


Abstract
Cytochrome P-450 3A4 (CYP3A4), the predominant cytochrome P-450 expressed in adult human liver, is subject to transcriptional induction by a variety of structurally unrelated xenobiotics, including the antibiotic rifampicin. The molecular mechanisms underlying this phenomenon are poorly understood. We transfected a human liver-derived cell line (HepG2) with various CYP3A4-luciferase reporter gene constructs containing a nested set of 5′-deletions of theCYP3A4 5′-flanking region. Rifampicin-inducible transcription of the reporter gene was observed only with the longest construct, which encompassed bases −13000 to +53 ofCYP3A4 (3-fold induction). The responsive region was functional regardless of its position or orientation relative to the proximal promoter of CYP3A4 and was capable of conferring rifampicin-inducible expression on a heterologous promoter. Further deletion mutants localized the induction to bases −7836 to −7607. In vitro DNase I footprint analysis of this region revealed four protected sites (FP1, FP2, FP3, and FP4). Two of these sites, FP3 (bases −7738 to −7715) and FP4 (bases −7698 to −7682), overlapped binding motifs for the orphan human pregnane X receptor (hPXR). Cotransfection of responsive constructs with a hPXR expression vector substantially increased the rifampicin-inducibility to ∼50-fold. In addition, the rifampicin-responsive constructs were strongly activated by a range of CYP3A inducers. Finally, we demonstrate cooperativity between elements within the distal enhancer region andcis-acting elements in the proximal promoter ofCYP3A4. Our results provide evidence for the existence of a potent enhancer module, 8 kb distal to the transcription start point, which mediates the transcriptional induction ofCYP3A4 by activators of hPXR.
Cytochromes P-450 (P-450) are a superfamily of hemoproteins that play a pivotal role in the oxidative metabolism of numerous endogenous and exogenous compounds (Nelson et al., 1996). The human P-450 3A subfamily contains three functional members: CYP3A4, CYP3A5, and CYP3A7. CYP3A4 is the predominant isoform expressed in adult human liver and is reported to be responsible for the metabolism of more than 60% of therapeutic drugs (Li et al., 1995). In addition, this enzyme is the primary catalyst of steroid 6β-hydroxylation (Waxman et al., 1991) and therefore has a central role in steroid hormone homeostasis. CYP3A4 is also involved in the bioactivation of anticancer drugs and environmental procarcinogens, including benzo(a)pyrene and other dihydrodiol derivatives of polycyclic aromatic hydrocarbons and carcinogenic mycotoxins (Li et al., 1995). Thus, CYP3A4 is considered a key enzyme in chemical carcinogenesis in both the liver and extrahepatic tissues.
CYP3A4 is transcriptionally regulated by a variety of hormones, including glucocorticoids, growth hormone, and triiodothyronine (Schuetz et al., 1993; Liddle et al., 1998), and xenobiotics such as phenobarbital, clotrimazole, mifepristone (RU486), and rifampicin (Daujat et al., 1991; Schuetz et al., 1993; Kocarek et al., 1995). Rifampicin, a macrocyclic antibiotic, is known to be one of the most potent inducers of CYP3A4 expression both in vivo and in cultured hepatocytes (Schuetz et al., 1993; Kocarek et al., 1995;Michalets, 1998). Induction of CYP3A4 expression by rifampicin and other xenobiotics underlies many reported drug interactions and is of considerable importance for patients subject to combination drug therapy such as for HIV/AIDS (Michalets, 1998).
Although the nucleotide sequence of the proximal 5′-flanking region ofCYP3A4 has been reported (Hashimoto et al., 1993), the molecular mechanisms underlying the transcriptional regulation of this gene have yet to be elucidated. The proximal promoter of theCYP3A4 gene (bases −172 to −149) contains two copies of an AG(G/T)TCA hexamer, the recognition sequence for the nuclear receptor family of transcription factors (Mangelsdorf et al., 1995). Barwick et al. (1996) demonstrated that these half-sites, organized as an ER-6 (everted repeat separated by six nucleotides), conferred rifampicin-responsiveness on heterologous reporter gene constructs when transfected into rabbit but not rat hepatocytes. These observations led to the hypothesis that the host cellular environment, rather than the structure of the gene, is responsible for the well-documented interspecies differences in CYP3A inducibility (Wrighton et al., 1985; Kocarek et al., 1995). Importantly, the magnitude of the rifampicin induction in this study (2- to 4-fold) was considerably less than would be expected from either the endogenous rabbitCYP3A6 gene or the human CYP3A4 gene (Daujat et al., 1991; Kocarek et al., 1995).
Recently, a number of studies have led to the isolation of a human orphan nuclear receptor, the pregnane X receptor [hPXR (NR1I2)]. This versatile receptor forms a heterodimer with the 9-cisretinoic acid receptor [RXR (NR2B1)] and is capable of promoting transcription of heterologous reporter gene constructs containing multimerized copies of the proximal ER-6 element of CYP3A4. The hPXR is activated by a wide variety of lipophilic compounds, including CYP3A inducers (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998). Interestingly, hPXR and mPXR (mouse PXR) exhibit divergent activation profiles. Thus, although both hPXR and mPXR are effectively activated by dexamethasonet-butylacetate, RU486, corticosterone, and 5β-pregnane-3,20-dione, hPXR but not mPXR is activated by rifampicin, clotrimazole, coumestrol, and diethylstilbestrol. Conversely, pregnenolone 16α-carbonitrile (PCN), dexamethasone, and 17α-hydroxypregnenolone are efficacious activators of mPXR but not of hPXR (Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998). The pharmacology of mPXR and hPXR activation correlates well with species-specific CYP3A induction profiles. The existence of a broad-specificity, low-affinity receptor acting as a sensor for hormones and xenobiotics and modulating their metabolism by transcriptionally activating P-450 genes is an attractive model (Blumberg et al., 1998).
In view of the relatively modest activation by rifampicin of reporter gene constructs containing the proximal PXR response element (prPXRE) alone (Barwick et al., 1996; Ogg et al., 1999), we attempted to define further regulatory regions of the CYP3A4 gene involved in transcriptional activation by xenobiotics. Here we report the isolation and characterization of a distal xenobiotic-responsive enhancer module (XREM) that mediates transactivation of the CYP3A4 gene by inducers that are hPXR activators. Moreover, we demonstrate cooperativity between response elements in the distal XREM and proximal promoter regions of CYP3A4. This study clearly establishes that hPXR is the major transactivating factor responsible for the induction of CYP3A4 by xenobiotics.
Experimental Procedures
Materials.
DNA-modifying enzymes, unless otherwise stated, were obtained from Boehringer-Mannheim Australia (Sydney, Australia). [γ-32P]dATP, [α-32P]dCTP (>3000 Ci/mmol), and Megaprime random prime DNA labeling kits were obtained from Amersham Pharmacia Biotech (Buckinghamshire, England). Cell culture media, fetal bovine serum, and media supplements were obtained from GIBCO BRL (Grand Island, NY). PCN and RU486 were provided by BIOMOL Research Laboratories (Plymouth Meeting, PA). Clotrimazole was purchased from Calbiochem-Novabiochem (San Diego, CA). All other chemicals, including rifampicin, were obtained from Sigma Chemical Co. (St. Louis, MO). Oligonucleotides were prepared by Bresatec (Adelaide, Australia).
Isolation of CYP3A4 Genomic Clone.
Two clones, encompassing bases +53 to −3099 of the CYP3A45′-flanking region, were generated from a commercially available system that facilitates the amplification of unknown DNA adjacent to known DNA sequence (PromoterFinder; Clontech Laboratories, Palo Alto, CA). These fragments were radioactively labeled with [α-32P]dCTP by random priming and used to screen a human genomic library contained in the pWE15 cosmid vector (Stratagene, La Jolla, CA), according to the manufacturer's instructions. A single positive clone (p3A4-C1) was isolated, and confirmatory sequencing was performed. This clone was used to generate all subsequent deletion mutants. The partial nucleotide sequence of p3A4-C1 (bases −10468 to +906 of CYP3A4) has been deposited with the GenBank/EMBL/DDBJ databases under accession numberAF185589. Putative regulatory motifs in the XREM region were identified manually and by screening the TRANSFAC database (http://transfac.gbf.de/TRANSFAC) with use of the PatSearch and MatInspector programs (Quandt et al., 1995).
Plasmid Constructs.
Chimeric CYP3A4 luciferase reporter plasmids were prepared as follows. First, a 1.13-kb fragment ofCYP3A4, encompassing bases −1084 to +53, was amplified by polymerase chain reaction (PCR) using the oligonucleotides CYP3A4UP1 (5′-CATTGCTGGCTGAGGTGGTT-3′; sense, bases −1084 to −1065) and CYP3A4GSP2 (5′-catggatccTGTTGCTCTTTGCTGGGCTATGTGC-3′; antisense, bases +29 to +53), creating a BamHI restriction site at the 3′-end. Construct p3A4-362 was prepared by digesting this 1.13-kb amplicon with BglII and BamHI and cloning the resultant 415-bp fragment (bases −362 to +53) into theBglII site of pGL3-Basic, a promoter-less luciferase reporter vector (Promega, Madison, WI), destroying the 3′-BglII site. Confirmatory sequencing was performed, and the fragment was found to be identical with the published sequence (Hashimoto et al., 1993). Second, a 1.89-kbKpnI/BglII fragment of p3A4-C1 (bases −3195 to −1310) was inserted into KpnI/BglII-digested p3A4-362. This construct was linearized with BglII, and a 948-bp BglII fragment of p3A4-C1 (bases −1310 to −362) was inserted, creating p3A4-3195. Third, p3A4-13000 was prepared by cloning a 10-kb KpnI fragment (bases −13000 to −3195) of p3A4-C1 into p3A4-3195 linearized with KpnI. The remaining clones illustrated in Fig. 1 were prepared according to standard techniques (Sambrook et al., 1989) with the restriction sites indicated. Enhancer deletion mutants were generated by inserting BamHI or BglII fragments of p3A4-C1 (encompassing bases −12.6 to −6 kb) into the BamHI site downstream of the luciferase reporter gene in the p3A4-1200 clone (Fig.3)

Identification of a rifampicin-responsive region in the 5′-flank of the CYP3A4 gene. ChimericCYP3A4-luciferase (Luc) reporter gene constructs were generated as described in Materials and Methods and transiently transfected into HepG2 cells. The position relative to the transcription initiation site (Hashimoto et al., 1993) and the restriction enzymes used in the generation of these constructs are indicated. Transfected cells were treated with rifampicin (5 μM) for 48 to 60 h before harvest and determination of luciferase and β-galactosidase activities on cell lysates. Control cultures received vehicle alone (0.1% DMSO). Data represent the mean ± S.D. from at least three independent experiments performed in triplicate. Luciferase values are normalized to β-galactosidase and expressed as percentage of control cells.

The rifampicin-responsive region functions regardless of orientation or position relative to the proximal promoter ofCYP3A4. Enhancer constructs were prepared as outlined inMaterials and Methods. Constructs were transfected into HepG2 cells, which were subsequently cultured in the presence of rifampicin (5 μM) or vehicle (0.1% DMSO) for 48 h before harvest. Data are derived exactly as described in legend to Fig. 1.
Constructs p3A4-362(7836/7208ins), p3A4-362(7797/7208ins), p3A4-362(7686/7208ins), and p3A4-362(7607/7208ins) were generated by PCR. The 5′-position of sense and antisense primers is delineated in the construct name. All PCR products were verified by sequencing. Each primer contained either an XbaI or a BamHI restriction site, facilitating cloning of the amplicon into theNheI or BglII sites in the multiple cloning site of p3A4-362. Site-directed mutagenesis of putative PXR response elements (PXREs) was performed on the p3A4-362(7836/7202ins) construct using the Transformer mutagenesis system (Clontech) with the following primers: dNR1 mut (bases −7746 to −7714, 5′-GATCTCAGCTGAAaGcAtTTGCTGACCCTCTGC-3′), dNR2 mut (bases −7697 to −7665, 5′-GGTGCCCTTGttATCATGTCGGaaCAAGCAGCC-3′), dNR3 mut (bases −7291 to −7258, 5′-AATATATTGTTATaGcAtTATCAAAGCCTTTTCC-3′), and prPXREmut (bases −181 to −144, 5′-CCTCATAGAATATGttCTCAAAGGAGaaCAGTGAGTGG-3′). Mutated constructs were sequenced and verified to be free of nonspecific base changes.
pCMVβ, an expression vector containing the β-galactosidase cDNA under the control of human cytomegalovirus promoter and enhancer, was obtained from Clontech. pGEM3Zf(+) was purchased from Promega. The hPXR expression vector pSG5-hPXRΔATG, p(ER6)3-tk-Luc control plasmid (Lehmann et al., 1998), and human 9-cisretinoic acid receptor-α expression vector (pSG5-hRXRα) were generously provided by Steven A. Kliewer (Glaxo Wellcome Research and Development, Research Triangle Park, NC). The human glucocorticoid receptor expression vector (hGR) and the p(GRE)2-tk-Luc control plasmid, which contains dimerized glucocorticoid response elements (GREs) from the rat tyrosine aminotransferase gene linked to a minimal thymidine kinase (tk) promoter and luciferase reporter gene, were a gift from Jan Carlstedt-Duke (Karolinska Institute, Stockholm, Sweden). All DNA used in temporary transfection studies was purified using an anionic-exchange resin (QIAGEN, Clifton Hill, Australia).
Cell Culture and Transfections.
The human hepatocellular carcinoma cell line HepG2 was obtained from the American Type Culture Collection (Rockville, MD) and cultured in antibiotic-free Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Murine fibroblast, NIH3T3, and simian kidney (COS7) cells were maintained under similar conditions. Cells (3 × 105) were inoculated onto 6-well plates (Nunc A/S, Roskilde, Denmark) 24 h before transfection with the cationic liposomesN-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate or FuGene-6 (Boehringer-Mannheim Australia) according to manufacturer's instructions. Transfection with 2 μg of luciferase reporter gene constructs, 0.8 μg of pCMVβ, and 0.1 μg of expression vector, when included, was allowed to proceed for 5 h in serum-free medium. The pSG5 expression vector (Stratagene) was used in control cotransfection experiments. Subsequently, cells were cultured for an additional 48 to 60 h in fresh medium supplemented with 10% fetal bovine serum in the presence of various inducers (see figure legends), added as a 1000× stock solution in dimethyl sulfoxide (DMSO). Control cultures received vehicle (0.1% DMSO) alone. Luciferase activities were determined on cell lysates using a commercially available assay system (Promega). β-Galactosidase assays were performed as described elsewhere (Foster et al., 1988).
Preparation of Nuclear Extracts and In Vitro DNase I Footprinting.
Nuclear extracts were prepared from adult male Wistar rats (220–250 g) according to the method of Gorski et al. (1986). All buffers were supplemented with 0.4 mM sodium orthovanadate, 1 mM sodium fluoride, 0.5 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 2 μg/ml aprotinin, and 1 mM dithiothreitol shortly before use. For in vitro DNase I footprint analysis, a 269-bp fragment encompassing bases −7836 to −7568 ofCYP3A4 was asymmetrically 5′-end-labeled with [γ-32P]CTP by PCR as outlined elsewhere (Krummel et al., 1990). The protein/DNA binding reaction contained the following components in a volume of 50 μl: 25 mM HEPES, pH 7.6, 60 mM KCl, 10% glycerol, 0.1 mM EDTA, 2 μg of poly(dI/dC), 5 mM MgCl2, 1 mM dithiothreitol, and 3 to 25 μg of nuclear protein extract. The binding reaction was incubated on ice for 10 min before the addition of radiolabeled probe (∼12,000 cpm) and allowed to proceed for am additional 80 min at the same temperature. DNase I (0.005–0.1 U, DPRF grade; Worthington Biochemical Corporation, Lakewood, NJ) was added to the binding reaction in 50 μl of 25 mM HEPES, pH 7.6, 60 mM KCl, 10% glycerol, 5 mM MgCl2, and 5 mM CaCl2. After digestion at room temperature for 2 min, samples were processed as previously described (Lichtsteiner et al., 1987). Purine-specific (G + A) sequence ladders were prepared according to the Maxam-Gilbert chemical sequencing method (Maxam and Gilbert, 1980).
Electrophoretic Mobility Shift Assay.
Electrophoretic mobility shift assays (EMSAs) were performed essentially as described elsewhere (Lehmann et al., 1998). hPXR and hRXRα were synthesized from pSG5-hPXRΔATG and pSG5-hRXRα expression vectors, respectively, using the TNT rabbit reticulocyte system (Promega). Unprogrammed lysate was prepared using the pSG5 expression vector (Stratagene). Typically, binding reactions contained 10 mM HEPES, pH 7.8, 60 mM KCl, 0.2% Nonidet P-40, 6% glycerol, 2 mM dithiothreitol, 2 μg of poly(dI/dC), and 1 μl each of synthesized hPXR or hRXRα. Control incubations received unprogrammed lysate alone. Reactions were preincubated on ice for 10 min before the addition of 32P-labeled double-stranded oligonucleotide probe (0.2 pmol). Samples were held on ice for an additional 20 min, and the protein/DNA complexes were resolved on a pre-electrophoresed 5% polyacrylamide gel in 0.5× TBE (45 mM Tris-borate, 1 mM EDTA) at room temperature. Gels were dried and autoradiographed at −70°C for 1 to 2 h. Oligonucleotides used as probes are indicated in Fig. 6A. Competitor oligonucleotides were added to the preincubation at 5-, 25-, and 75-fold molar excess.
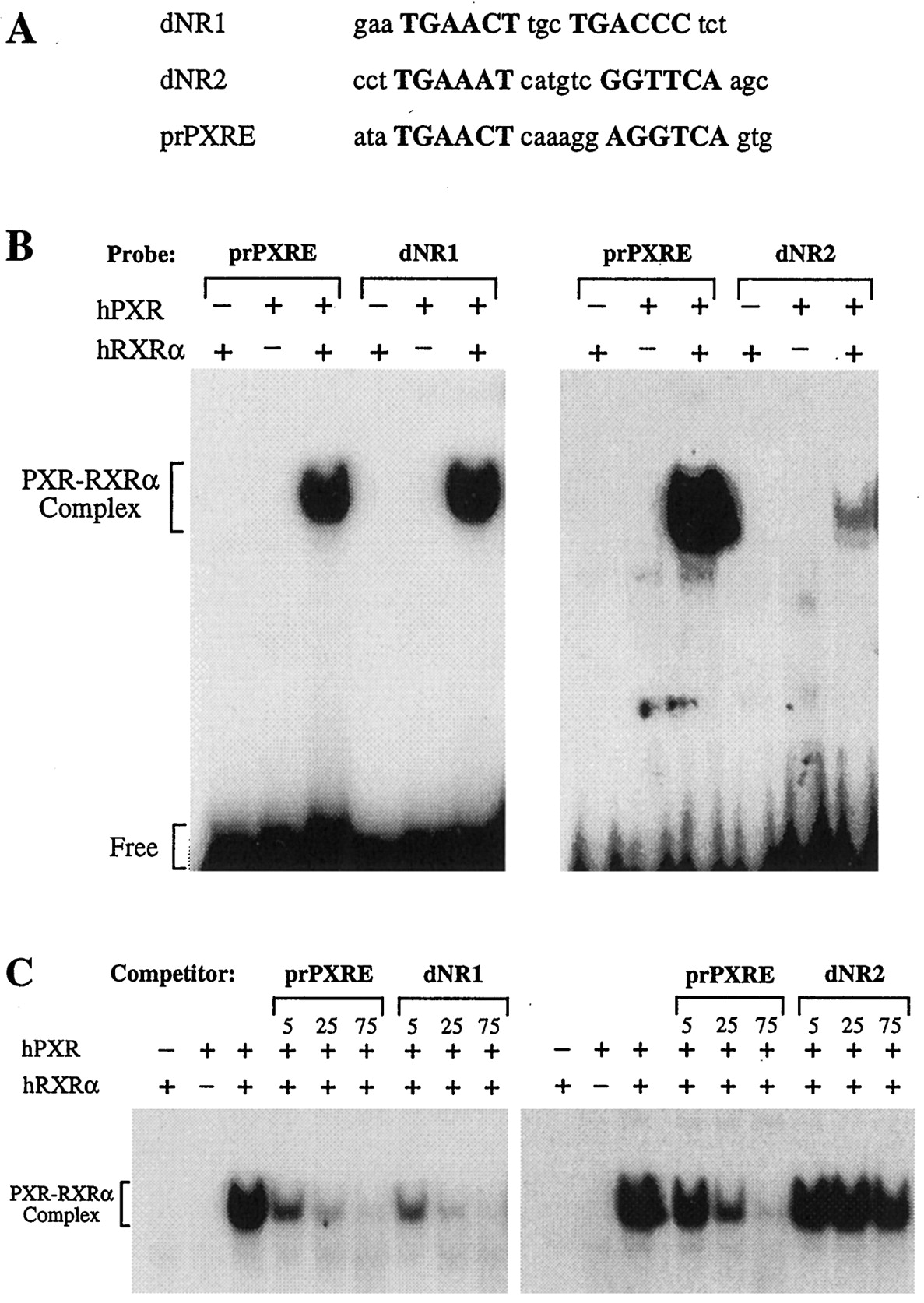
EMSA of putative PXREs. The ability of putative PXREs to bind hPXR-hRXRα was investigated with the use of EMSA. A, oligonucleotides were used as probes and competitors. Putative PXRE half-sites are shown in uppercase. B, EMSA with radiolabeled dNR1 (left) and dNR2 (right). The CYP3A4 prPXRE (ER-6) is included for reference. Incubations received recombinant hPXR, RXRα, or both as indicated. C, competition studies were performed using radiolabeled prPXRE as probe. Unlabeled competitor oligonucleotides corresponding to dNR1 (left) and dNR2 (right) were added at 5-, 25-, and 75-fold molar excess.
Results
Functional Analysis of CYP3A4 5′-Flanking Region.
A single genomic clone (bases −20 kb to +15 kb of CYP3A4, approximately) was isolated from a cosmid library using two probes corresponding to bases −1084 to +53 and −3099 to −1062. An 11.4-kbBamHI fragment of p3A4-C1 encompassing bases −10468 to +906 of CYP3A4 was subcloned into pGEM-3Zf(+), and the nucleotide sequence was derived. Nucleotides +14 to +174, corresponding to exon 1, were identical with the reported cDNA sequence (accession number M18907). Comparison with the previously published sequence of the CYP3A4 5′-flanking region (bases −1105 to +175) revealed 5-bp mismatches localized between bases −725 and −714 (Hashimoto et al., 1993). No further differences were observed. We have not determined the functional significance, if any, of these base changes.
To analyze the transcriptional regulation of CYP3A4 by rifampicin, deletion mutants encompassing bases −13000 to +53 ofCYP3A4 attached to a luciferase reporter gene were constructed (Fig. 1) and transiently transfected into HepG2 cells. Induction of endogenous CYP3A genes in rifampicin-treated HepG2 cells has been previously demonstrated (Schuetz et al., 1993;Sumida et al., 1999). In line with observations by others (Hashimoto et al., 1993), the basal activity of all CYP3A4-luciferase reporter gene constructs was low (data not shown). Rifampicin treatment of cells transfected with constructs containing bases −3195 to +53 failed to induce luciferase reporter gene expression (Fig. 1). However, treatment of cells containing the p3A4-13000 construct (bases −13000 to +53) resulted in a 3-fold induction of reporter gene activity (Figs.1 and 2, A and B). Additional deletion mutants demonstrated that removal of an XbaI/SpeI fragment, encompassing bases −7836 to −7493, destroyed the rifampicin response (Fig. 1).
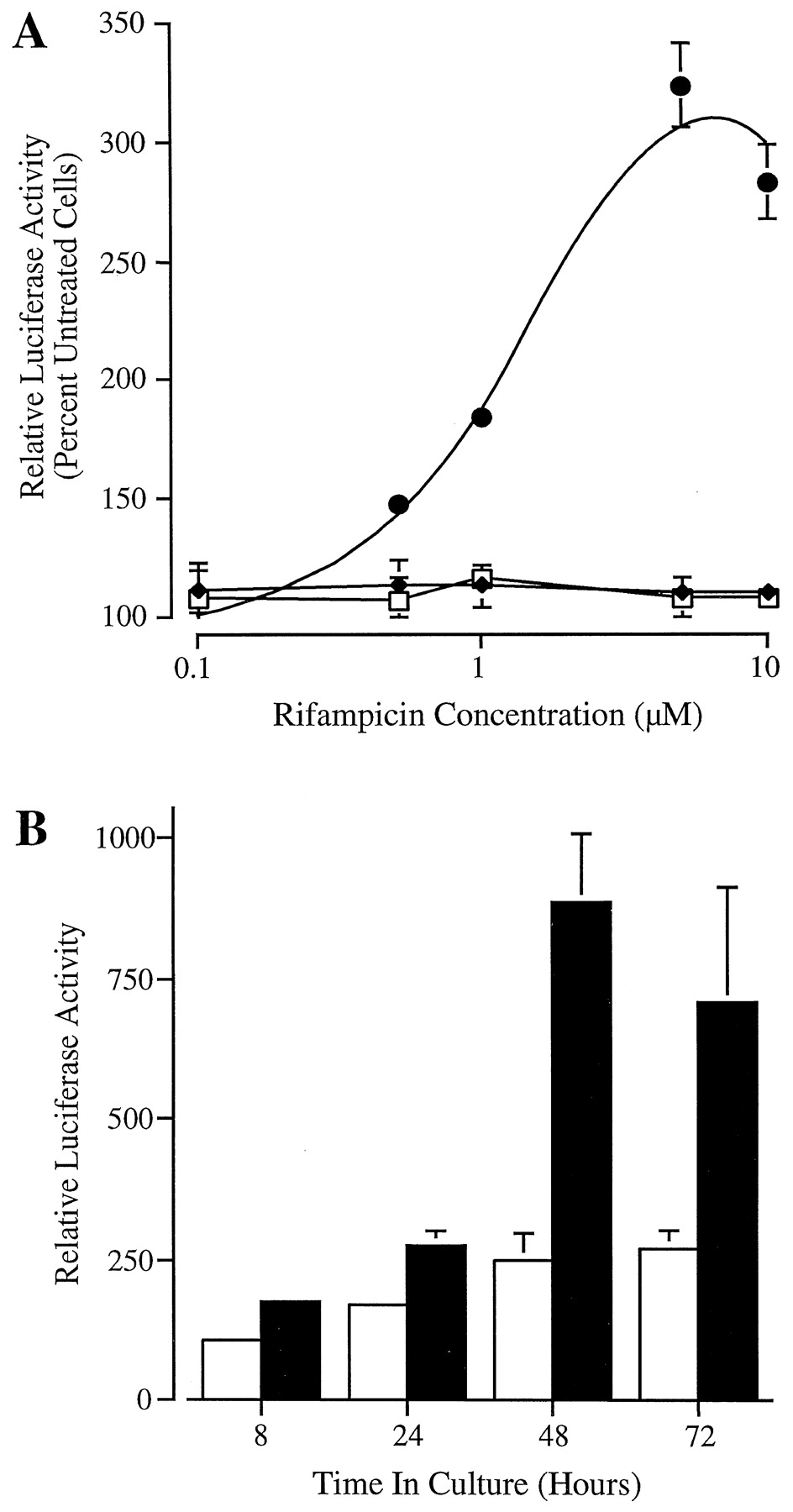
Dose- and time-dependent activation ofCYP3A4-luciferase reporter gene constructs by rifampicin. A, constructs p3A4-13000 (●), p3A4-3195 (♦), encompassing base −13000 to +53 and −3195 to +53 ofCYP3A4, respectively, and the promoterless pGL3-Basic (■) were transfected into HepG2 cells. Cells were treated with the indicated concentration of rifampicin for 48 h before luciferase and β-galactosidase determination on cell lysates. Luciferase values are normalized to β-galactosidase and expressed as percentage of control cells. B, time-dependent activation of p3A4-13000 by rifampicin. Transfected HepG2 cells were exposed to the drug for the time indicated before harvest. Relative luciferase activity is expressed as a percentage of that in untreated cells after 8-h culture. All data are mean ± S.D. of triplicate transfections from a single representative experiment.
The activation of p3A4-13000 by rifampicin was dose dependent with maximal induction observed at 5 to 10 μM and an estimated half-maximal effective concentration (EC50) of ∼1 to 2 μM (Fig. 2A). In addition, the time-dependent transactivation of p3A4-13000 by rifampicin (5 μM) was examined. Although luciferase activity was marginally enhanced after 8- and 24-h exposure to the drug (1.6- to 1.8-fold), maximal induction of reporter gene activity (3- to 4-fold) was observed at 48 h in culture. Continued rifampicin treatment did not significantly enhance luciferase reporter gene expression (Fig. 2B). The time-dependent activation of these chimeric CYP3A4-luciferase constructs is identical to that of endogenous CYP3A genes in HepG2 cells (Sumida et al., 1999). Moreover, the dose-dependent induction of p3A4-13000 by rifampicin is comparable to that of endogenous human CYP3Agenes (Schuetz et al., 1996). Finally, the magnitude of the rifampicin response (3- to 4-fold) is in agreement with that of CYP3Agenes in HepG2 cells (Schuetz et al., 1993; Sumida et al., 1999). Taken together, these data indicate that activation ofCYP3A4-luciferase reporter gene constructs by rifampicin closely resembles that of endogenous CYP3A genes and, as such, represents a valid model for the examination of CYP3A4transcriptional regulation.
To examine whether the rifampicin-responsive domain exhibited enhancer properties, fragments of the 5′-flanking region were inserted downstream of the CYP3A4 promoter proximal and luciferase reporter gene (Fig. 3). Inducible expression was observed regardless of the position or orientation of the responsive region relative to the proximal promoter ofCYP3A4 (Fig. 3). Thus, a 3.8-kb BglII fragment, corresponding to bases −9751 to −6043, imparted rifampicin-responsiveness on the promoter proximal region (bases −1252 to +53) when inserted in either orientation downstream of the luciferase reporter gene (Fig. 3). Furthermore, the distal region (bases −7836 to −7208) was capable of conferring inducibility on a minimal tk promoter (Fig. 8). Importantly, in this system, the transcriptional activity of constructs containing the CYP3A4proximal promoter region alone was not enhanced by rifampicin (Figs. 1,3, and 4A; see also Fig. 8A).

Mutational analysis of putative PXR response elements in the XREM. A, PXREs in the p3A4-362(7836/7208ins) construct were mutated as described in Materials and Methods, and their rifampicin-responsiveness was analyzed in temporary transfection studies. ■, wild-type PXREs. ▪, mutated PXREs. B, identification of an additional functional PXRE (dNR3) required for maximal rifampicin responsiveness. The sequence of the putative PXRE is shown, and the repeated nuclear receptor half-sites within this motif are indicated by horizontal arrows. The mutated bases are indicated. Transfected HepG2 cells were cultured in the presence of rifampicin (5 μM) for 48 h before harvest and determination of luciferase and β-galactosidase activity. Activation of constructs by the drug is expressed as fold induction and as a percentage of the wild-type rifampicin response, which is nominally that obtained from p3A4-362(7836/7208ins). All transfections contained pSG5-hPXRΔATG, the hPXR expression vector. ND indicates no induction of reporter gene activity. Data are mean ± S.D. of triplicate transfections.
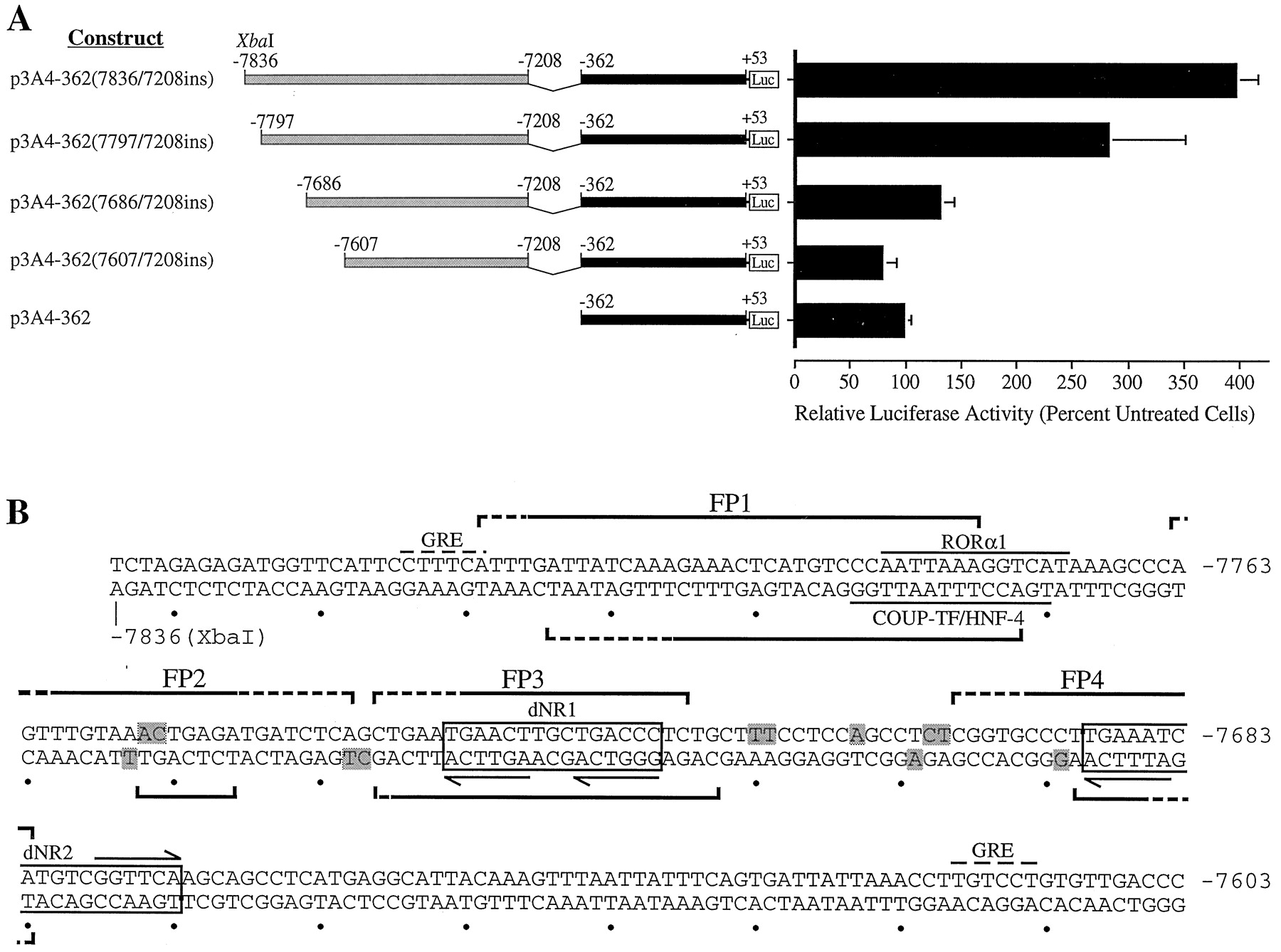
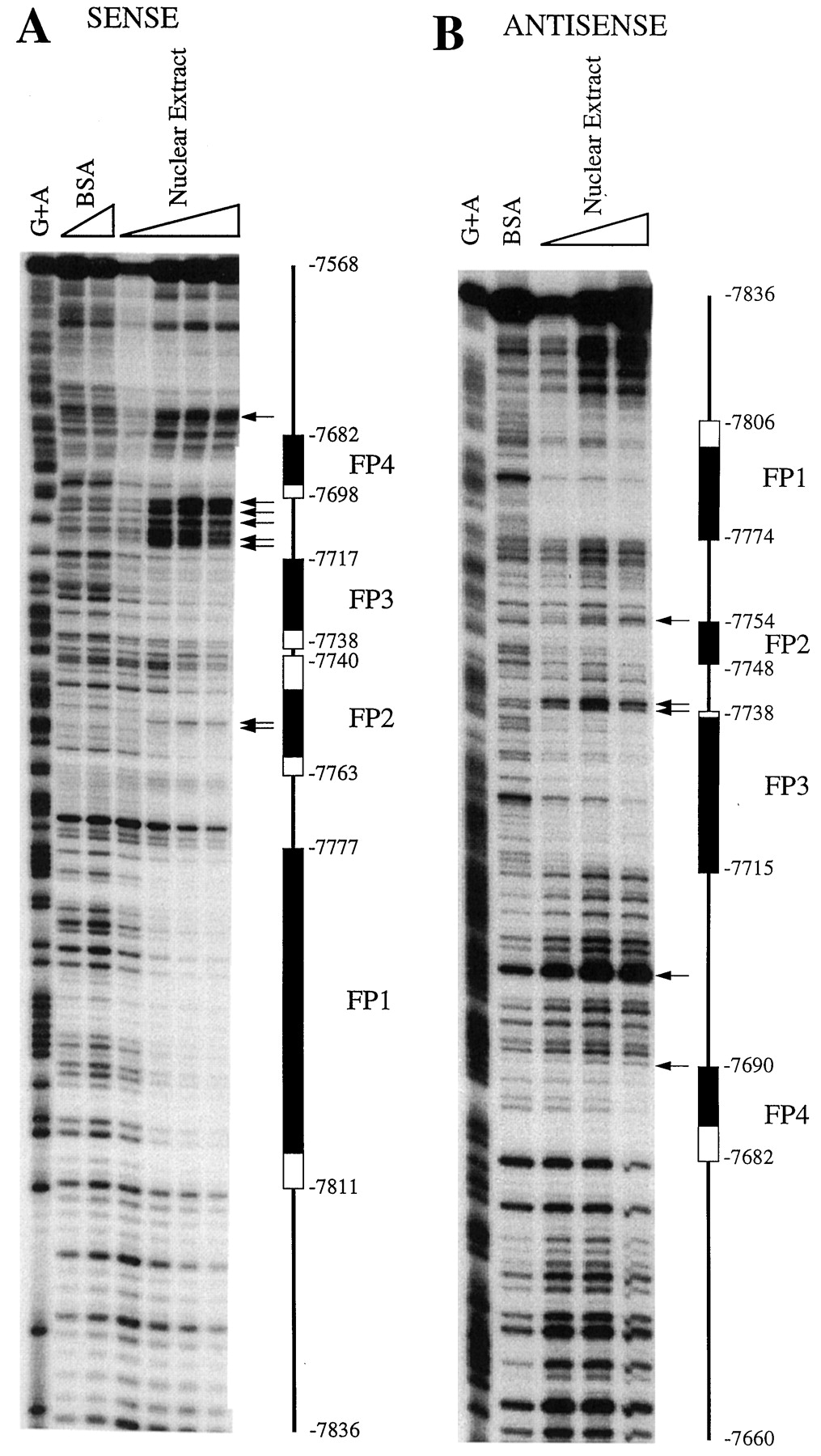
A, localization of the rifampicin-responsive region to bases −7836 to −7607. Deletion analysis ofCYP3A4-Luc constructs transfected into HepG2 cells. Transfected cells were cultured in the presence of rifampicin (5 μM) for 48 h. Relative luciferase activities are derived as described in legend to Fig. 1. B, nucleotide sequence analysis of the rifampicin-responsive region. Regions protected in DNase I footprint on sense and antisense strands are delineated by upper and lower brackets, respectively. Brackets with dashed lines indicate weakly protected regions. DNase I-hypersensitive sites are shaded. Putative PXR response elements (dNR1 and dNR2) are boxed, and the repeated nuclear receptor half-sites within these motifs are indicated by horizontal arrows. Putative binding sites for RORα1 and COUP-TF/HNF-4 are overlined and underlined, respectively. Potential GRE half-sites are denoted by dashed overlines (Jantzen et al., 1987).
A second set of deletion mutants were prepared and transiently transfected into HepG2 cells (Fig. 4A). The removal of bases −7836 to −7797 marginally reduced the rifampicin response, whereas the deletion of bases −7797 to −7686 removed 70% of the wild-type activity (Fig.4A). The deletion of bases −7836 to −7607 completely destroyed rifampicin inducibility (Fig. 4A). These data suggested that the rifampicin induction was dependent on the integrity of a number of distinct transcription factor-bindin sites contained within the −7836 to −7607 region of CYP3A4. We have referred to this core region as an XREM.
DNase I Footprint Analysis of Rifampicin-Responsive region.
DNase I footprint analysis of the responsive region (bases −7836 to −7568) using rat liver nuclear extract revealed two strongly (FP1 and FP3) and two weakly (FP2 and FP4) protected regions (Figs. 4B and5). Footprint coordinates on the sense strand (Fig. 5A) were as follows: FP1, −7811 to −7777; FP2, −7763 to −7740; FP3, −7738 to −7717; and FP4, −7698 to −7682. Footprint coordinates on the antisense strand (Fig. 5B) were as follows: FP1, −7806 to −7774; FP2, −7754 to −7748; FP3, −7738 to −7715; and FP4, −7690 to −7682. No other regions were consistently protected on either the sense or antisense strands. In addition, a number of DNase I-hypersensitive sites were identified. These results are summarized in Figs. 4B and 5. A similar pattern of nuclease protection was observed with nuclear extracts prepared from control and rifampicin-treated HepG2 cells (data not shown).

Binding of liver nuclear proteins to the rifampicin-responsive region. In vitro DNase I footprint analysis was performed using 32P-end-labeled sense (A) or antisense (B) strands as described in Materials and Methods with increasing amounts of rat liver nuclear extract (3–25 μg). Protected regions FP1–FP4 are delineated by filled columns. Partially protected regions are outlined by open columns, and DNase I-hypersensitive sites are indicated by horizontal arrows. The position relative to the transcription initiation site of CYP3A4 is shown at the border of each protected region. BSA denotes reactions that were performed in the absence of nuclear extract. G + A indicates the Maxam-Gilbert chemical sequence ladder generated from the probe.
Potential regulatory elements within the footprinted regions were identified by inspection and by screening the TRANSFAC database, a compilation of transcription factors and their binding motifs, with the MatInspector and PatSearch search engines (Quandt et al., 1995). A number of putative cis-acting elements were identified within or overlapping the FP1 region. First, a response element highly homologous to the recognition sequence for the orphan retinoic acid receptor-related receptor α-1 [RORα1 (NR1F1)] is located between bases −7783 to −7771 (Giguère et al., 1994). Second, a site with significant identity to a binding motif in the ornithine transcarbamylase gene that was recognized by both chicken ovalbumin upstream promoter-transcription factors and hepatocyte nuclear factor-4 [COUP-TF/HNF-4 (NR2F1/NR2A1)] is situated between bases −7785 to −7772 (Kimura et al., 1993). Both the RORα1 and COUP-TF/HNF-4 sites overlap the 5′ terminus of FP1 and contain an AGGTCA hexamer, the recognition sequence for members of the nuclear receptor superfamily (Fig. 4B; Mangelsdorf et al., 1995). We were unable to identify any potential transcription factor-binding sites within the FP2 domain, although significant homology with a previously reported CCAAT/enhancer-binding protein-α motif was evident (Roman et al., 1990).
Examination of FP3 and FP4 revealed potential binding sites for the orphan hPXR. Although members of the nuclear receptor family that heterodimerize with RXRα typically bind to repeats of the AGGTCA hexad (Mangelsdorf et al., 1995), the PXR-RXRα complex preferentially binds the variant AGTTCA half-site (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998). Thus, FP3 contained an imperfect DR-3 (direct repeat with a three-nucleotide spacer) of the AGTTCA hexamer. The structure of this element was almost identical to the PXRE in the proximal promoter of CYP3A23(Kliewer et al., 1998). FP4 overlapped an ER-6-like element that was highly homologous to the previously characterized hPXR-bindin motif in the proximal promoter of CYP3A4 (bases −172 to −149). The hPXR-binding motifs encompassed by FP3 and FP4 are nominally dNR1 (distal nuclear receptor-binding element 1) and dNR2, respectively, whereas the ER-6 motif in the CYP3A4 proximal promoter is designated prPXRE (proximal PXRE). These elements are illustrated in Figs. 4B and 6A. Finally, two potential GRE half-sites were evident in this region (Fig. 4B; Jantzen et al., 1987).
hPXR-RXRα Heterodimers Bind Elements within Responsive Region.
The ability of the dNRs to bind the hPXR-hRXRα heterodimer was investigated by EMSA (Fig. 6). As reported elsewhere (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998), the prPXRE (ER-6) of CYP3A4 formed a complex with hPXR-hRXRα. dNR1 was capable of binding hPXR-hRXRα with high affinity and effectively competing with prPXRE for binding of the hPXR-hRXRα heterodimer (Fig. 6, B and C). The affinity of dNR1 for hPXR-hRXRα was comparable to that of prPXRE and a PXRE in the promoter proximal region of the rat CYP3A23 (Fig. 6, B and C; data not shown; Kliewer et al., 1998). dNR2 was also capable of forming a complex with hPXR-hRXRα, albeit with significantly lower affinity (Fig. 6B). In addition, dNR2 was only capable of competing with prPXRE for protein binding at high molar excess (Fig. 6C).
Cotransfection of hPXR with Rifampicin-Responsive Constructs Substantially Augments Induction.
Recent reports have demonstrated that hPXR is activated by rifampicin in transfection studies (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998). A heterologous reporter gene construct [p(ER6)3-tk-Luc] containing multiple copies of the prPXRE element was capable of conferring rifampicin-responsiveness on a minimal tk promoter in the presence of hPXR. Furthermore, Kliewer and coworkers demonstrated that rifampicin promotes the association of hPXR with the steroid receptor coactivator 1 (Lehmann et al., 1998). Taken together, these studies indicate that rifampicin can serve as a ligand for hPXR.
In this study, the EC50 value of ∼1 to 2 μM for rifampicin-mediated induction of p3A4-13000 (Fig. 2A) was in excellent agreement with published EC50 values for activation of hPXR by this drug (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998). This observation, in conjunction with the ability of hPXR-RXRα heterodimers to bind elements within the XREM region, strongly suggested that hPXR may mediate the rifampicin induction. Therefore, we examined the ability of hPXR to augment the rifampicin-responsiveness of p3A4-362(7836/7208ins). Cotransfection of liver-derived HepG2 cells with p3A4-362(7836/7208ins) and an hPXR expression vector substantially increased the rifampicin response. Typically, a 40- to 50-fold rifampicin-mediated induction of luciferase activity was observed in cotransfection experiments (Fig.7A). This level of induction is similar to that of the endogenous CYP3A4 gene in primary cultures of human hepatocytes (Daujat et al., 1991). However, rifampicin treatment of non-liver-derived simian kidney (COS7) or murine fibroblast (NIH3T3) cell lines cotransfected with p3A4-362(7836/7208ins) and hPXR resulted in a modest 3- to 5-fold induction of reporter gene activity (Fig. 7, B and C), suggesting that other transcription factors, present in HepG2 but not COS7 or NIH3T3 cells, are required for maximal responsiveness. In agreement with earlier experiments (Figs. 1-3 and 4A), cultures of HepG2 cells transfected with empty expression vector exhibited a 2- to 4-fold induction after treatment. The effect of overexpressed hPXR on the basal expression of the rifampicin-responsive constructs was minimal (∼1.5- to 2-fold increase in activity). The hPXR-responsive control plasmid p(ER6)3-tk-Luc exhibited a 2- to 4-fold rifampicin activation in the presence of cotransfected hPXR regardless of cell type (Fig. 7A, inset; data not shown).

Rifampicin inducibility is mediated by hPXR. A, hPXR, but not hGR, substantially augments the rifampicin response. The ability of hPXR, hGR, or hPXR and hGR to modulate the responsiveness of the XREM construct, p3A4-362(7836/7208ins), and proximal promoter (p3A4-362) was investigated. HepG2 cells were transfected as described in Materials and Methods, and the cells were exposed to rifampicin or dexamethasone at the concentrations indicated for 48 h before determination of luciferase and β-galactosidase. p(GRE)2-tk-Luc, which contains dimerized copies of a GRE from the rat tyrosine aminotransferase gene, was included as a control for hGR activation. The hPXR control plasmid, p(ER6)3-tk-Luc, contains three copies of the proximal PXRE from CYP3A4 (Lehmann et al., 1998) and was transfected in the presence of pSG5-hPXRΔATG (A, inset). The effect of cotransfection of the hPXR expression vector and p3A4-362(7836/7208ins) into COS7 (B) and NIH3T3 (C) cells. Control cultures were transfected with p3A4-362(7836/7208ins) and the pSG5 expression vector. Cells were cultured in the presence of vehicle alone (0.1% DMSO; open columns) or 5 μM rifampicin (filled columns) for 48 h before harvest. All data are mean ± S.D. of triplicate transfections from a single representative experiment. Luciferase activities are normalized to β-galactosidase.
Recently, Calleja et al. (1998) reported that rifampicin was a nonsteroidal ligand and activator of the hGR. The presence of putative GREs in the proximal promoter of CYP3A4 (Hashimoto et al., 1993) and the rifampicin-responsive region (Fig. 4B) led us to examine the role of hGR in the induction. Rifampicin induction of p3A4-362(7836/7208ins) was not augmented by cotransfection of an hGR expression vector. Indeed, overexpression of hGR partially abrogated the rifampicin response (Fig. 7A; data not shown). The presence of ligands for both hPXR and hGR, namely rifampicin and dexamethasone (1 nM to 10 μM), did not augment the hPXR-mediated induction of p3A4-362(7836/7208ins; data not shown). In parallel transfections, a hGR control plasmid, p(GRE)2-tk-Luc, which contains dimerized GREs from the tyrosine aminotransferase gene, was dose-dependently activated by dexamethasone (Fig. 7A). However, in line with observations by others (Lehmann et al., 1998; Ray et al., 1998;Jaffuel et al., 1999), we were unable to activate this construct with rifampicin in the presence of overexpressed hGR, hPXR, or both hPXR and hGR (Fig. 7A). The proximal promoter construct p3A4-362 was not activated by rifampicin under any conditions.
Cooperative Interaction of dNRs and prPXRE in Rifampicin Response.
We next examined the effect of mutation of potential PXREs in the p3A4-362(7836/7208ins) construct on the rifampicin response. Thus, mutation of dNR1 reduced the wild-type response by 40 to 50% (Fig. 8A), where the fold activation of the p3A4-362(7836/7208ins) construct is nominally 100%. Interestingly, mutation of prPXRE removed ∼50% of the wild-type response. A similar reduction in rifampicin-responsiveness was observed when the proximal promoter of CYP3A4 was replaced by a minimal tk promoter (bases −105 to +52), suggesting that there are no further CYP3A4 promoter-specific elements required for maximal activity of the XREM region (Fig. 8A). Although prPXRE has no inherent ability to promote a rifampicin response in the context of the p3A4-362 construct, it is evidently capable of a cooperative interaction with elements within the XREM. Surprisingly, mutation of dNR2 did not reduce rifampicin inducibility. Indeed, disruption of this element resulted in a significant increase (20–30%) in responsiveness (Fig. 8A). Furthermore, disruption of both dNR2 and prPXRE (73% of wild-type activity) resulted in significantly higher rifampicin responsiveness than mutation of prPXRE alone (54% wild-type activity). However, mutation of dNR2 in the context of a construct containing the mutated dNR1 did not enhance induction relative to a construct harboring a disrupted dNR1 alone. Thus, factors bound at dNR2 exerted a partially suppressive effect on the activity of dNR1. In vitro, dNR2 was capable of weakly binding hPXR-RXRα heterodimers; however, in HepG2 cells, this site may bind additional factors, possibly other members of the nuclear receptor superfamily. The significance of these latter observations is unclear, but taken together, these data support the idea of complex cooperative behavior between proteins bound at distal response elements and elements in the proximal promoter. It is important to note that the cooperativity between dNRs was retained when the XREM deletions and mutations were placed upstream of a heterologous tk promoter (data not shown).
Interestingly, simultaneous mutation of dNR1, dNR2, and prPXRE did not completely abolish the rifampicin-mediated reporter gene activation (Fig. 8A), implying that additional functional PXREs were present in the XREM or proximal promoter regions of CYP3A4. The observation that the XbaI/SpeI fragment (bases −7836 to −7493), the deletion of which destroyed inducible reporter gene expression (Fig. 1), did not confer wild-type rifampicin-responsiveness on either the CYP3A4 proximal promoter or a minimal tk promoter (Fig. 8B; data not shown) suggested that the region encompassing bases −7492 to −7208 contained accessory sites required for maximal activity. Nucleotide sequence analysis of this region revealed a third putative nuclear receptor-bindin motif (dNR3; Fig. 8B). This element, an imperfect DR-3 of the AGTTCA hexamer (bases −7290 and −7270), was capable of weakly binding hPXR-RXRα heterodimers (data not shown). In addition, mutation of this motif significantly reduced rifampicin responsiveness by 15 to 20% (Fig.8B).
Rifampicin-Responsive Constructs Are Strongly Activated by a Number of CYP3A4 Inducers.
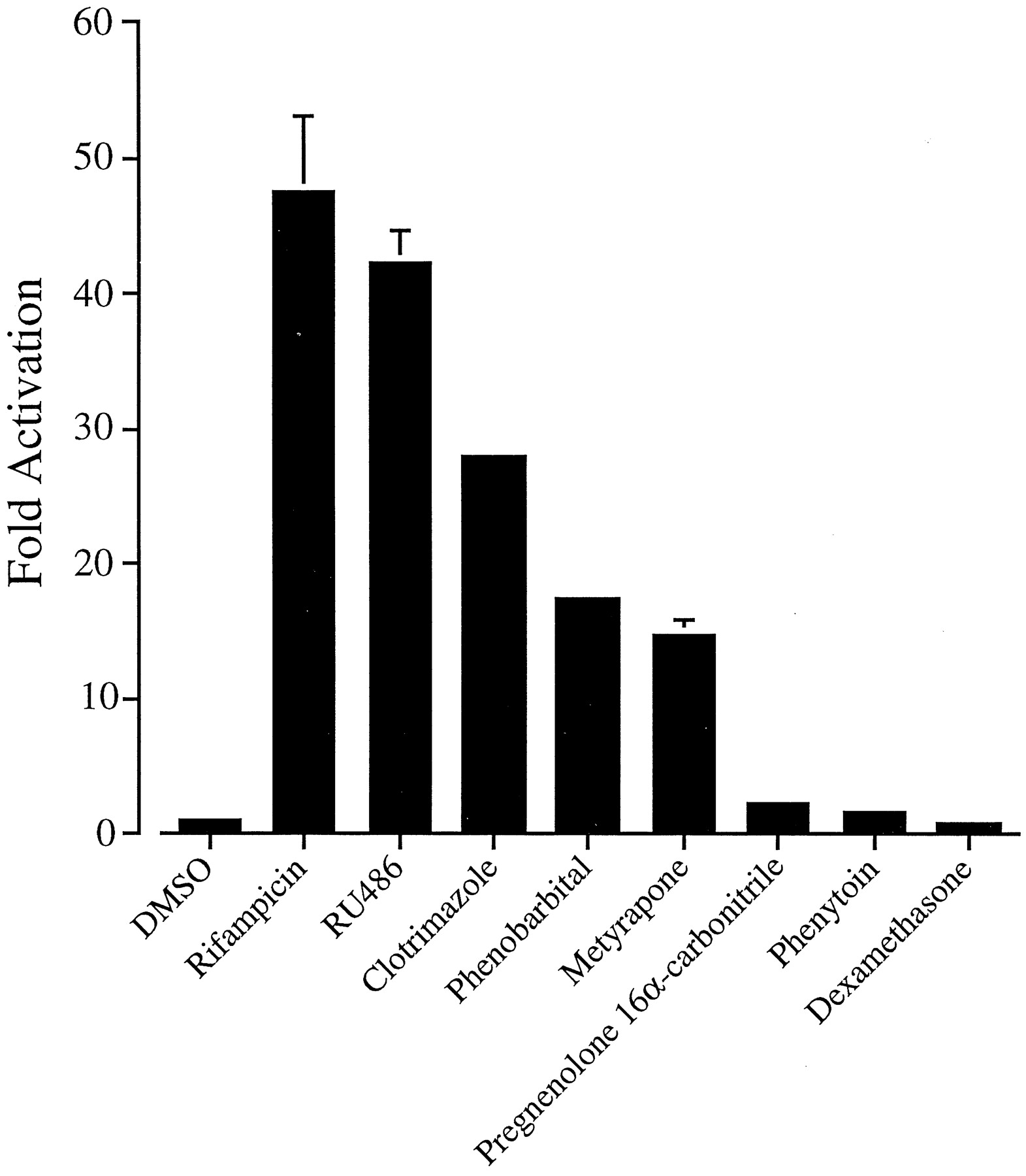
In parallel transfections, the p3A4-362(7836/7208ins) construct was potently activated by a number ofCYP3A inducers, including RU486 (40-fold), clotrimazole (20- to 30-fold), phenobarbital (15-fold), and metyrapone (10- to 15-fold; Fig. 9). Weak activation by phenytoin (2-fold) and PCN (3- to 4-fold) was also observed (Fig. 9). In the absence of exogenously expressed hPXR, these xenobiotics were poor inducers of p3A4-362(7836/7208ins): maximum induction was observed with 5 μM rifampicin (2- to 4-fold; Figs. 1 and 2; data not shown). The induction of the XREM region by these and other compounds correlates well with detailed activation profiles of the hPXR published elsewhere (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998). None of these drugs were able to induce expression of reporter gene constructs containing the CYP3A4 proximal promoter regions alone (data not shown).

Activation of the XREM by CYP3Ainducers. The ability of various CYP3A inducers to activate the XREM region was tested. Constructs p3A4-362(7836/7208ins) and pSG5-hPXRΔATG were transiently transfected into HepG2 cells. Cells were treated with 0.1% DMSO (vehicle), 500 μM metyrapone, 1 mM phenobarbital, 5 μM rifampicin, or 10 μM phenytoin, pregnenolone 16α-carbonitrile, dexamethasone, clotrimazole, and RU486 for 48 h after transfection. Data are derived as described in the legend to Fig. 1.
Discussion
CYP3A4, the predominant P-450 expressed in adult human liver, is known to be transcriptionally regulated by a number of structurally unrelated compounds. Transcriptional activation of this gene is at the center of many clinically important drug interactions. However, the mechanisms underlying this induction have remained elusive. Recently, a number of independent studies have described an orphan nuclear receptor, hPXR, which interacts with an ER-6 (prPXRE, bases −172 to −149) element in the proximal promoter of CYP3A4. This receptor is activated by a variety of lipophilic compounds, many of which, including rifampicin, are CYP3A inducers (Bertilsson et al., 1998; Blumberg et al., 1998; Lehmann et al., 1998).
In this study, we defined regulatory regions of CYP3A4involved in its transcriptional activation by rifampicin and other xenobiotics. To dissect functional elements in both proximal and distal regions, we endeavored to use the proximal CYP3A4 promoter (bases −362 to +53) as a minimal promoter. Here, we report the characterization of a distal enhancer module (XREM) that, in conjunction with elements in the proximal promoter region, directs the hPXR-mediated transactivation of CYP3A4.
The XREM region, located ∼7800 bp upstream of the CYP3A4transcription initiation site, is a complex array of transcription factor-binding sites that includes at least two elements (designated dNR1 and dNR2) that are capable of binding hPXR-RXRα heterodimers. In addition, a third putative PXRE (dNR3), located several hundred base pairs downstream of the core rifampicin-responsive region, appears to be critical for maximal xenobiotic responsiveness. Two additionalcis-acting elements (designated FP1 and FP2), situated immediately upstream of the hPXR-binding motifs, are critical for full functionality of the XREM region: deletion of these sites substantially reduces the rifampicin responsiveness of the XREM region. The precise nature of protein/DNA interactions at these accessory sites is under investigation; however, binding motifs for other members of the nuclear receptor superfamily, notably RORα1 and COUP-TF/HNF-4, were evident in FP1. Interestingly, a PXRE in the proximal promoter of the ratCYP3A23 gene is located between functional COUP-TF- and HNF-4-binding sites (Huss and Kasper et al., 1998; Ogino et al., 1999), the mutation or deletion of which substantially abrogates PXR-mediated transactivation (Huss and Kasper et al., 1998).
The ability of hPXR-RXRα heterodimers to interact with elements within the XREM region strongly suggests hPXR mediated rifampicin induction of reporter gene constructs. Indeed, cotransfection of rifampicin-responsive constructs with a hPXR expression vector increased induction by the drug from 2- to 4-fold to >40-fold, a level of activation similar to that of the endogenous CYP3A4 gene in primary cultures of human hepatocytes. The dose- and time-dependent activation of reporter gene constructs, in both the presence and absence of exogenously expressed hPXR, paralleled that of endogenousCYP3A genes (Schuetz et al., 1996; Sumida et al., 1999). The modest rifampicin induction of these constructs in the absence of cotransfected PXR is almost certainly due to reduced expression of thehPXR gene in HepG2 cells. Support for this observation is provided in a report by Miyata et al. (1995). Thus, binding of nuclear proteins to a probe corresponding to the promoter proximal PXRE from the rat CYP3A2 gene was substantially lower in extracts prepared from HepG2 cells than in those isolated from rat liver (Miyata et al., 1995). Interestingly, the rifampicin response was markedly lower in cells of extrahepatic lineage, suggesting that liver-enriched transcription factors, in addition to hPXR, may be required for full functionality. Indeed, putative binding sites for multiple liver-enriched transcription factors are evident in theCYP3A4 proximal promoter (Hashimoto et al., 1993), as well as the XREM region. Identification of the additional liver-specific factors required for xenobiotic inducibility of these constructs may provide insights into the developmental and tissue-specific regulation of CYP3A4.
In the present study, we were unable to elicit any rifampicin induction from CYP3A4-reporter gene constructs containing solely prPXRE. This contradicts an earlier report by Ogg et al. (1999) that demonstrated that a plasmid containing 1 kb of the 5′-flanking region of CYP3A4 conferred xenobiotic responsiveness on a reporter gene. Importantly, these studies used the potent cytomegalovirus promoter as a minimal promoter, whereas we have used the nativeCYP3A4 proximal promoter throughout. However, the isolated and multimerized prPXRE was capable of mediating activation of heterologous reporter gene constructs by hPXR. In line with observations by Hashimoto et al. (1993), luciferase activity from the p3A4-362 construct was very low, and an attractive hypothesis to account for this would be the existence of silencer elements in this region of the CYP3A4 promoter that repress both basal transcriptional activity and the previously characterized activity of the isolated prPXRE (Bertilsson et al., 1998; Blumberg et al., 1998;Lehmann et al., 1998). However, mutation or removal of the prPXRE reduced the wild-type rifampicin response by ∼50%. This indicates that the prPXRE cooperatively interacts with either the distal nuclear receptor-binding elements themselves or with other elements within the XREM. Disruption of prPXRE and dNR1 destroyed 80 to 90% of xenobiotic responsiveness, demonstrating that these two elements are central to transactivation by ligand-activated hPXR. Although the exact role of each PXRE remains to be elucidated, it is now clear that there is a considerable level of complexity to both the XREM and the proximal promoter regions of CYP3A4. In this respect, the XREM is analogous to hormone response regions in genes such as the mouse mammary tumor virus and tyrosine aminotransferase gene, where synergism between hormone response elements and other transcription factors is well documented (Jantzen et al., 1987; Tsai and O'Malley et al., 1994). Similarly, the induction of CYP2B genes by phenobarbital and other xenobiotics is mediated by distal enhancer modules that contain multiple nuclear receptor-bindin motifs (Honkakoski et al., 1998; Stoltz et al., 1998; Sueyoshi et al., 1999).
Finally, we demonstrated that rifampicin-responsive constructs were potently activated by a number of well-documented CYP3Ainducers, including phenobarbital. Recently, Negishi and coworkers reported that transactivation of CYP2B genes by this drug was mediated by the orphan constitutive androstane receptor [CAR (NR1I4); Honkakoski et al., 1998; Sueyoshi et al., 1999). Moreover, the multimerized CYP3A4 prPXRE was shown to be capable of conferring CAR-RXRα-meditated phenobarbital inducibility on a tk promoter. The ability of CAR-RXRα and hPXR-RXRα heterodimers to interact with the same nuclear receptor-binding motif suggests that interplay between orphan receptors is likely to play a role in the regulation of CYP3A4 expression.
In summary, a potent xenobiotic-responsive enhancer module controls the transcriptional induction of CYP3A4 by compounds that are activators for the hPXR. Further characterization of this region should provide a framework for understanding the molecular basis of many clinically important drug interactions. Moreover, it is now clear that the transcriptional regulation of P-450 genes by diverse xenochemicals and endogenous compounds, such as steroid hormones, bile acids, cholesterol, and fatty acids, is predominantly mediated by orphan members of the nuclear receptor superfamily. The elucidation of the molecular mechanisms underlying the regulation of P-450 expression should provide valuable insights into the biology of the orphan nuclear receptors.
Acknowledgments
We gratefully acknowledge Graham R. Robertson and Geoffrey C. Farrell for their patient advice and support throughout this project.
Footnotes
- Received July 6, 1999.
- Accepted August 26, 1999.
-
Send reprint requests to: Dr. Christopher Liddle, Department of Clinical Pharmacology and Storr Liver Unit, University of Sydney at Westmead Hospital, Westmead, NSW 2145, Australia. E-mail: chrisl{at}westgate.wh.usyd.edu.au
-
This work was supported by a grant from the National Health and Medical Research Council (Australia) and the Robert W. Storr bequest to the Medical Foundation, University of Sydney. B.G. was a recipient of the National Health and Medical Research Council (Australia) Dora Lush Biomedical Scholarship.
-
1 Three independent studies have implicated an orphan nuclear receptor in CYP3A regulation. The human PXR (hPXR; Lehmann et al., 1998) and human pregnane activated receptor (hPAR; Bertilsson et al., 1998) are identical in the derived amino acid sequence. The human steroid and xenobiotic receptor (hSXR; Blumberg et al., 1998) contains a single base-pair insertion at position 1225 and a single base deletion at 1279, relative to hPXR, which results in a shift in the reading frame for amino acid residues 215–233. However, hPXR, hPAR, and hSXR almost certainly represent products of the same gene. In this study, we used the “hPXR” nomenclature.
Abbreviations
- P-450
- cytochrome P-450
- PXR
- pregnane X receptor
- RXRα
- 9-cis retinoic acid receptor-α
- PXRE
- pregnane X receptor response element
- prPXRE
- proximal pregnane X receptor response element
- HNF
- hepatocyte nuclear factor
- EMSA
- electrophoretic mobility shift assay
- RORα1
- retinoic acid receptor-related receptor α-1
- COUP-TF
- chicken ovalbumin upstream promoter-transcription factor
- PCR
- polymerase chain reaction
- PCN
- pregnenolone 16α-carbonitrile
- tk
- herpes simplex virus thymidine kinase
- hGR
- human glucocorticoid receptor
- GRE
- glucocorticoid-responsive element
- XREM
- xenobiotic-responsive enhancer module
- DMSO
- dimethyl sulfoxide
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}