to the receptor has prompted the pursuit of ligands that selectively activate only a subset of the available partners. A biased or functionally selective ligand may be able to distinguish between different active states of the receptor, and this would result in the preferential activation of one signaling cascade more than another. Although application of the “standard” operational model for analyzing ligand bias is useful and suitable in most cases, there are limitations that arise when the biased agonist fails to induce a significant response in one of the assays being compared. In this article, we describe a quantitative method for measuring ligand bias that is particularly useful for such cases of extreme bias. Using simulations and experimental evidence from several κ opioid receptor agonists, we illustrate a “competitive” model for quantitating the degree and direction of bias. By comparing the results obtained from the competitive model with the standard model, we demonstrate that the competitive model expands the potential for evaluating the bias of very partial agonists. We conclude the competitive model provides a useful mechanism for analyzing the bias of partial agonists that exhibit extreme bias.
Introduction
It is now broadly recognized that G protein–coupled receptors are able to stimulate the activity of multiple signaling cascades. These signaling cascades extend beyond heterotrimeric G proteins to βarrestins and other scaffolding proteins (including numerous kinases). Furthermore, it has been demonstrated, both in vitro and in vivo, that some ligands are able activate one pathway while lacking agonism in responses mediated by other pathways; this effect has been termed agonist trafficking, agonist-directed trafficking, functional selectivity, and biased agonism (Kenakin, 1995, 2003; Berg et al., 1998; Mottola et al., 2002; Wisler et al., 2007; Urban et al., 2007). Because the propensity for a receptor to respond to an agonist can be a function of not only the agonist-receptor interaction but also the context of the system, the analysis of such activation profiles requires simultaneous consideration of multiple parameters, prompting an expansion of the 2-state model of receptor activation.
The 2-state model of receptor activation is a useful mechanistic model for understanding receptor-mediated signaling. According to this model, the receptor isomerizes between two states; one state is the inactive state, and the other is the active state of the receptor (del Castillo and Katz, 1957; Monod et al., 1965; Leff, 1995). The positive efficacy of an agonist is defined by the degree of preference it exhibits for the active state over the inactive state. One outcome of this relationship is that agonists may present different degrees of agonist activity in different systems but the model predicts that the rank order of agonist activity will remain constant (Leff, 1995). The demonstration that this rank order of agonist activity is not constant resulted in the adaptation of the 2-state model to the 3-state and the n-state models of receptor activation (Kew et al., 1996, Leff et al., 1997; Berg et al., 1998). These models incorporate multiple active receptor states to provide a mechanism for explaining differences in the rank order of agonist potency and permit the state model to accommodate agonist bias.
Not only did the observation of changes in the rank order of agonist activity lead to the evolution of the multistate models, but it also inspired the search for and development of biased agonists. Biased agonists are able to distinguish between different active states and, in the extreme case, produce selective agonist activity in only one pathway. Several biased agonists are currently in various stages of preclinical and clinical development, including those targeting the angiotensin IIA receptor as well as the μ and δ opioid receptors (DeWire et al., 2013; Soergel et al., 2013; Valant et al., 2014). Recent efforts in our laboratory have explored biased agonism at the κ opioid receptor (KOR) (Schmid et al., 2013, Zhou et al., 2013). For example, 6′-guanidinonaltrindole (6′-GNTI) was initially characterized as a potent partial KOR agonist (Sharma et al., 2001). It was subsequently shown to stimulate G protein coupling yet induced very little detectable βarrestin2 recruitment when compared with U69,593 [(+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide], a KOR agonist that displays high efficacy in both assays (Rives et al., 2012; Schmid et al., 2013). Using the method of bias analysis described by Ehlert (2008) and Kenakin et al. (2012), referred to herein as the “standard” model, 6′-GNTI was determined to be a biased agonist for G protein signaling over βarrestin2 recruitment (Schmid et al., 2013).
There is considerable interest in developing pharmacologically diverse ligands at the KOR as the KOR modulates dopamine levels and thereby affects mood and perception. KOR antagonists hold promise for treating drug addiction (Carroll et al., 2004; Walker and Koob, 2008) whereas KOR agonists produce antinociception and suppress itch. However, the appeal of KOR agonists is limited by the dysphoria they produce (Pfeiffer et al., 1986; Togashi et al., 2002; Land et al., 2008; Van’t Veer and Carlezon, 2013). Interestingly, recent studies suggest that the dysphoric component of KOR agonism may be mediated by arrestins inspiring interest in developing G protein signaling–biased KOR agonists (Bruchas et al., 2006; Redila and Chavkin 2008). This opportunity for the development of biased KOR agonists is accompanied by the challenge of elucidating the properties of bias with confidence. This is especially true in the cases where the agonists display extremely partial agonist activity.
The first pharmacologic characterization of nalmefene suggested it was a competitive KOR antagonist with the ability to block the antinociceptive response in mice (Heilman et al., 1975). However, nalmefene was shown in human studies to have agonist properties for stimulating prolactin release and therefore its pharmacologic properties are likely context dependent (Bart et al., 2005). This apparent complex pharmacology suggests that nalmefene may be a candidate for further exploration of ligand bias. Importantly, nalmefene is clinically used in the treatment of alcoholism; therefore, a better understanding of this ligand’s pharmacologic properties may provide important guidance for future therapeutic development (Mason et al., 1994; Boening et al., 2001; Walker and Koob, 2008; Zindel and Kranzler, 2014).
In the studies presented herein, we describe a “competitive” method for analyzing ligand bias that is particularly useful for agonists that display minimal agonist activity. We apply both the competitive and standard models of bias analysis to mathematic simulations and experimental data from select KOR agonists (the standard model referred to earlier and defined in Materials and Methods is from Kenakin et al., 2012). Specifically, we compare nalmefene and two structurally related morphinan KOR partial agonists that exhibit different degrees of agonism compared with nalmefene, 6′-GNTI, and nalbuphine with the full agonist U69,593, which serves as the reference agonist. We find that the standard model can be used to estimate bias with confidence for agonists that stimulate a response significantly greater than baseline. However, as the agonist activity of the ligand approaches baseline in the assay, the competitive method allows for a more accurate estimate of bias. From these findings, we conclude that, although the standard method is well suited to analyze the bias of agonists with considerable agonist activity, the competitive model of bias analysis is a useful mechanism for assessing the bias of very partial agonists.
Materials and Methods
Reagents.
All cell culture reagents were purchased from Life Technologies (Carlsbad, CA). 6′-GNTI ditrifluoroacetate was purchased from Tocris Bioscience (Ellisville, MO). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO). The reference compound U69,593 was purchased from Sigma-Aldrich and was prepared as a 10 mM stock in ethanol. All other ligands were prepared as 10 mM stocks in dimethylsulfoxide (DMSO) (Fisher Scientific, Fair Lawn, NJ). Each ligand was diluted in serial dilutions in DMSO. [35S]GTPγS [guanosine 5′-O-(3-[35S]thio)triphosphate] was purchased from PerkinElmer (Waltham, MA). DMSO was added to assay media to maintain the DMSO concentration at 2%. The DiscoveRx PathHunter assay detection reagent was purchased from DiscoveRx (Fremont, CA).
Cell Lines and Cell Culture.
Chinese hamster ovary (CHO) cells expressing recombinant human κ opioid receptor (CHO-hKOR cell line) were generated as described previously (Schmid et al., 2013). The CHO cell lines were maintained in Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium from Invitrogen (Carlsbad, CA) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 500 µg/ml geneticin. All cells were grown at 37°C (5% CO2 and 95% relative humidity).
[35S]GTPγS Binding.
Membranes were prepared according to a modified procedure of Schmid et al., (2013). Briefly, CHO-hKOR cells were serum starved for 1 hour in Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium, collected with 5 mM EDTA, washed with phosphate-buffered saline, and stored at −80°C. For each assay, cell pellets were homogenized via Teflon-on-glass homogenizer in buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA), centrifuged twice at 20,000g for 30 minutes at 4°C, and resuspended in assay buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, and 3 µM GDP). For each reaction, 15 μg of membrane protein were incubated in assay buffer containing ∼0.1 nM of [35S]GTPγS and increasing concentrations of compounds in a total volume of 200 µl for 1 hour at room temperature. The reactions were terminated by rapid filtration over GF/B filters using a 96-well plate harvester (Brandel Inc., Gaithersburg, MD). Filters were dried overnight, and radioactivity was determined with a TopCount NXT HTS microplate scintillation and luminescence counter (PerkinElmer).
βArrestin2 Recruitment.
A commercial enzyme fragment (β-galactosidase) complementation assay (DiscoveRx PathHunter; DiscoveRx) was used to assess βarrestin2 recruitment as previously described elsewhere (Zhou et al., 2013). U2OS-hKOR-βarrestin2-DX cells were incubated for 60 minutes with the ligands indicated. Each condition was measured in duplicate in each assay at least three times. After the 60-minute incubation, the manufacturer’s substrate and protocol were used to assess βarrestin recruitment. Assay plates were read using a Molecular Devices SpectraMax M5e multimode microplate reader (Sunnyvale, CA).
Simulations.
All simulations were generated using the “simulate XY data” function built into GraphPad Prism version 6.0f (GraphPad Software, La Jolla, CA). Two different forms of the operational model were used to produce response simulations. Stimulatory dose–response curves were generated using the operational model (Black and Leff, 1983):
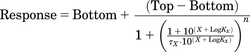 (1)
(1)In eq. 1, Top represents the maximum response of the system, and Bottom represents the basal level of response present in the system. X represents the log of the agonist concentration, n the transducer slope factor of the system, and LogKX the log of the affinity constant of the agonist. In the equation, τ represents a parameter proportional to the intrinsic efficacy of each agonist. For all simulations, the values for Top and Bottom were set equal to 1 and 0, and the value for n, the transducer slope factor, was set equal to 1. The values for each of the other parameters in the equation were specifically defined for each agonist. The parameter values used for these simulations are provided in Supplemental Table 1.
A second form of the operational model was used to simulate the incomplete inhibition of response produced by a partial agonist (Kenakin, 2009; Ehlert, 2014):
 (2)
(2)Several parameters in this equation share identical names and definitions with eq. 1 (Top, Bottom, and n). The noticeable difference between the two equations is that eq. 2 incorporates parameter definitions for two different agonists. The log concentration, affinity, and intrinsic activity of one agonist are presented by one set of parameters (X, LogKX, and τX). The same attributes are also presented for a second agonist (B, LogKB, and τB). The incorporation of two different agonist-specific parameters and the relationship of these parameters in the equation permit this model to accommodate competition between the two agonists for a common binding site. In the simulations produced with eq. 2, a specific log concentration value for the reference agonist (−8.0) was imposed as the X parameter in the equation. The inhibition curves were simulated using increasing concentrations of each test agonist. The parameter definitions and values used for these inhibition curves were identical to the dose–response curves described earlier and stated in Supplemental Table 1.
Both equations were used to simulate concentration-response and concentration-inhibition curves for two different responses. The distinction of one response versus another was made by selecting different τ values for the agonists in each response. In one response, the agonists were more efficacious; in the second response, the agonists were less efficacious. In both responses, the simulated reference agonist was much more efficacious than each of the test agonists. The data sets were simulated three times, with each simulated data set intended to describe a single experiment. For both the stimulatory and inhibitory simulations, 10 X values were produced in triplicate for each simulated data set. Random error was superimposed in each simulation by setting the standard deviation to 0.07.
Monte Carlo simulations were performed using the simulation feature in Prism 6.0f. Equations 3, 4, and 7 (expressed in the Data Analysis section) were used to construct simulated concentration-response curves based on the parameter values defined in Supplemental Table 1. The simulated data were generated with an equal number of data points for each data set, and a random error with standard deviation of 0.07 was imposed. The resulting data were fit with either the standard or competitive model. This process was repeated for 1000 iterations, and the resulting LogRA value of each test agonist was recorded. The frequency distribution of the LogRA values was then plotted, and the standard deviation of the distribution was calculated.
Data Analysis.
All curves were fitted by nonlinear regression using GraphPad Prism version 6.0f. Two different methods of analysis are presented herein, and both methods use a derivation of the operational model (eq. 1). The first method of analysis, defined here as the standard method, employs eqs. 3 and 4 (Griffin et al., 2007; Ehlert, 2008):
 (3)
(3)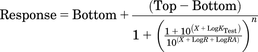 (4)
(4)These equations are used to fit the response produced by the reference agonist (eq. 3) and the test agonist(s) (eq. 4). Each of these equations is derived from the operational model, and the definitions for the parameters Top and Bottom are identical. It should be noted that the equation is written in a form that accommodates a transducer slope factor, defined as n, different from 1. This form of the equation has previously been used to analyze responses with slope factors different from 1 (Griffin et al., 2007; Kenakin et al., 2012). In eq. 3, X represents the log of the concentration of the reference agonist whereas in eq. 4 X represents the log of the concentration of the test agonist. Similarly, LogKReference is the log affinity constant (inverse molar units, M−1) of the reference agonist in eq. 3 and LogKTest is the test agonist’s log affinity constant in eq. 4.
The τ parameter is noticeably absent from these equations. In fact, the equations are reparameterizations of the operational model that combine the τ and LogK parameters for each agonist (where possible) into a single LogR value. When the two equations are fitted to a reference and test agonist(s) simultaneously, the LogR parameter defines the log τ⋅K value of the reference agonist.
The LogR of the test agonist is subject to further reparameterization in eq. 4. Instead of determining the LogRTest of the test agonist directly, the equation is written in a form where the LogRA is determined (this parameter shares the same definition with the ∆LogR; Kenakin et al., 2012 and see Discussion). The LogRA is defined as the difference between the LogRTest (the LogR of each test agonist) and the LogRReference (the LogR of the reference agonist):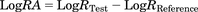 (5)The resulting LogRA of each test agonist is then compared between different assays to produce the ∆LogRA:
(5)The resulting LogRA of each test agonist is then compared between different assays to produce the ∆LogRA: (6)This is a measure of the bias of an agonist for Response 1 or Response 2. Specifically, a ∆LogRA value greater than 0 indicates a bias toward Response 1; a value less than 0 indicates a bias toward Response 2.
(6)This is a measure of the bias of an agonist for Response 1 or Response 2. Specifically, a ∆LogRA value greater than 0 indicates a bias toward Response 1; a value less than 0 indicates a bias toward Response 2.
The second method of bias analysis also uses eq. 2 to fit the reference agonist. The distinct difference in this method, defined as the competitive method of bias analysis, is the type of experimental design it requires and the form of the operational model it employs. The experimental design required in this method leverages the competitive relationship between the reference agonist and test agonist. The results of these experiments demonstrate a functional reversal of the response, produced by the reference agonist, upon application of increasing concentrations of the test agonist. Equation 2 can be reparameterized to provide an equation that is adequately suited to analyze the effects produced by partial agonist competition for the binding site occupied by a full agonist:
 (7)
(7)In this form of the equation, the parameters defined are identical to the parameters in eqs. 3 and 4. Specifically, A defines the log concentration of the reference agonist, and LogKReference refers to the log affinity constant of the reference agonist. The LogR corresponds to the same parameter in eq. 3. Similarly, the parameter identities are the same for the test agonist. X in eq. 7 refers to the log concentration of the test agonist, and LogKTest represents the affinity of the test agonist. As expressed in eq. 4 and defined in eq. 5, the LogRA defines the difference between the LogR of the test agonist and the LogR of the reference agonist. The resulting LogRA values for each response were used to determine a ∆LogRA value for each test agonist. The ∆LogRA value is produced by the same calculation stated in eq. 6.
In order for the equations to fit the data and produce results for comparison, it was appropriate to set specific values and constraints for some parameters in the equations. For instance, it has been shown that an infinite number of combinations of τ and Kx will produce identical concentration-response curves for a full agonist but, nonetheless, a unique LogR value (i.e., log τ⋅KX). For this reason, it was reasonable to constrain the LogKX of the reference agonist to a large specific value. We constrained the affinity of the reference agonist to 100 mM (LogKReference = −1).
Two other constraints were imposed to limit the parameter space available and thereby produce appropriate parameter estimates for all the data sets in both methods of bias analysis. The affinity of each test agonist was constrained to lie in a range from 1 fM to 1 M. Additionally, the LogRA of each test agonist was constrained to have an absolute value between 0 and 10. The resulting parameter values, from each experiment, were pooled to produce logK and LogRA estimates (mean and 95% confidence interval [CI]) for each test agonist. An unpaired Student’s t test of the results from the two systems was then used to calculate the ∆LogRA mean and 95% CI for each test agonist.
Results
Simulation of Dose-Response Curves of Biased Ligands.
The initial comparison of the two methods of analysis is presented using several sets of simulated data. (The construction of the simulations is described in detail in Materials and Methods). The simulations are designed to mimic the responses produced by a full reference agonist and two test agonists in two different systems. In each system, the full agonist is more efficacious than the two test agonists. Each of the responses is independently simulated 3 times with each data point produced in triplicate. One of the simulated responses is intended to mimic a highly coupled system (Response 1) whereas the other response is made to resemble a more lowly coupled system (Response 2). This produces test agonists with greater maximal response in the first response than in the second response.
Fitting Response Curves with the Standard Model That Permits Variability in the Test Ligand’s Affinity Produces Poorly Defined Parameter Estimates.
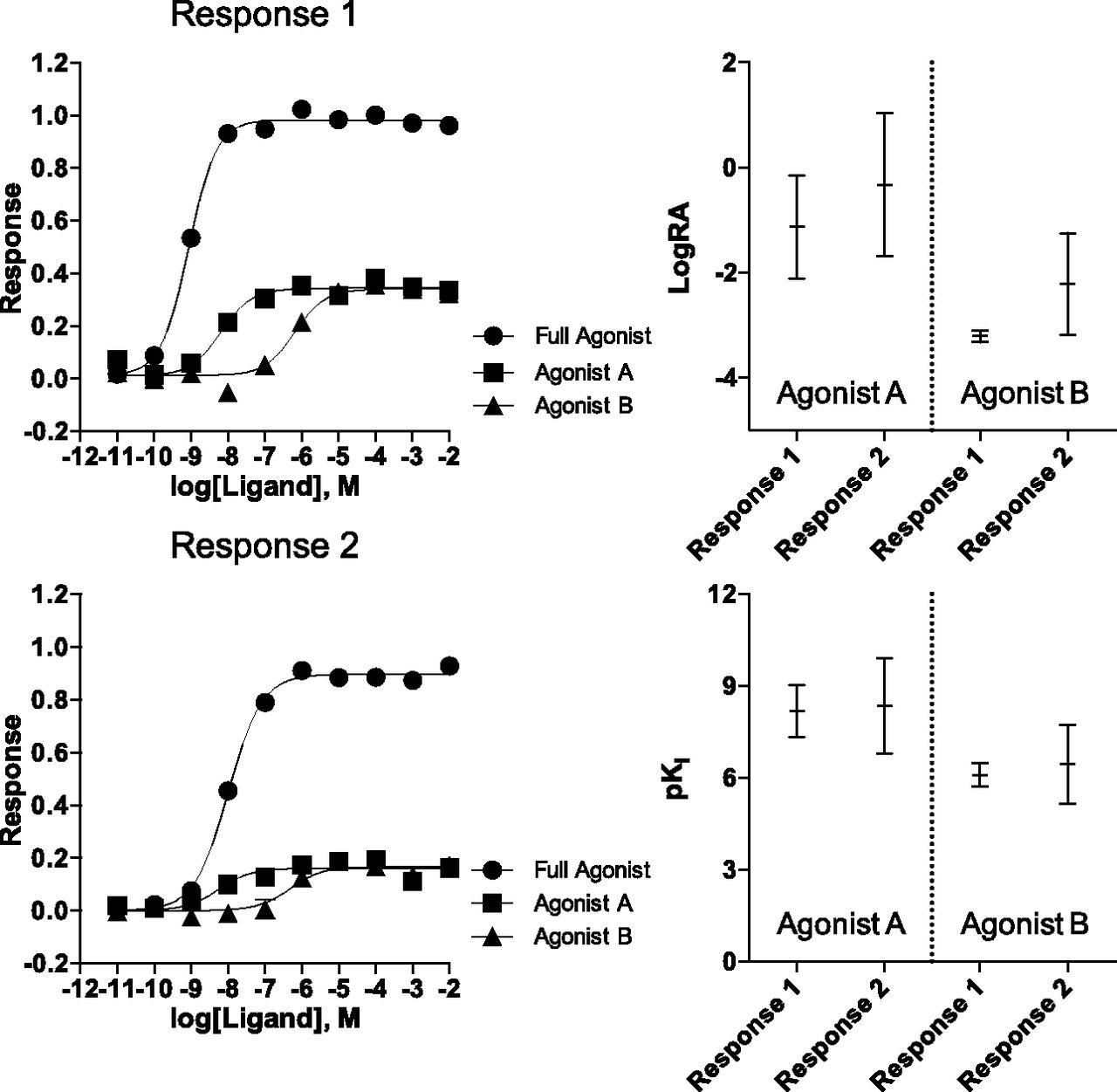
In the simulation of Response 1, presented in Fig. 1, the response produced by the full agonist and each of the test agonists is apparent. That is, the midpoint of each response curve is well defined. This is an outcome of Response 1 being defined as a highly coupled system. In contrast, the EC50 of the full agonist in Response 2 is easily discernible, but the midpoint of each test agonist response curve is much harder to estimate. Although these responses are subject to different degrees of amplification, it is possible to normalize the response produced by the test agonists to the response produced by the full agonist in each system. The normalization is accomplished by fitting the experimental data with eqs. 3 and 4 (derived by Griffin et al., 2007). For clarity, we refer to this method as the standard method throughout this article. An important parameter, the intrinsic relative activity (LogRA), is produced by this fit. This parameter defines the difference between the log(τ⋅K) values of the test and reference agonists in the system analyzed (eq. 5). When the LogRA values are compared across assays, an estimate of each test ligand’s bias is produced (∆LogRA). The simulations presented in Fig. 1 are analyzed using the standard method.

The standard bias analysis of simulated dose–response curves for a reference agonist and two test agonists in two different systems are presented in the panels Response 1 and Response 2. Each data point in the curve is the mean ± S.E.M. of three simulated data sets; curves are produced using the standard method. The values derived from each individual simulation were pooled and averaged to produce the LogRA and pKi values in Supplemental Tables 2 and 4. The graphical representations of LogRA and pKi are produced from the supplemental tables and demonstrate the wide 95% confidence intervals that are produced with the standard method.
In addition, a second parameter estimate, the affinity of the test agonists (pKi), is also obtained with the standard method analysis. The mean LogRA and pKi values for these simulations are presented in Supplemental Tables 2 and 4 and are plotted with 95% CI in Fig. 1. We have chosen to represent the mean ±95% CI as error throughout this manuscript because this representation provides a useful measure of how well the estimated mean compares with the true mean of the population (Motulsky, 2010). Upon inspection of the widths of these 95% CIs, for each parameter estimate, there is a large degree of uncertainty for both parameters seen in Response 2 (the lowly coupled system). For example, the pKi for the two test agonists in Response 2 span two to three orders of magnitude (Agonist A pKi = 9.9–6.8; Agonist B pKi = 7.7–5.2). In the case of Response 1, the highly coupled system, the fit to the model produces more reasonable estimates of the parameter values, with relatively narrow 95% CIs. These estimates still exhibit significant error, with the 95% CI of the pKi for Agonist A spanning close to two orders of magnitude (9.0–7.3).
Fitting Response Curves with the Competitive Model Can Produce More Defined Parameter Estimates.
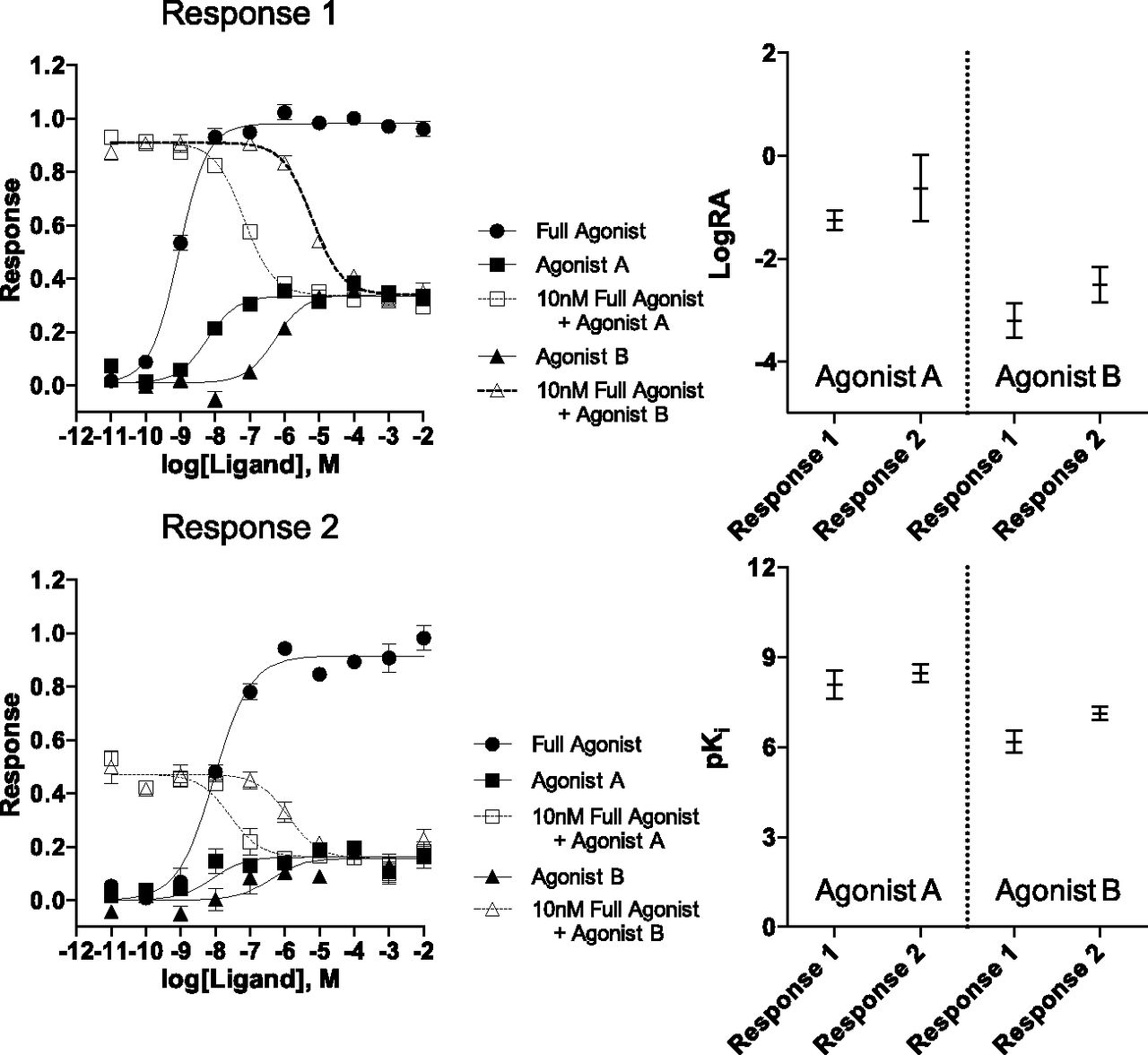
In Fig. 1, there is a large separation between the maximal response produced by the full agonist and the maximal responses produced by each test agonist. Because this submaximal response is a characteristic of partial agonism, there arises the opportunity to investigate the ability of the test agonist to inhibit the response produced by a preselected (∼EC50–EC80) concentration of the full agonist. In fact, using an equation derived from a form of the operational model that accommodates competition (eq. 7), all the data can now be analyzed to produce a global estimate of the LogRA and pKi values of each test agonist. This method of analysis is referred to herein as the competitive method.
Specifically, in contrast to the standard method, this competitive method simultaneously uses both stimulatory and inhibitory curves to characterize the test agonists. Figure 2 presents the stimulation produced by each agonist, the inhibition of response that each test agonist produced in the presence of 10 nM of the full agonist, and the competitive fit analysis parameters with 95% CI. When compared with the error obtained for the LogRA and pKi values using the standard model (Fig. 1), the competitive method of analysis demonstrates a substantial reduction in error in all cases except for the estimate of the LogRA of Agonist B in Response 1 wherein the error remains very similar (standard: −3.2 ± 0.1; competitive: −3.2 ± 0.3), suggesting that this difference in error is unremarkable.

The competitive model analysis of simulated dose–response curves representing the response produced by a reference agonist and two test agonists in two different systems. In addition, simulations where each partial agonist, relative to its affinity, produces a reversal of the response stimulated by the full agonist are presented for each response, titled Response 1 and Response 2. Shown are the mean ± S.E.M. of three simulated data sets; values derived from individual simulations were pooled and averaged to produce the LogRA and pKi values in Supplemental Tables 3 and 5. The graphical representations of LogRA and pKi are produced from the supplemental tables and demonstrate that the competitive method produces narrow 95% confidence intervals.
For Very Partial Agonists, the Competitive Fit Produces More Precise Test Ligand LogRA and ∆LogRA Values Bias Estimates Compared with the Standard Fit.
To compare the ability of the two methods to correctly estimate the LogRA parameter value of a population, we produced a series of Monte Carlo simulations. From these simulations, we compiled frequency distributions of the LogRA of the two simulated test agonists in Response 2 from both the standard and competitive methods. These distributions are presented in Fig. 3 along with the standard deviation for each distribution. It can be seen, both visually and by comparison of the standard deviation values, that the competitive model produces LogRA values that lie closer to the actual (known) LogRA value of each test agonist. For Agonist A, the mean and standard deviation for the standard model were −0.71 and 0.36 whereas the mean and standard deviation for the competitive model were −0.7 and 0.19. (The actual LogRA for Agonist A in these simulations was −0.7.) For Agonist B, the mean and standard deviation for the standard model were −2.59 and 0.79 whereas the mean and standard deviation for the competitive model were −2.71 and 0.20. (The actual LogRA for Agonist B in these simulations was −2.7.) From these simulations, we are able to conclude that the competitive model provides a distinct advantage when producing parameter estimates for extremely partial agonists.

The frequency distribution of the LogRA values produced by Monte Carlo simulation of the standard and competitive method. The distributions were produced based on the same values used to simulate the two test agonists in the Response 2 of Supplemental Table 1. Each bin of the distribution is 0.1 units wide, and the dotted line intersecting each distribution indicates the true LogRA value of the agonist. For Agonist A the true LogRA value is −0.7 and for Agonist B the true LogRA value is −2.7. The associated standard deviation is presented in the text box above each distribution.
The degree of agonist bias is described by the term ∆LogRA (defined in Materials and Methods as the difference between the LogRA of each agonist in the two responses). If the mean ∆LogRA is equal to 0 (or the 95% CI of the mean overlaps 0) then that test ligand is defined as unbiased (Kenakin et al., 2012). (It is worth noting that the reference agonist, by definition, has a ∆LogRA value equal to 0, ∆LogRA ≡ 0.) The distance of the ∆LogRA value from 0 defines the degree of bias displayed by the agonist between two systems relative to the reference agonist. The value of ∆LogRA can be either positive or negative, indicating that the agonist is biased toward or away from one or the other response.
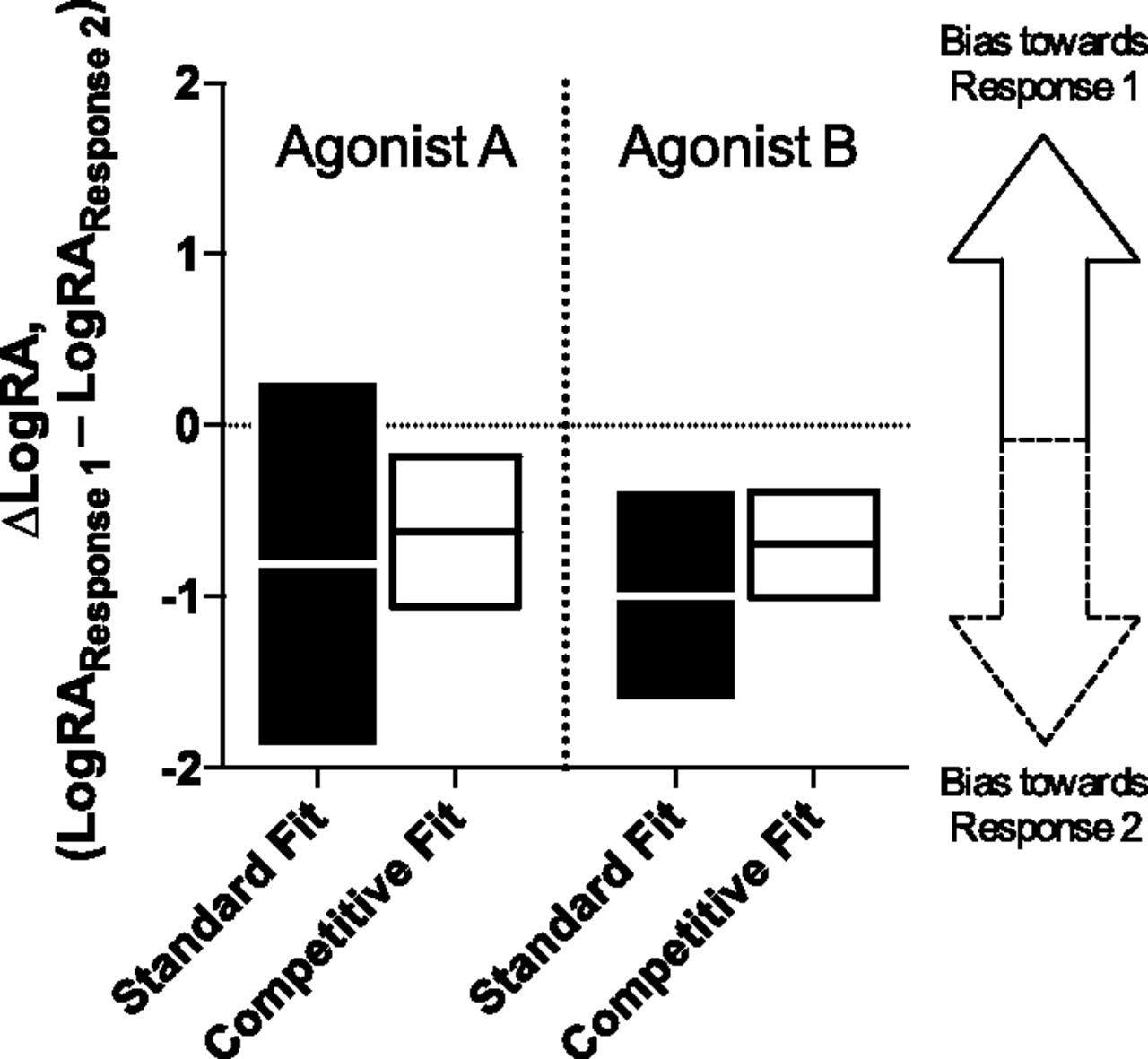
Using the LogRA values produced from the two different methods we have discussed, the ∆LogRA of each test agonist is calculated using eq. 6 (these values are presented in Supplemental Tables 6 and 7). The resulting bias values from each method are plotted in Fig. 4. The figure shows that there is a significant reduction in error in the bias estimate produced using the competitive fit versus the standard fit of the data. In fact, the estimate of the ∆LogRA for Agonist A includes 0 when the LogRA values for the standard fit are used, but it does not when the LogRA values for the competitive fit are used. This finding is important because it indicates that the reduction in the error of the LogRA produced by the competitive estimate can result in both a qualitative and quantitative difference in the demonstration of ligand bias.

The bias factors derived from the standard model and competitive model for each simulated test agonist are presented as ∆LogRA. The bars indicate the 95% confidence interval of the ∆LogRA determination, and the midline represents the mean. If the bar overlaps 0, the ligand is considered unbiased. A bar that does not touch 0 is considered biased, and the degree of bias is defined as the distance from 0. Calculating the difference-of-mean of the LogRA, according to eq. 5, produces the ∆LogRA estimate for each method. The standard method and the competitive method are calculated separately, and the figure presents a graphical representation of the values in Supplemental Tables 6 and 7. The results of the competitive method produce a decrease in the width of the confidence interval for each test agonist compared with the results of the standard method.
Analyzing the [35S]GTPγS Coupling Stimulation of Partial Agonists with the Competitive Method Produces a Reduction in Error for Some of the Parameter Estimates Compared with the Standard Method.
To validate the findings presented for the simulated data, we analyzed a series of experimental data using both methods of bias analysis. For the first response, agonist stimulation of KOR is measured using a [35S]GTPγS binding assay (Fig. 5) by running three partial agonists with varying response profiles in parallel with U69,593, a full agonist in the assay. Using U69,593 as the reference agonist, the responses produced by the three test agonists are fit using the standard method (eqs. 3 and 4), and the LogRA and pKi values with 95% CI are presented in Tables 1 and 2.
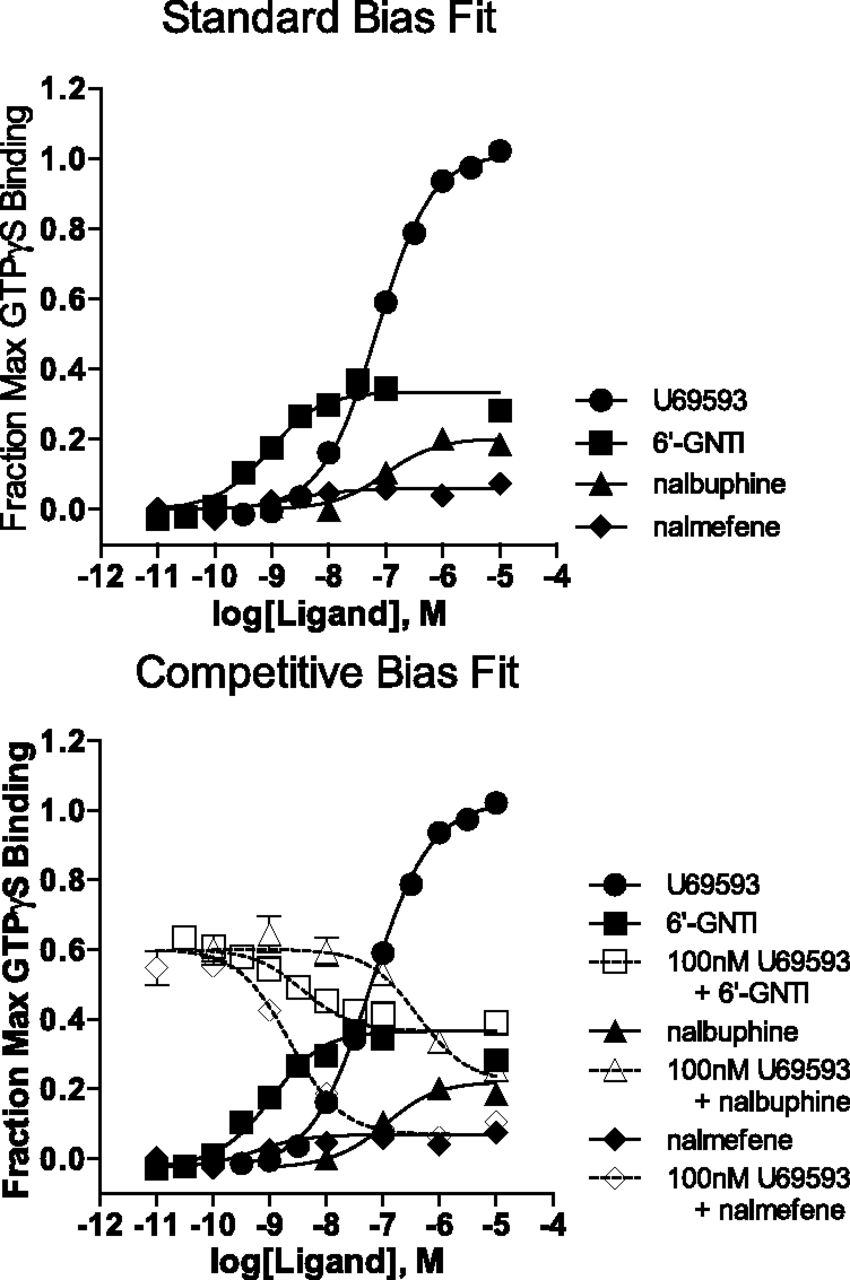
G protein signaling via KOR is measured by stimulation of [35S]GTPγS binding in CHO-hKOR cell membranes. U69,593, the reference agonist, was used for comparing relative responses of nalmefene, nalbuphine, and 6′-GNTI. Data were analyzed using both the standard and competitive methods, as indicated. For each data set, both equations produced good fits of the data. In the case of the competitive method, the inhibition curves are fit to a top equal to the response produced by 100 nM U69,593. Each data point in the curve is the mean ± S.E.M. of more than three independent experiments.
pKi values from standard fit
LogRA values from the standard fit
It is apparent from the stimulation curves, that nalmefene generates such a low amount of stimulation that fitting the curve with nonlinear regression is not ideal. Therefore, to gain greater confidence in the relative potency of the compounds in the assay, inhibition curves, presented in the lower panel of Fig. 5, were run in parallel to the stimulation curves (against 100 nM U69,593). Together, these data were used to calculate the competitive fit parameters (presented in Tables 3 and 4).
pKi values from competitive fit
LogRA values from the competitive fit
To compare the two methods of analysis, the resulting parameter estimates from each fit are plotted in Fig. 6 (with 95% CI). For the competitive model, we observed a slight increase in the size of the LogRA confidence interval for 6′-GNTI when comparing with the standard model. For nalbuphine, there was no difference observed between the two models. The benefit of the competitive fit became apparent when comparing the results for nalmefene. There was a substantial reduction in the width of the 95% CI for nalmefene with the competitive model compared with the standard model.

The competitive and standard models produce differences in the estimates of LogRA and pKi. The graphs present the parameter estimates for each method as mean ± 95% confidence interval. The results of each individual experiment were pooled and averaged to produce the LogRA and pKi values for [35S]GTPγS binding in Tables 1–4. The graphical representation of both LogRA and pKi in this figure are produced from the values in these tables. The graphs demonstrate the competitive method of normalization produced 95% confidence intervals that are similar to, or more narrow than, the standard fit.
The Competitive Method of βArrestin2 Recruitment Studies Also Produces Error Reduction, in Some Cases, Compared with the Standard Method.
A second response that we use to measure agonist stimulation of the KOR is the βarrestin2 recruitment enzyme complementation assay (DiscoveRx PathHunter). U69,593 is also used as the reference agonist in this response, allowing for the comparison of the relative agonist activity of the test agonists in the two different responses. Fig. 7 shows that U69,593 produces a maximum response that is much greater than each of the test agonists in this response. The upper panel of Fig. 7 shows only the stimulation curve data that is applied to the standard fit analysis; it is important to note that nalmefene has extremely low efficacy in this assay.
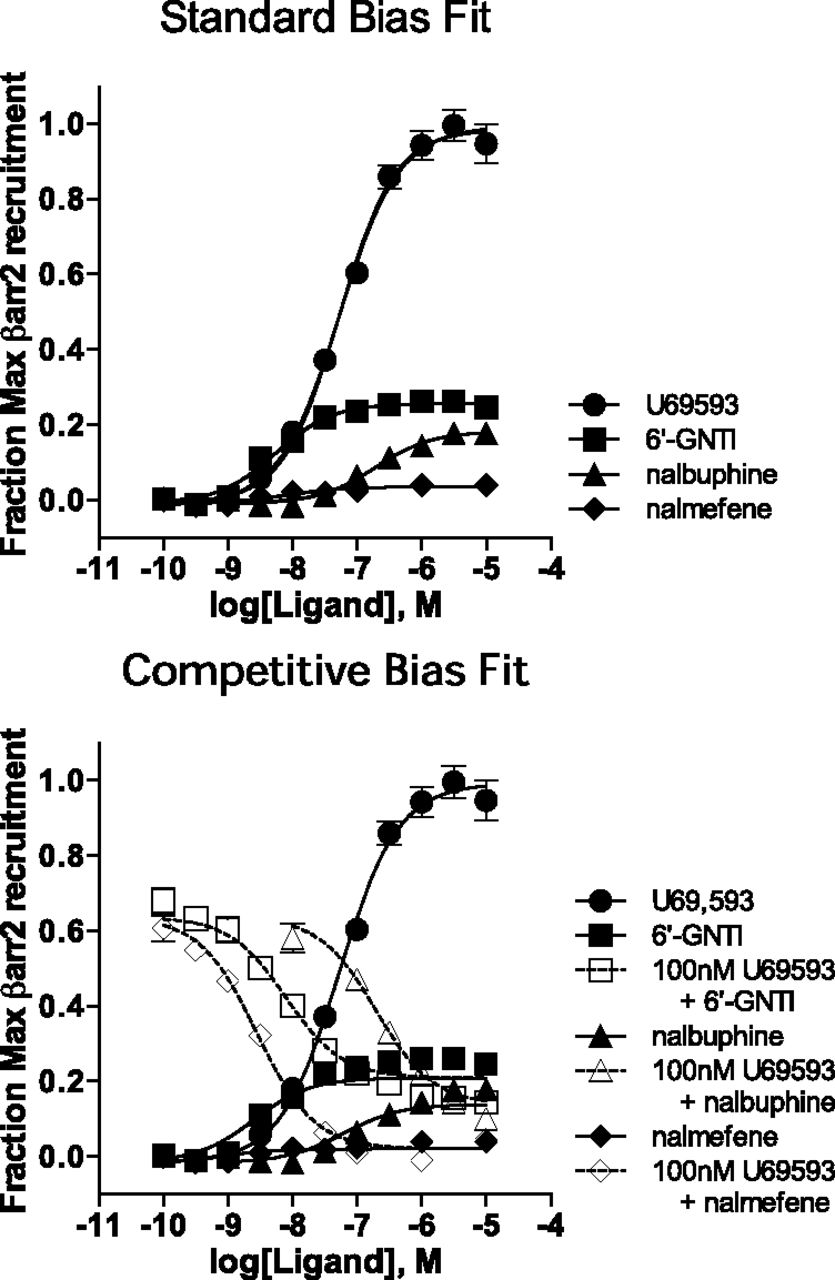
Stimulation of βarrestin2 recruitment to KOR is measured for U69,593 and each of the three test agonists. These graphs show the data analyzed using both the standard and competitive methods. For each data set, both equations produced good fits of the data. In the case of the competitive method, the inhibition curves are fit to a top equal to the response produced by 100 nM U69,593. Each data point in the curve is the mean ± S.E.M. of at least three independent experiments.
To model the data for the competitive fit, both stimulatory and inhibitory curves are produced for 6′-GNTI, nalmefene, and nalbuphine using 100 nM U69,593 for the competitive curve analyses. The fit of the competitive method is shown in the lower panel of Fig. 7. The resulting estimates for the LogRA and pKi (with 95% CI) from each method are presented together in Fig. 8 for comparison. (The values for each fit are also provided in Tables 1–4.) In each case, the competitive method produces a LogRA with less than or equal error compared with the standard method.
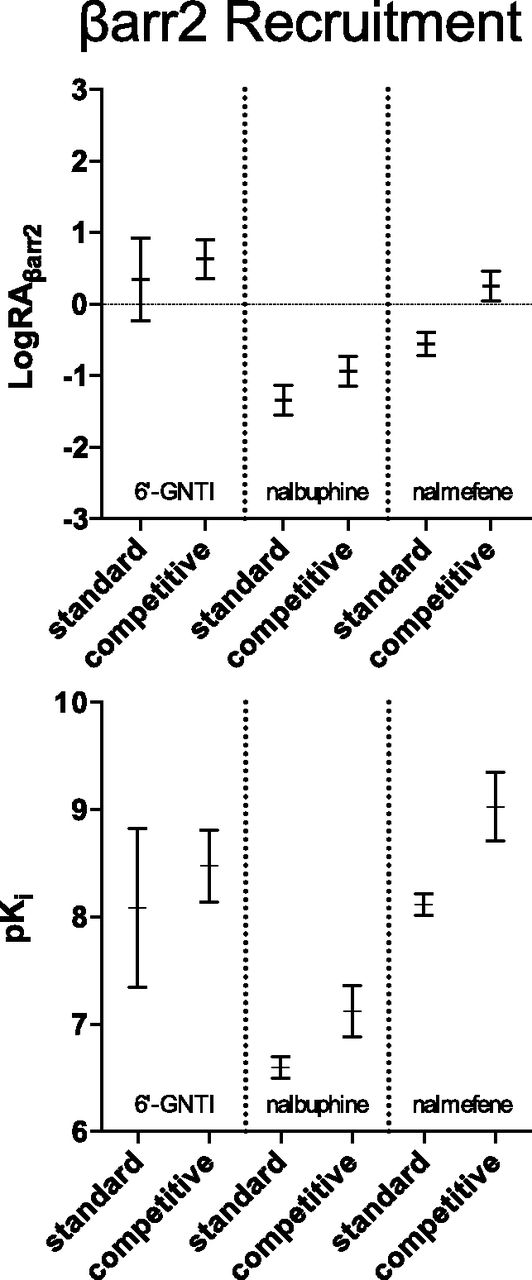
The competitive and standard methods of analysis produce pronounced differences in the estimates of LogRA and pKi for βarrestin2 recruitment. The graphs present the parameter estimates for each method as mean ± 95% confidence interval. The results of each individual experiment were pooled and averaged to produce the LogRA and pKi values for βarrestin2 recruitment in Tables 1–4. These graphs demonstrate that the competitive method of normalization produces 95% confidence intervals for LogRA that are similar to, or more narrow than, the standard fit.
It is important to note that the lack of increase in error for the LogRA values supports the conclusion that there is little downside risk (of greater error) to employing the competitive method when analyzing agonist bias. For the predicted pKi values, there is error reduction for 6′-GNTI, but there is a slight increase in error for nalbuphine and nalmefene. Although the widths of the pKi estimates do increase for both nalbuphine and nalmefene, Fig. 8 demonstrates that the predicted 95% CIs for these agonists do not overlap or even touch. This indicates that the affinity values of nalbuphine and nalmefene, predicted by the competitive method, lie in a region where the standard method predicts the true affinity of the population would be found by chance 5% of the time. This striking difference between the methods suggests that the estimates of LogRA, produced by the competitive method, are substantially more accurate than those estimates resulting from the standard method.
The Competitive Method May Assist in Establishment of Bias for Some Partial Agonists.
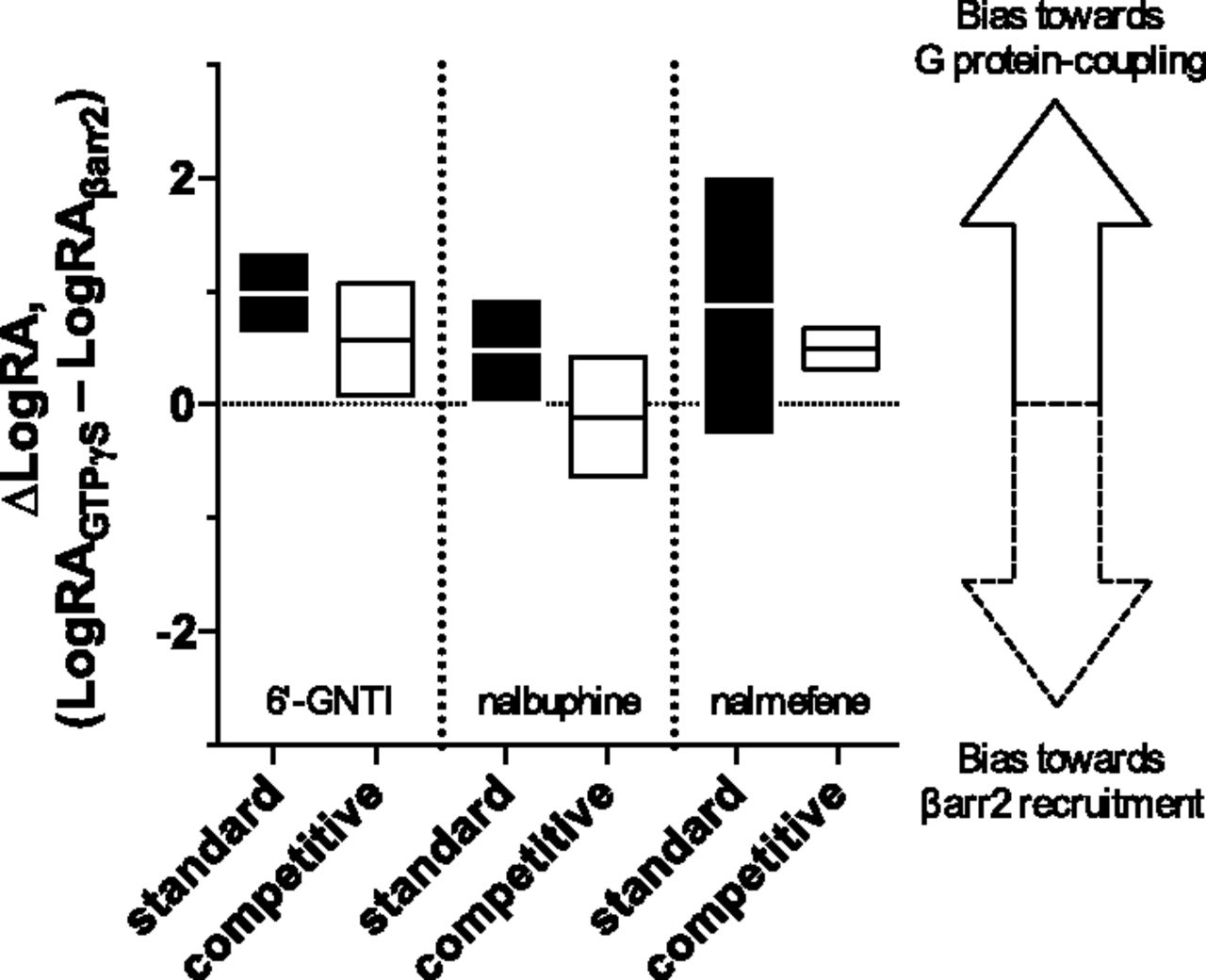
The ∆LogRA values for each method are presented in Fig. 9 (and Tables 5 and 6) and, in some cases, the two methods produce quite different bias predictions. In the case of 6′-GNTI, both methods indicate a bias toward G protein coupling. The previous estimate of ∆LogRA for 6′-GNTI was calculated, using the standard method, as 0.8; this resulted in a bias factor of 5.82 (the bias factor was calculated as the antilog of ∆LogRA in Schmid et al. (2013)). The results of the standard and competitive method are both in agreement with this conclusion. Specifically, we found that the ∆LogRA value of 6′-GNTI is 0.6 (bias factor of 4.0) using the competitive method and 1.0 (bias factor of 10) using the standard method. Moreover, in both methods, the 95% CI of ∆LogRA does not overlap zero indicating that the agonist is biased toward G protein coupling.

The bias factors derived from the standard model and competitive model for each test agonist are presented as ∆LogRA. The bars define the 95% confidence interval of the ∆LogRA determination, and the midline defines the mean, for each agonist produced by the two methods of analysis. As in Fig. 3, a bar overlapping 0 defines an unbiased agonist, and a bar that does not touch 0 is a biased agonist. The distance of the mean from 0 defines the degree of bias for G protein coupling or βarrestin recruitment. Calculating the difference-of-mean of the LogRA, according to eq. 5, results in the ∆LogRA estimate for each method. The standard method and the competitive method are calculated separately, and the figure presents the values in Tables 5 and 6. The results of the competitive method produce a decrease in the width of the confidence interval along with a change in the mean. The disparity between these intervals provides a striking difference in the conclusion of bias for the test agonists.
∆LogRA values from the standard fit
∆LogRA values from the competitive fit
For nalbuphine and nalmefene, the standard fit produces ∆LogRA values that suggest nalbuphine may be slightly biased toward G protein coupling and nalmefene is unbiased. That is, consideration of the 95% CI of the bias factors reveals greater uncertainty is obtained with the standard method than the competitive method. Specifically, in the case of nalbuphine, the 95% CI in the competitive fit overlaps 0, indicating that there is no bias observed, whereas analysis using the standard model suggests this agonist is biased. More interestingly, the width of the 95% CI of the ∆LogRA exhibited by nalmefene appears to decrease by close to an order of magnitude from a ∆LogRA range of −0.28 to 2.04 for the standard fit to a range of 0.32 to 0.67 for the competitive fit. This reduction in the width of the 95% CI from the competitive fit supports the conclusion that nalmefene does exhibit a degree of bias toward G protein coupling similar to 6′-GNTI [6′-GNTI ∆LogRA = 0.58 (0.08–1.07); nalmefene ∆LogRA = 0.50 (0.32–0.67)]. Overall, these findings demonstrate that the bias estimates produced by the two methods can be considerably different and that, in some cases, the competitive method produces a large reduction in error for LogRA, pKi, and ∆LogRA when compared with the standard method.
Discussion
In this study, we introduce a modification of the operational model that can be used to improve confidence in the calculation of bias parameters in cases of extreme bias. Because it takes advantage of estimating affinities from the competitive nature of partial agonists, the model is referred to as the competitive model. Based on the simulations and the original data provided herein, we demonstrate that the use of the competitive model can improve the estimates of bias factors in cases where the agonist response approaches baseline. In these cases of minimally efficacious agonists, the competitive model can provide conclusive evidence of the bias of the ligand for or against a specific signaling pathway where the standard operational model is unable to resolve certain parameters and thus unable to confidently calculate ligand bias.
We have used three KOR ligands that are potent partial agonists with different degrees of efficacy. Nalmefene, which produces the least stimulation in each assay, acts as a potent competitive partial agonist and allows for a confident assessment of affinity in each assay. We demonstrate that, by measuring both agonism and competitive agonism, this increased confidence in the affinity estimate can be used to determine that nalmefene is a KOR agonist with bias for G protein signaling. However, as the confidence for estimating intrinsic efficacy over baseline increases, the benefit of using the competitive model is no longer apparent. This can be seen using 6′-GNTI, which produces a clear and distinct activation of response in both assays that can easily be resolved from the baseline. Although the competitive model does not confer a distinct benefit over the standard model for 6′-GNTI, both the standard and competitive methods provide estimates of 6′-GNTI bias toward G protein signaling that are in agreement with previous studies (Rives et al., 2012; Schmid et al., 2013). In this case, the data obtained by calculating relative affinity using the competitive analysis does not further resolve the estimation of bias; moreover, this approach may actually introduce error into the analysis, as evidenced by an increase in the width of the 95% CIs. This finding suggests that fitting to the standard method produces a confident estimate of bias if the test agonist exhibits significant agonist activity in both responses (as previously suggested by Kenakin et al., 2012).
In this study, we have used the βarrestin2 recruitment assay based on enzyme fragment complementation. A hallmark of this system is that the receptors are generally highly overexpressed. These cell lines have been optimized for high-throughput screening and produce very robust responses. In a sense, the choice of this assay may “bias” the signaling response to seem more robust in the βarrestin2 assay. Nonetheless, the estimate of RAi is unaffected by receptor expression because this parameter is ultimately a relative estimate of affinity for the active receptor state.
Another issue relates to the assumption of a logistic dependence of arrestin recruitment on receptor occupancy (operation model) when the relationship is more likely to be a proportional one (Bohn and McDonald, 2010). Although applying the operational model in the latter case may introduce an unnecessary complication, Griffin et al. (2007) demonstrated that the operational model and a null method produce equivalent estimates of relative intrinsic activity. Therefore, although the operational model may incorrectly assume the occupancy-response relationship, it will produce an accurate measure of RAi regardless of this relationship.
The benefit of using the competitive analysis is evident when the ligand exhibits minimal agonism in a system (i.e., approach baseline). The estimate of ∆LogRA produced for nalbuphine using the standard model appears to be biased, although the 95% CI comes very close to overlapping zero (Table 5). However, the estimate of intrinsic activity provided by the competitive model supports the conclusion that this agonist is not biased. Whether the estimation of efficacy captured in these cell-based assays is predictive of the activity of the ligands in vivo remains to be determined. However, the information gained by applying the competitive method of analysis may be very helpful in binning and prioritizing ligands in drug development efforts, particularly when it is difficult to distinguish agonism over the baseline response.
The quantification of bias begins by recognizing that the product of intrinsic efficacy and affinity of a (test) agonist can always be estimated relative to the same efficacy:affinity product observed for a standard agonist. This estimate is known as intrinsic relative activity (RAi). (In this article, the RAi is expressed as the logarithm of the RAi value (denoted LogRA) to facilitate comparison between responses.) To use this information, it is only necessary to produce a “measurement of the concentration-response curve” of each agonist (Ehlert et al., 1999; Griffin et al., 2007). This ability provides a useful means of comparing agonist activity without the need to directly access the affinity of the agonist or the degree of receptor reserve present in a system. Specifically, it provides a relative measure of the active state affinity constant of the agonist for the particular signaling pathway under investigation (Tran et al., 2009).
Agonist activity can be addressed at different hierarchical levels relating to the output of single receptors (level 3), a population of receptors (level 2), and a response downstream in the signaling pathway (level 1). At a given level, drug action is defined by unique pharmacologic parameters, which can be expressed mathematically in terms of the next deeper level parameters (Tran et al., 2009). For example, the EC50 and Emax values of agonists (level 1 parameters) can each be expressed in terms of observed affinity and intrinsic efficacy (level 2 parameters). Analogously, the observed affinity and efficacy values of an agonist can be expressed in terms the affinity and isomerization constants of receptor states (level 3 parameters). Ultimately, the parameters of receptor states are independent parameters for drug receptor interactions.
There are implications of these relationships with regard to the estimation of RAi. Depending upon the available information, it may be impossible to estimate the observed affinity (KTest) and τTest values, or perhaps even the product (τTest ∙ KTest), from the concentration-response curve of an agonist. Nonetheless, it is always possible to estimate the RAi value of a test agonist, expressed relative to a reference agonist, because this parameter is equivalent to the corresponding ratio of active state affinity constants, which are independent parameters, unlike the K and τ values. Thus, if the operational model is used to analyze concentration-response curves, the observed affinity may be constrained to different values causing an inverse effect on the estimate of τ. This inverse relationship between the estimates of KA and τ results in no change in the estimate of RAi (Griffin et al., 2007). If it is known that the reference agonist is a full agonist, then it is possible to obtain a reasonable estimate of the product of τ ⋅ K of the test and reference agonists, even though the τ or K values may not be well defined.
This theory is relevant to a recent discussion in the literature regarding two methods of quantifying agonist bias (Kenakin and Christopoulos, 2013a; Rajagopal, 2013). Both methods produce a measure of the RAi of each test agonist by normalizing to a full reference agonist. This approach was elaborated by Griffin et al. (2007) and uses the operational model (Black and Leff, 1983). In one method (which we refer to as the standard method), the bias of an agonist is defined as the change in its RAi value (∆∆LogR) between assays (Kenakin et al., 2012; Kenakin and Christopoulos, 2013b). A slightly different adaptation proposed by Rajagopal et al. (2011) employs radioligand-binding studies to estimate the observed dissociation constant of each test agonist. The equilibrium dissociation constant is assumed to be the same for every measure of response. And, for this reason, the dissociation constant is then used to constrain the fit of the operational model and produce estimates of each agonist’s intrinsic efficacy. The difference between the intrinsic efficacy of each test ligand and the reference agonist is determined in each assay (eq. 3; LogRA), and the difference of differences is determined for the two assays (eq. 4; ∆LogRA). Aside from the scaling factor (√2) used by Rajagopal et al. (2011) to produce β-lig (β-lig is defined as a direct comparison of the ratio of efficacy of the test agonist to the reference agonist in the two systems), both methods yield similar estimates of bias because of the independent nature of RAi and the inverse correlation between estimates of K and τ described earlier.
One of the major differences between the two methods of calculating bias is the way the ligand affinity is evaluated when fitting with the model (reviewed in Kenakin and Christopoulos, 2013b). In the standard method (Kenakin et al., 2012), the affinity estimate for each test agonist is produced by assuming that the test agonists are partial compared with the reference agonist. In this method, the affinity of the ligand is a variable parameter value that is fit by the model instead of an accessible parameter to be measured in each response. The unconstrained nature of the affinity in this method permits it to take a dynamic role in facilitating the fit instead of providing a useful constraint for producing an appropriate measure of bias. Conversely, the method used by Rajagopal et al. (2011) assumes that the affinity of the test ligand is constant and directly correlates with the results of radioligand binding studies. However, this assumption may introduce error into the estimate of the test ligand’s coupling efficacy. Specifically, it was demonstrated that ligands may have different affinities for the same receptor in different measures of response (Berg et al., 1998; Perez and Karnik, 2005). Although each method of bias analysis may yield different estimates of affinity and efficacy, the error in the estimates of these parameters are correlated and hence have little effect on the bias estimate so long as the potency and maximal response of the test agonist are adequately defined.
The competitive method we present in this article incorporates assumptions from each of the two other methods. Specifically, the competitive method treats the test agonist’s affinity as a finite and accessible parameter value. The estimate produced by the competitive fit is based on both the stimulatory and inhibitory curves generated with each test agonist. This estimate permits the affinity value to differ between responses (an assumption used by the comparison of transduction coefficients). However, the agonist’s affinity value in each assay is defined by, and must satisfy, the predicted occupancy function produced by the inhibition curves (fundamentally equivalent to the affinity value used to calculate the βlig, and determined by competition of radioligand binding). In this way, the competitive fit produces the most predictive estimate of the test agonist’s EC50 in each system. In the case of nalbuphine and nalmefene, the EC50 of the response curve was not well defined in each assay (Figs. 7 and 8) because each of these agonists produces a marginal stimulation for βarrestin2 recruitment that approaches baseline measures. We show that by experimentally determining an appropriate estimate of the IC50 of each of these compounds, the competitive method can produce highly accurate estimates of the bias of each agonist. This is compelling evidence to support the conclusion that, in cases of very partial agonism, accurately estimating the affinity of a partial agonist can drastically improve the estimate of bias.
One of the most notable advantages of the competitive method is the large reduction of error produced for the parameter values. This is particularly interesting in cases where the test agonist demonstrates extreme bias. For agonists with extreme bias, an accurate estimate of occupancy provides invaluable confirmation that the agonist has retained affinity for the receptor in each response. (The currently available methods of bias analysis have not been optimized to verify the position of the response curve for extremely biased ligands [Kenakin, 2011].) It is also possible that comparison of affinity values between responses could lead to the discovery of multiple, highly distinct, active states. In fact, it may be appropriate to compare in the 3-state or n-state models (Kew et al., 1996; Leff et al., 1997).
In summary, we have presented a modification of the operational model that has utility for confidently assessing bias in cases of extreme partial agonism. This proves to be particularly useful in the cases of apparent “extreme bias” wherein the response profile appears to be flat. It is reasonable to suggest that this newly described route to accessing the affinity of biased ligands may lead to new uses for this parameter in receptor modeling, including the means to assess the relative potential for antagonism across assays. Furthermore, the competitive method produces a marked benefit when estimating LogRA and pKi as well as a reduction in the error propagated to the ∆LogRA. From these findings, we conclude that for extremely partial agonists this competitive method provides a conservative yet highly relevant means of demonstrating agonist bias. Additionally, based on the establishment of occupancy, we conclude that the calculated bias of the agonist produced by the competitive model is an innate property of the ligand-receptor complex and is not an artifact imposed by a limit of response amplification or threshold.
Authorship Contributions
Participated in research design: Stahl, Zhou, Bohn.
Conducted experiments: Stahl, Zhou.
Contributed new reagents or analytic tools: Stahl, Ehlert.
Performed data analysis: Stahl.
Wrote or contributed to the writing of the manuscript: Stahl, Zhou, Ehlert, Bohn.
Footnotes
- Received October 21, 2014.
- Accepted February 13, 2015.
This work was supported by a grant from the National Institutes of Health National Institute on Drug Addiction [Grant R01-DA031927].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- CHO
- Chinese hamster ovary
- CI
- confidence interval
- DMSO
- dimethylsulfoxide
- 6′-GNTI
- 6′-guanidinonaltrindole
- KOR
- κ opioid receptor
- [35S]GTPγS
- guanosine 5′-O-(3-[35S]thio)triphosphate
- U69,593
- (+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}