


Abstract
The human β2-adrenergic receptor (βAR) is rapidly desensitized in response to saturating concentrations of agonist by G protein-coupled receptor kinases (GRKs) and cAMP-dependent protein kinase A (PKA) phosphorylation of the βAR, followed by β-arrestin binding and receptor internalization. βAR sites phosphorylated by GRK in vivo have not yet been identified. In this study, we examined the role of the carboxyl terminal serines, 355, 356, and 364, in the GRK-mediated desensitization of the βAR. Substitution mutants of these serine residues were constructed in which either all three (S355,356,364A), two (S355,356A and S356,364A), or one of the serines (S356A and S364A) were modified. These mutants were constructed in a βAR in which the serines of the PKA consensus site were substituted with alanines (designated PKA−) to eliminate any PKA contribution to desensitization, and they were stably transfected into human embryonic kidney 293 cells. Treatment of the PKA−mutant with 10 μM epinephrine for 5 min caused a 3.5-fold increase in the EC50 value and a 42% decrease in theV max value for epinephrine stimulation of adenylyl cyclase. Substitution of all three serines completely inhibited the epinephrine-induced shift in the EC50. Both double mutants, S355,356A and S356,364A, showed a nearly complete loss of the EC50 shift, whereas the single substitutions, S356A and S364A, caused only a slight decrease in desensitization. None of the mutations altered the epinephrine-induced decrease inV max, which seems to be downstream of the receptor. The triple mutation caused a 45% decrease in epinephrine-induced internalization and a 90 to 95% reduction in phosphorylation of the βAR relative to the PKA−(1.9 ± 0.2- and 16.6 ± 3.8-fold phosphorylation over basal, respectively). The double mutants caused an intermediate reduction in internalization (20–21%) and phosphorylation (43–52%). None of the serine mutations altered the rate of βAR recycling. Our data demonstrate that the cluster of serines within the 355 to 364 βAR domain confer the rapid, GRK-mediated, receptor-level desensitization of the βAR.
The β2-adrenergic receptor (βAR) is rapidly inactivated after exposure to epinephrine. Rapid βAR desensitization at high concentrations of epinephrine results from phosphorylation of the receptor by cAMP-dependent protein kinase (PKA) and one or more members of the G protein-coupled receptor kinase (GRK) family (Clark et al., 1989, 1999; Kunkel et al., 1989; Krupnick and Benovic, 1998;Lefkowitz et al., 1998). Considerable recent evidence supports the proposal that GRK-mediated phosphorylation of receptors greatly promotes their binding to β-arrestin, leading to receptor internalization through a clathrin-mediated mechanism (Tsuga et al., 1994; Ferguson et al., 1995, 1996; Goodman et al., 1996). At high occupancy with strong agonists, it is clear that these events cause the majority of the desensitization of the βAR (Clark et al., 1999).
The current model of G protein-coupled receptor desensitization was first developed through studies of rhodopsin. Phosphorylation of the light-activated rhodopsin increased its affinity for visual arrestin (Kuhn et al., 1984). Arrestin binding is thought to be the most critical step in desensitization, serving to uncouple rhodopsin from the G protein transducin (Wilden et al., 1986; Bennett and Sitaramayya, 1988). Desensitization of the βAR is hypothesized to occur similarly, requiring receptor phosphorylation followed by β-arrestin binding (Lohse et al., 1990; Palczewski and Benovic, 1991). Unlike rhodopsin, βAR interaction with β-arrestin leads to receptor internalization through a clathrin-coated pit-dependent pathway (Ferguson et al., 1996;Goodman et al., 1996). Mutagenesis studies have suggested that multiple domains within visual arrestin (Gurevich and Benovic, 1993; Gurevich, 1998; Vishnivetskiy et al., 1999) and the β-arrestins (Kovoor et al., 1999) interact with receptor sites. The complementary domains within either rhodopsin or the βAR have not been identified, although they are thought to include regions that undergo agonist-induced conformational changes and phosphorylation (Kovoor et al., 1999).
Overwhelming evidence indicates that βAR phosphorylation and subsequent β-arrestin binding are important for desensitization. Despite this, identification of the receptor sites at which GRK phosphorylation occurs and that are required for desensitization has proven difficult. In the first study of putative GRK sites, it was shown that substitution of all 11 carboxyl-terminal serines and threonines reduced GRK-mediated desensitization without affecting regulation by PKA (Bouvier et al., 1988). Relative to the wild-type (WT) βAR, phosphorylation of this mutant in response to a high concentration of agonist was reduced by half but its internalization was unaffected. Sequence analysis of βAR phosphorylated in vitro by GRK suggested that the critical residues were in the distal portion of the carboxyl tail (Fredericks et al., 1996). However, our recent mutagenesis studies of these sites demonstrated that they were not required for in vivo desensitization (Seibold et al., 1998), suggesting that other regions of the receptor carboxyl tail, namely the 355–364 domain, may be important for GRK regulation. Interestingly, Hausdorff et al. (1991) found that substitution of four residues, S355, S356, T360, and S364, in the proximal portion of the βAR carboxyl tail (a subset of the 11 carboxyl tail serines and threonines previously described), eliminated rapid desensitization mechanisms, both PKA- and GRK-mediated. The effect on desensitization was not specific, because phosphorylation and internalization of the mutant were also completely blocked. The discrepancy between this study and the previous work in which all 11 serines and threonines were mutated led to the conclusion that the four amino acid substitutions caused an altered receptor conformation that prevented normal regulation. Adding further complexity, Yu et al. (1993) showed that substitution of serines 356 and 364 did not alter desensitization, but eliminated βAR internalization and resensitization. The inability of this mutant to resensitize after agonist removal led to the proposal that receptor internalization was required for the reversal of desensitization.
The inconsistencies in these reports coupled with our demonstration of the lack of effect of mutating the more distal six serines and threonines in the carboxyl tail prompted the studies presented in this article on the potential role of the S355-S364 domain in GRK-mediated desensitization. Mutants in this domain were constructed in which one (or more) of the three serine residues was substituted with alanine. After stable transfection into human embryonic kidney (HEK) 293 cells, the mutant receptors were examined for coupling efficiency, epinephrine-induced desensitization, internalization, recycling, and phosphorylation. To focus specifically on the role of GRK-mediated desensitization, the mutants were constructed in a βAR in which the PKA consensus sites were ablated by substitution of serines 261, 262, 345, and 346 with alanine (designated PKA−). Ablation of the serines of the PKA consensus sites aided analysis because previous studies have shown that PKA effects contribute to the level of overall desensitization but do not affect the component attributed to GRK-mediated homologous desensitization (Green et al., 1981; Clark et al. 1988; Hausdorff et al., 1989; Yuan et al., 1994). The data reported here show that mutation of all three serines (S355,356,364) in the C-terminal domain was required for complete elimination of homologous receptor-level desensitization and for a 90 to 95% reduction in phosphorylation of the βAR, although mutation of only two amino acids in this cluster resulted in a dramatic reduction of desensitization. We conclude that these serines are the likely sites for GRK-meditated desensitization.
Materials and Methods
Description of Mutant βARs.
Mutations were introduced into the βAR in amino acid region 355 to 364, as shown in Table1 and in Fig.1. All of the mutant βARs contain alanine substitutions for the serines of the two consensus PKA sites. Mutagenesis was performed using the polymerase chain reaction as described previously (Seibold et al., 1998). The mutants were sequenced through the entire βAR coding region and epitope tags to ensure accuracy of the mutagenesis procedure. All of the βARs in Table 1include the hemagglutinin (HA) epitope at the amino terminus and the 6HIS tag at the carboxyl tail, as described previously (January et al., 1997). Recycling data also were obtained for the untagged WTβAR, and desensitization data were obtained for both the untagged WTβAR and for the amino-terminally HA-tagged WTβAR. All of the plasmids were stably transfected into HEK 293 cells, as described in Table 1.
Summary of the βAR substitution mutants

Diagram of the human βAR. The figure shows the carboxyl-terminal portion of the βAR. The amino acids substituted for alanine in the mutant receptors used in this study are highlighted in black. The six-histidine epitope tag is represented at the carboxyl terminus.
Transfection of HEK 293 Cells.
The HEK 293 cells were cultured at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum. The plasmids were linearized by PvuI digestion and transfected into subconfluent HEK 293 cells using the CaPO4method. Sixteen hours later, the cells were shocked with 25% glycerol in DMEM and placed in media containing 0.4 mg/ml G418. Stable transfectants expressing βAR were identified using an intact cell [125I]iodocyanopindolol (125ICYP) binding assay described below.
Membrane Preparation.
Cells were plated into 100-mm dishes that had been precoated with poly-l-lysine. Pretreatment with epinephrine or carrier was performed in 5% CO2 at 37°C and was stopped by removal of media followed by six 5-ml washes with ice-cold HME buffer (20 mM HEPES, pH 8.0, 2 mM MgCl2, 1 mM EDTA, 1 mM benzamidine, 10 μg/ml trypsin inhibitor, 0.1 mg/ml BSA). The final concentration of the carrier components were 0.1 mM ascorbate and 1 mM thiourea (AT), pH 7.0. The cells were scraped into HME buffer containing 10 μg/ml leupeptin, 2 mM tetrasodium pyrophosphate and 0.1 μM okadaic acid and homogenized with seven strokes in a type B Dounce homogenizer. The homogenates were layered onto sucrose step gradients (23 and 43%) prepared in HE buffer (20 mM HEPES, pH 8.0, 1 mM EDTA) and centrifuged at 25,000 rpm in a Beckman SW28.1 rotor for 35 min. The fraction at the 23/43% sucrose interface was taken, frozen in liquid nitrogen, and stored at −80°C.
Measurement of Receptor Levels.
Cells were cultured in 12-well dishes for measurement of intact cell receptor number by125ICYP binding. The125ICYP was prepared as described previously (Barovsky and Brooker, 1980; Hoyer et al., 1984). The cells were rinsed in serum-free DMEM and then removed from the plates by pipetting up and down with 0.5 ml of serum-free DMEM. Aliquots (25–50 μl) of the resuspended cells were used in triplicate binding reactions, each containing about 200 pM 125ICYP. Nonspecific binding was measured in triplicate reactions containing 1 μM alprenolol. The reactions were performed on ice for 50 min and were terminated by the addition of 2.5 ml of ice-cold 50 mM Tris·HCl, pH 7.5, and 10 mM MgCl2. The125ICYP-bound βAR was collected by filtration through Whatman GF/C filters. The filters were rinsed three times with 2.5 ml of the cold Tris/MgCl2 buffer and then counted using a Beckman 4000 gamma counter (Beckman Instruments, Columbia, MD). Protein was measured using a Bio-Rad dye reagent.
Receptor levels in cell membranes were measured in binding reactions using 5 μg of membrane protein, 0.1 mM phentolamine, 40 mM HEPES, pH 7.2, 2 mM EDTA, 0.2 mM ascorbate, 2 mM thiourea, and about 200 pM125ICYP. Nonspecific binding was determined in the presence of 1 μM alprenolol. Reactions were performed at 30°C for 50 min and terminated as described for the intact cell-binding assay.
Measurement of Equilibrium Binding Constants for ICYP and Epinephrine
The K dvalues for 125ICYP and epinephrine were determined for each of the mutant βARs using methods described previously (January et al., 1997, 1998). The range of 125ICYP concentrations used for the K d measurement was 1 to 150 pM. Reactions were performed in triplicate with 1 μg of membrane protein, and nonspecific binding was measured with the inclusion of alprenolol at 1 μM. The K d value was estimated by fit of the data to a rectangular hyperbola. The assays to measure epinephrine K d included 40 to 50 pM125ICYP, 10 μM guanosine 5′-3-O-(thio)triphosphate (GTPγS), and concentrations of epinephrine ranging from 0.1 to 100 μM. Reactions were performed with triplicate points using 1 μg of membrane protein. Graph-Pad (San Diego, CA) analysis was used to fit the data to a one-component sigmoidal curve with a Hill coefficient of −1. Because the mutant βARs had 125ICYP K d values similar to one another, an average value of 7.7 pM was used in the Cheng-Prusoff calculation of the epinephrineK d.
Adenylyl Cyclase Assay.
Adenylyl cyclase activity was assayed using a modification of the method described by Salomon et al. (1974). Membranes were diluted to a final protein concentration of 0.1 to 0.2 mg/ml, to achieve 5 to 10 μg per reaction. Incubation was carried out at 30°C for 10 min with 40 mM HEPES, pH 7.7, 6 mM MgCl2, 1 mM EDTA, 100 μM ATP, 1 μM GTP, 0.1 mM 1-methyl-3-isobutylxanthine, 8 mM creatine phosphate, 16 U/ml creatine kinase, and 2 μCi of [α-32P]ATP (30 Ci/mmol; NEN Life Science Products, Boston, MA) in a total volume of 100 μl. Each point was assayed in triplicate, with six to eight concentrations of epinephrine bracketing the EC50. The [32P]cAMP produced in the assay was collected using Dowex and Alumina columns as described previously (Clark et al., 1988). Graph-Pad software was used to estimate the EC50 and theV max values.
Quantification of Coupling Efficiency and Desensitization.
We have previously described the equation for coupling efficiency, given below (Whaley et al., 1994)
Two other equations from Whaley et al. (1994) describe the changes inV
max (eq. 4) and EC50(eq. 5) as a function of receptor level, r. Equation 5Eqs. 4 and 5 can be used to predict the changes in EC50 and V
max that occur with decreases in k1r as a result of desensitization or experimental manipulation of r. Whaley et al. (1994)showed that at high receptor density (>200 fmol/mg) where the EC50 ≪ K
d, a decrease in k1r resulted in a large change in EC50 and almost no decrease inV
max, a finding predicted from eqs. 4 and5. Therefore at the high receptor densities used here, receptor-level desensitization will be represented primarily through EC50 shifts. Thus, any significant decrease inV
max for epinephrine stimulation after agonist-induced desensitization can be attributed to effects on components downstream of βAR/Gs coupling.
Equation 5Eqs. 4 and 5 can be used to predict the changes in EC50 and V
max that occur with decreases in k1r as a result of desensitization or experimental manipulation of r. Whaley et al. (1994)showed that at high receptor density (>200 fmol/mg) where the EC50 ≪ K
d, a decrease in k1r resulted in a large change in EC50 and almost no decrease inV
max, a finding predicted from eqs. 4 and5. Therefore at the high receptor densities used here, receptor-level desensitization will be represented primarily through EC50 shifts. Thus, any significant decrease inV
max for epinephrine stimulation after agonist-induced desensitization can be attributed to effects on components downstream of βAR/Gs coupling.
Measurement of Receptor Internalization by [3H]CGP-12177 Binding.
Epinephrine-stimulated βAR internalization was measured as described previously (January et al., 1997; Seibold et al., 1998). Cells were plated into 12-well dishes that had been precoated with poly-l-lysine to aid cell adhesion. The cells were pretreated with either carrier or epinephrine from 100× stocks. Pretreatment was performed at 37°C in 5% CO2 for various times and was stopped by removal of media followed by 6 ice-cold DMEM rinses. To each well, 1 ml of serum-free DMEM was added containing 10 nM [3H]CGP-12177 (CGP) to measure surface receptor number. Incubations were performed on ice for 1 h. The assays included triplicate points, and nonspecific binding was determined by inclusion of 1 μM alprenolol. Some assays included 0.2% digitonin during the CGP incubation to measure total receptor number, including the internalized pool. After incubation, the CGP mixture was removed and the wells were washed twice with ice-cold PBS. The cells were scraped into 0.5 ml of trypsin and liquid scintillation counting was performed. Internalization data are plotted as the percentage of surface receptor number measured in carrier (AT)-treated samples. The data were fit to the curve for monoexponential decay and Graph Pad software was used to estimate the apparent rate of internalization.
βAR Recycling Assay.
Cells were seeded in 12-well dishes coated with poly-l-lysine and grown to confluence. The cells were pretreated with either carrier or 1 μM epinephrine for 20 min. The concentration of 1 μM epinephrine permitted more complete washout compared with 10 μM and still provided about 70% receptor occupancy. At 20 min, the medium was removed and the cells were rinsed three times with 2 ml of warm (37°C) DMEM plus 10% fetal bovine serum and then refed with the same. The cells were incubated at 37°C for 0 to 60 min to allow recycling. Recycling was stopped by removal of media and two rinses with ice-cold PBS. Serum-free DMEM containing about 10 nM CGP was then added with and without 1 μM alprenolol and the cells incubated on ice for 1 h. The CGP mixture was removed and the cells rinsed twice with ice-cold PBS. The cells were scraped into 0.5 ml of trypsin and liquid scintillation counting was performed. Surface receptor number is reported as a percentage of that found in the carrier-treated control. The return of receptors to the cell surface was fit to the curve for monoexponential decay and the rate of recycling determined. The rate of endocytosis was calculated according to eq. 6, described by Koenig and Edwardson (1994).

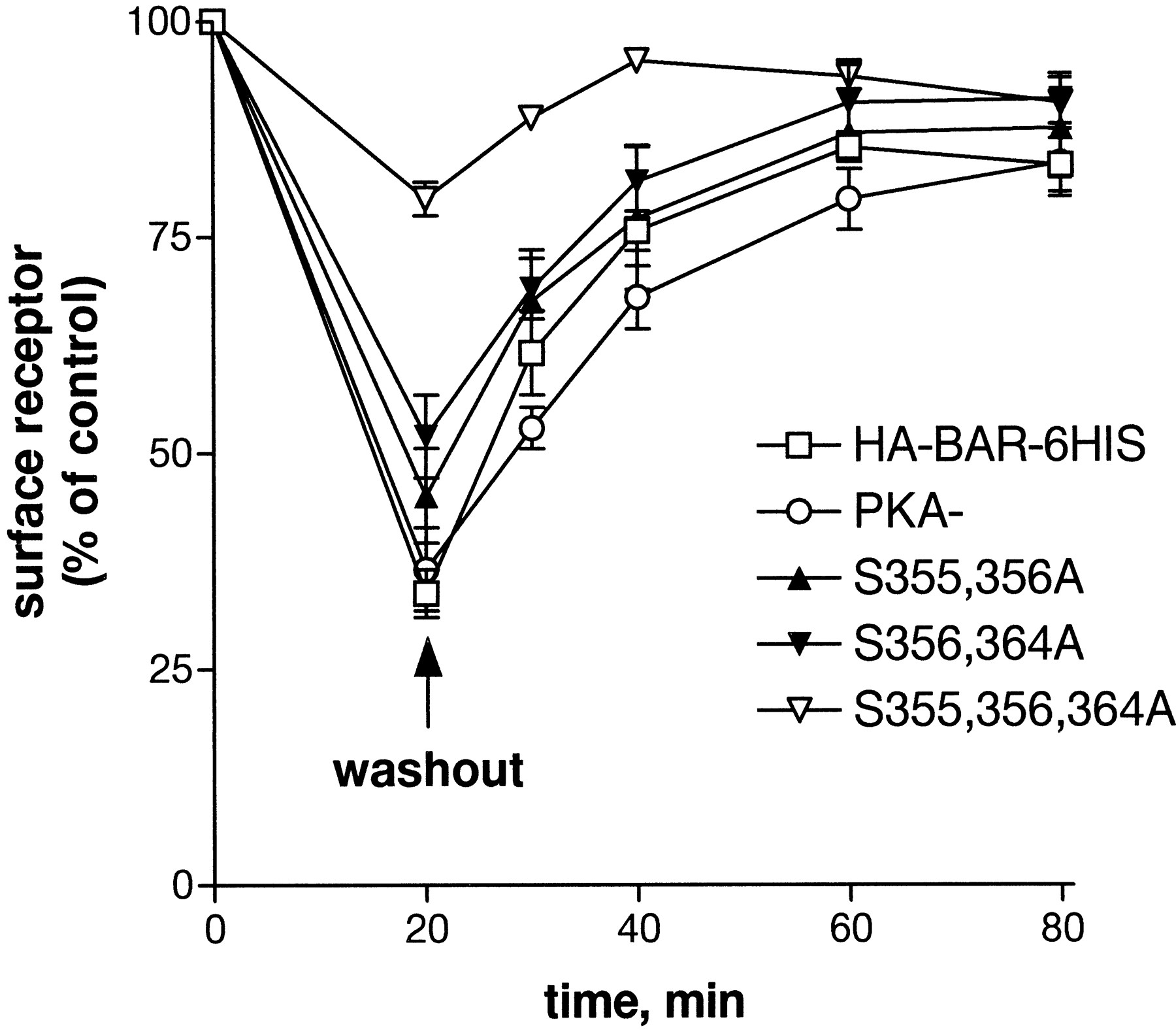
Time course of βAR recycling to the cell surface after washout of epinephrine. Cells expressing the HA-βAR-6HIS (■), PKA−(○), S355,356A (▴), S356,364A (▾), or S355,356,364A (▿) were pretreated with 1 μM epinephrine for 20 min. Epinephrine was washed out as described under Materials and Methods and the cells incubated for an additional 10 to 60 min to allow return of βARs to the surface. The number of surface receptors was measured using [3H]CGP-12177 binding and is expressed relative to carrier-treated control. Each point represents the mean of at least two experiments with triplicate points.
Observation of βAR Internalization by Immunofluorescence.
Stably transfected cells were plated on poly-d-lysine-coated #1 glass cover slips in 35-mm culture dishes, grown to 50 to 80% confluence, then chilled on ice. Monoclonal antibody mHA.11 (Berkeley Antibody Co., Berkeley, CA) was added to 2 μg/ml and the incubation on ice continued for 60 min. The monolayers were washed three times with ice-cold medium, then warmed to 37°C for 5 min or 30 min in the presence or absence of 10 μM isoproterenol. The monolayers were then rapidly chilled, washed once with PBS containing 1.2% sucrose (PBSS) and fixed at 4°C for 10 min with PBSS containing 4% paraformaldehyde (Electron Microscopy Sciences, Ft. Washington, PA). The fixed cells were incubated in 0.34%l-lysine, 0.05% Na-m-periodate in PBSS for 20 min, washed, and permeabilized with 0.2% Triton X-100 for 5 min, then blocked for 15 min with 10% heat-inactivated goat serum. Goat anti-mouse IgG conjugated with Alexa Fluor 488 (Molecular Probes, Eugene, OR), diluted to 5 μg/ml in PBSS with 0.2% heat-inactivated goat serum and 0.05% Triton X-100, was added to the cells and left overnight in the dark. The cover slips were mounted in Mowiol (Calbiochem, La Jolla, CA) and imaged with a DeltaVision Restoration Microscopy System (Applied Precision, Issaquah, WA). The digitized images were then deconvoluted with a DeltaVision workstation running DVSoftWoRx and assembled using Adobe Photoshop 5.5 (Adobe Systems, Mountain View, CA).
Determination of βAR Phosphorylation.
The methods used are modifications of those described previously (January et al., 1997). Cells were plated into 100-mm dishes precoated with poly-l-lysine and grown to confluence. Cells were rinsed once with phosphate-free DMEM and then incubated with 0.5-mCi [32P]orthophosphate in phosphate-free DMEM containing 1% fetal bovine serum for 3 h at 37°C in 5% CO2. The labeling medium was removed and replaced with 5 ml of bicarbonate- and phosphate-free DMEM containing 10% FBS. After 30 min equilibration the cells were treated with 10 μM epinephrine or AT carrier for the indicated times. The medium was removed and the cells rinsed with 5 ml ice-cold PBS. The dishes were placed on ice and scraped into 3 ml of PBS containing 10 μg/ml leupeptin and 100 nM okadaic acid. The cells were collected by centrifugation at 2000 rpm in an IEC DPR 6000 centrifuge. The cell pellet was solubilized by vortexing in buffer containing 20 mM HEPES, pH 7.4, 300 mM NaCl, 0.8%n-dodecyl-β-d-maltoside (DBM), 5 mM EDTA, 3 mM EGTA, 20 mM sodium pyrophosphate, 10 mM sodium fluoride, 25 mM imidazole, 10 μg/ml benzamidine, 10 μg/ml trypsin inhibitor, 100 nM okadaic acid, 10 μg/ml leupeptin, and 14 mM β-mercaptoethanol. After 30 min rocking at 4°C, the solubilized cells were centrifuged for 30 min at 45,000 rpm in a Beckman 50 Ti rotor.
After solubilization, the βAR was purified using either of two procedures. Procedure 1 used for the majority of experiments consisted of a Ni-NTA affinity step followed by either wheat germ agglutinin-agarose (WGA) chromatography or immunoprecipitation. The solubilized supernatant was applied to Ni-NTA superflow resin (Qiagen, Valencia, CA) packed into disposable columns (Bio-Rad), 0.8 ml of 2× slurry per column. The eluate was collected and recycled onto the columns. The columns were washed once with 5 ml of buffer containing 0.05% DBM, 20 mM HEPES, pH 7.4, 300 mM NaCl, 25 mM imidazole, 4 M guanidine HCl, and 1 M LiCl. After a rinse with 5 ml of Ni2+ column buffer (0.05% DBM, 20 mM HEPES, pH 7.4, 300 mM NaCl, 25 mM imidazole), the βAR was eluted in a single step with 4 ml of buffer containing 0.05% DBM, 20 mM HEPES, pH 7.4, 300 mM NaCl, and 100 mM imidazole. The βAR was further purified using either WGA or immunoprecipitation. Similar results were obtained with both procedures. The WGA step was performed as described previously (January et al., 1997; Seibold et al., 1998). For immunoprecipitation, each 4-ml Ni2+ column eluate was precleared with 50 μl of a 2× protein A Sepharose slurry (Pharmacia, Piscataway, NJ). Preclearing was performed in a 1 h incubation at 4°C with rocking. Samples were centrifuged at 2000 rpm in the IEC DPR 6000, the supernatants were transferred to fresh tubes, and 20 μl (4 μg) of anti-βAR antibody added (antibody SC-569 directed against the C-terminal 20 amino acids; Santa Cruz Biotechnology, Santa Cruz, CA). The samples were incubated for 90 min at 4°C with rocking, after which 50 μl of Protein A Sepharose was added and the samples further incubated for 1 h at 4°C with rocking. The immune complexes were centrifuged for 5 min in the IEC DPR 6000, the supernatants aspirated, and the pellets washed twice with 2 ml of Ni column buffer. To each pellet was added 125 μl of SDS sample buffer (50 mM Tris, pH 6.8, 2% SDS, 0.025% bromphenol blue, 6 M urea, and 14 mM β-mercaptoethanol). The samples were incubated at 60°C for 15 min with frequent vortexing. The samples were transferred to Eppendorf tubes, briefly spun in a Microfuge and loaded onto 7.5% SDS-polyacrylamide gels along with prestained molecular mass markers. After electrophoresis, the proteins were transferred from the gel to 0.22-μm polyvinylidene difluoride (PVDF) membranes.
In the course of these studies, a substantially modified procedure was developed that allowed better recovery and considerably improved quantification and will be referred to as procedure 2. In outline, this procedure consisted of a C-tail antibody affinity column,N-glycosidase F treatment of the antibody column eluate, and a Talon affinity resin step (Co2+-carboxymethylaspartate-agarose; Clontech, Palo Alto, CA). Solubilized βAR (equal amounts from control and epinephrine-treated based on βAR levels in the extracts) was applied to a 100-μl packed volume of antibody resin in a column (SC-569 C-tail antibody from Santa Cruz linked to agarose) that had been prewashed with PBS, pH 7.0. After recycling the extract three times through the column, it was washed once with 3 ml of 10 mM phosphate buffer, pH 6.8, containing 0.05% DBM. The βAR was eluted with 1 ml of 100 mM glycine buffer, pH 2.5, plus 0.05% DBM and the eluate collected in 0.3 ml 1 M phosphate buffer, pH 8.0 for neutralization. The eluate was digested with 1500 units of N-glycosidase F (New England Biolabs, Beverly, MA) for 2 h at 37°C, and applied to the Talon Co2+-carboxymethylaspartate-agarose column (0.5 ml packed resin) that had been prewashed with Talon buffer (0.05% DBM, 20 mM HEPES, pH 7.4, 150 mM NaCl) and recycled through the resin two times. The column was washed twice with Talon buffer, once with 4 ml of 10 mM imidazole, and finally with 0.25 ml of 20 mM imidazole, all in the same buffer. The βAR was eluted with 0.75 ml of Talon buffer containing 100 mM imidazole and concentrated to 50 μl in a centricon (Amicon; 30-kDa cutoff). SDS-sample buffer was added to the 50 μl of eluate and heated at 60°C for 15 min. Samples were run on SDS-polyacrylamide gel electrophoresis (PAGE; 12% gel) and transferred to nitrocellulose.
PhosphorImager analysis was performed on the PVDF or nitrocellulose membranes from either procedure using a Molecular Dynamics Storm PhosphorImager model 860 and ImageQuant software (Molecular Dynamics, Sunnyvale, CA). Western blotting was performed using the anti-HA antibody or the anti-carboxyl terminal βAR antibody as the primary antibody as described previously (Seibold et al. 1998). After incubation with the secondary antibody, a horseradish peroxidase-conjugated goat anti-rabbit (Bio-Rad) antibody, enhanced chemiluminescence was performed.
Results
Determination of the Coupling Efficiency for Epinephrine Activation of Adenylyl Cyclase for the HA-βAR-6HIS and Mutant βARs.
To eliminate the possibility that the mutations we introduced into the βAR caused nonspecific effects on coupling, we determined the coupling efficiency value for each mutant using eq. 1. Calculation of coupling efficiency requires measurement of receptor number, the low-affinity K d value for agonist binding, and the EC50 value for adenylyl cyclase activation (Whaley et al., 1994). We calculated the coupling efficiency for at least two clones of each mutant. The coupling efficiencies and the experimentally determined values used in its calculation are summarized in Table 2. None of the mutant receptors showed a significant alteration of theK d value for epinephrine binding relative to the HA-βAR-6HIS or the PKA−. The variation we observed in coupling efficiencies in this study are typical and reflect the variation in the three parameters used in the calculation of coupling efficiency.
Characterization of βARs stably expressed in HEK 293 cells
Desensitization of the HA-βAR-6HIS and Mutant βARs.
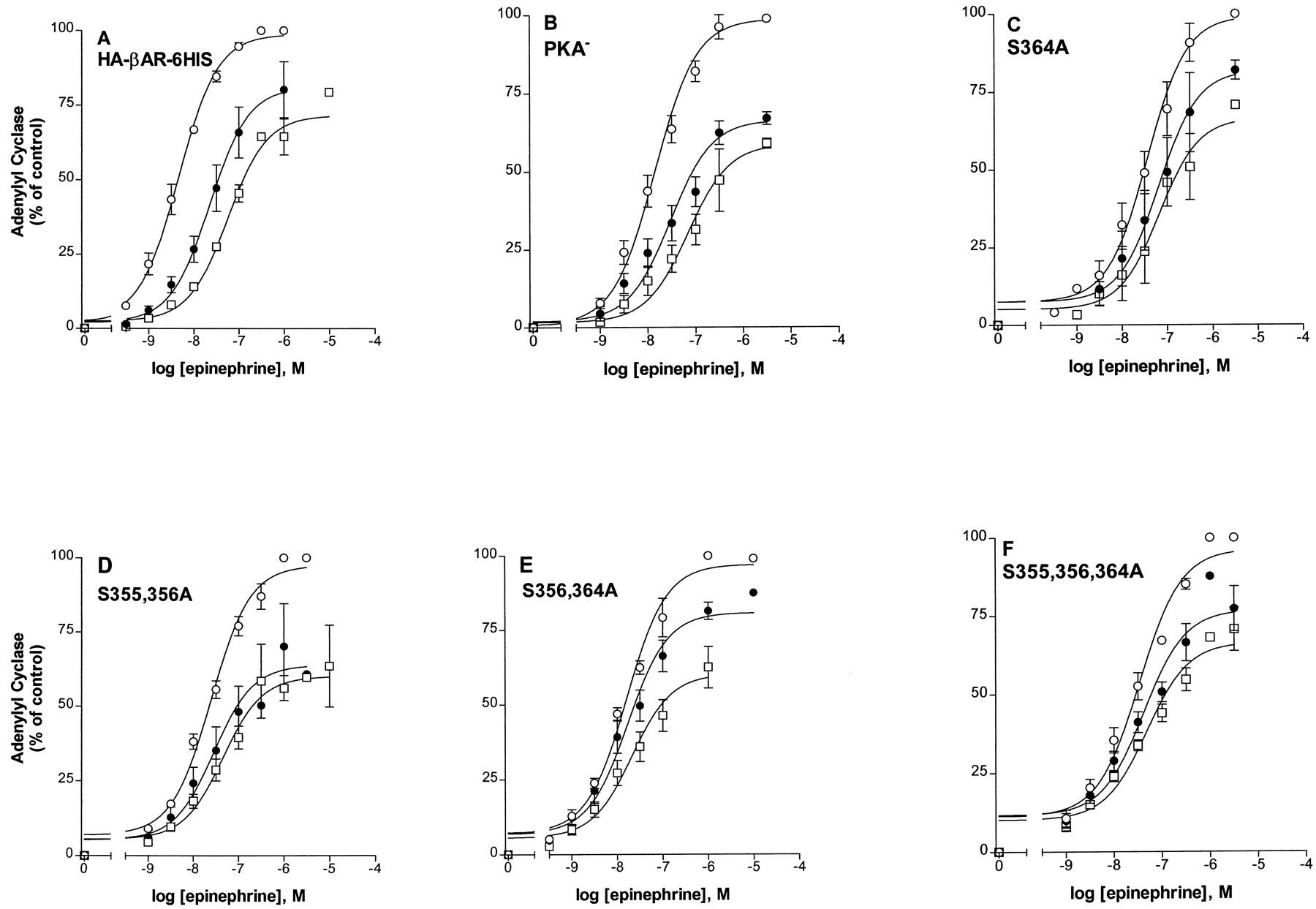
To measure desensitization, cells expressing the mutant βARs were pretreated with 10 μM epinephrine or carrier for various times. Membranes were prepared and assayed for activation of adenylyl cyclase with a range of epinephrine concentrations. Figure2 shows typical data from epinephrine dose-response curves in which adenylyl cyclase activity was measured in membranes prepared from cells pretreated with carrier (control) or 10 μM epinephrine for 2 or 5 min. The increase in EC50 value and the decrease in theV max value with 10 μM epinephrine pretreatment were measured in many experiments, such as those in Fig.2, and used to calculate desensitization as fraction activity remaining for the various pretreatment times (1 to 30 min), as summarized in Fig.3. Compared with the HA-βAR-6HIS and the PKA−, the triple and double mutants showed greatly reduced desensitization. Desensitization was measured in at least two clones for each mutant. For the 30-min pretreatment with 10 μM epinephrine, the percentage of activities remaining for the HA-βAR-6HIS, PKA−, S355,356,364A, S355,356A, and S356,364A, respectively, were 3% ± 0.3, 8% ± 1.0, 72% ± 8, 28% ± 4, and 64% ± 15. Relative to PKA−, these values are 9 (S355,356,364A), 3 (S355,356A), and 8 (S356,364A) times greater and highly significant (P < .001). Compared with HA-βAR-6HIS, these values are 24, 9, and 21 times greater for the triple- and double-serine mutants. The PKA− mutant provides the most appropriate comparison because all of the mutant βARs described here contain alanine substitutions for the serines of consensus PKA sites. The two single-serine substitutions, S356A and S364A, were not as effective as the double mutants in impairing desensitization. However, the percentage of activities remaining after 30-min desensitization were 16% ± 3 and 20% ± 1, which differed significantly from the PKA− (P < .05).
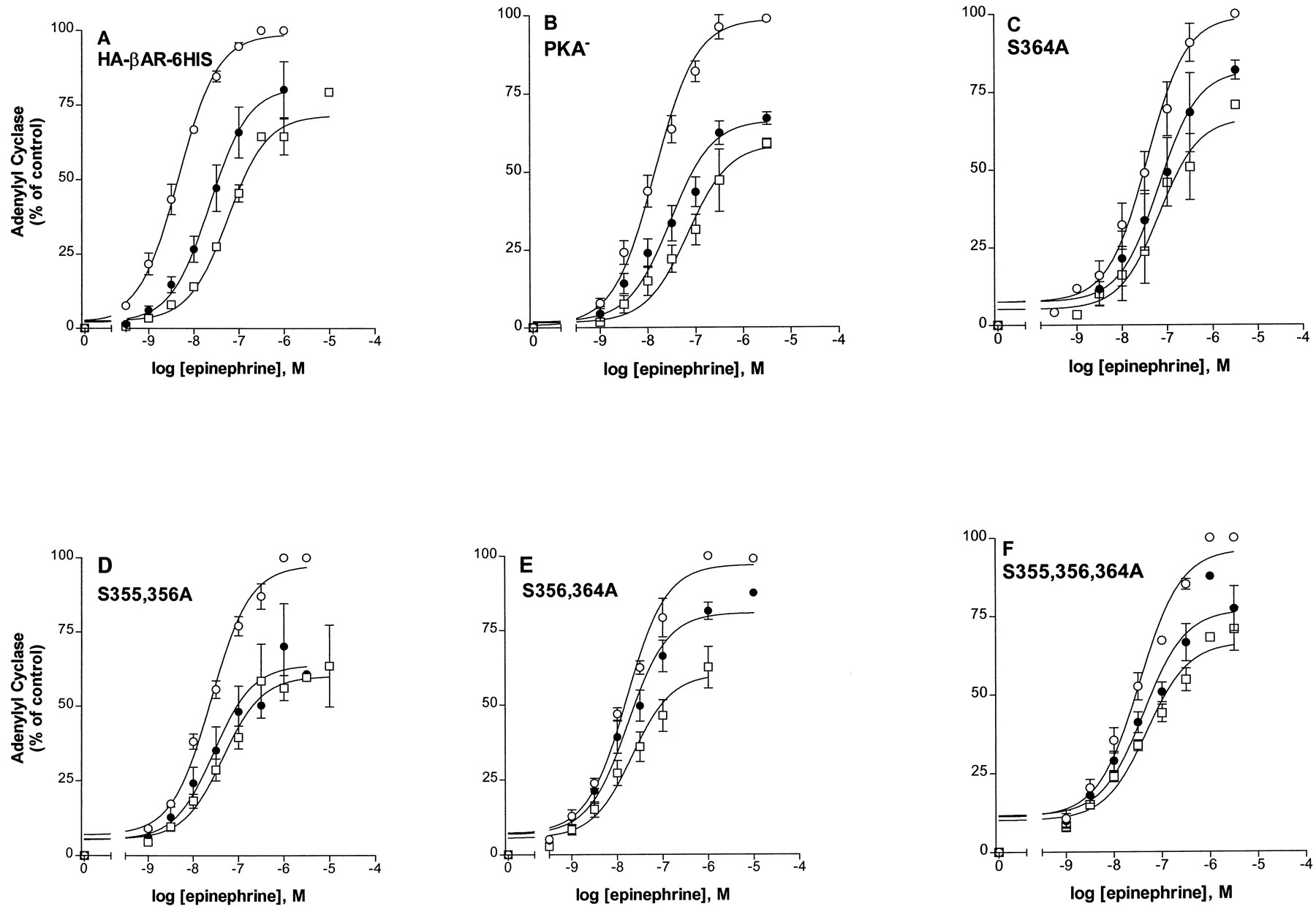
Epinephrine-induced desensitization of the HA-βAR-6HIS and mutant βARs. Cells expressing the HA-βAR-6HIS (A), PKA− (B), S364A (C), S355, S356A (D), S356, S364A (E), or S355,356,364A (F), were pretreated with carrier (○) or with 10 μM epinephrine for 2 (●) or 5 min (■). Membranes were prepared and assayed for adenylyl cyclase activity in triplicate with the indicated epinephrine concentrations. The results are shown normalized to the V max for the carrier-treated sample (set to 100%) after subtraction of basal. The data summarize at least two representative experiments for each receptor type.

Time course of HA-βAR-6HIS and mutant βAR desensitization in response to 10 μM epinephrine. Cells expressing the HA-βAR-6HIS (■), the PKA− (○), S355,356A (▴), S356,364A (▾), S356A (♦), S364A (▪), or S355,356,364A (▿) were pretreated with carrier or 10 μM epinephrine for various times from 1 to 30 min. Membranes were prepared and assayed for epinephrine-stimulated adenylyl cyclase activity as described underMaterials and Methods. The values for fraction activity remaining were calculated according to eq. 3. Each point was calculated using data from at least three experiments, except for the 10 min data for S355,356,364A and the 30 min data for the single mutants, S356A and S364A, where n = 2. Each experiment included separate adenylyl cyclase dose response curves for the epinephrine treated and carrier (control) pretreated samples. Each point in the adenylyl cyclase dose response curves was the average of triplicates.
Although the various serine substitutions caused reductions in the right shift of the EC50 for epinephrine stimulation of adenylyl cyclase after epinephrine-induced desensitization, the mutations had no significant effect on the epinephrine-induced decrease in the V max(Figs. 2 and 3). To show more clearly the differential effect of the mutants on these two parameters, the fold increase in EC50 and the percentage decrease inV max after 5 min of epinephrine pretreatment are given in Fig. 4 and Table 3. The triple serine mutation completely eliminated the epinephrine-induced increase in the EC50, whereas the EC50 of the PKA− and HA-βAR-6HIS increased 3.4-fold and 10.8-fold, respectively. For the S355,356A and S356,364A mutants the EC50 after desensitization increased 1.5 and 1.2-fold, respectively. In contrast to the EC50changes, the extent of the epinephrine-induced decrease inV max was similar for the mutant βARs, PKA−, and HA-βAR-6HIS (Table 3), ranging between 31.0 and 47.7%. The data show that the reduced desensitization measured for the 355–364 domain mutants resulted from inhibition of EC50 shifts rather thanV max effects.

EC50 changes in response to epinephrine pretreatment in the HA-βAR-6HIS and mutant βARs. The data summarized in Fig. 3 were analyzed for the increase in EC50value that occurred in response to the 5-min pretreatment with 10 μM epinephrine. The fold increase in EC50 value measured with epinephrine treatment was calculated relative to the EC50value of the carrier-treated control. A 1-fold increase indicates no change. The data are given as the mean ± S.E.M. For the S356A mutant, n = 3. For all the other βARs,n ≥ 6. * indicates significantly different from PKA−, as determined by an unpaired t test at P < .0004. The ** indicates significantly different from PKA−, as determined by an unpairedt test at P < .0001.
Vmax changes in response to epinephrine pretreatment
This lack of effect of the mutants on theV max is consistent with the predictions from eqs. 4 and 5 under Materials and Methods and the data in Whaley et al. (1994), that changes in the EC50value rather than changes in V max values are the primary effects of receptor level desensitization at the high receptor densities used in these experiments. The data strongly suggest that the V max changes we observe are downstream of receptor/Gs coupling. To explore this possibility, we examined the effects of 10 μM epinephrine-induced desensitization (5-min pretreatment) on prostaglandin E1 (PGE1), GTPγS, and forskolin stimulation. We found this treatment of the PKA− cells caused decreases in both PGE1 (10 μM) and GTPγS (10 μM) stimulation of adenylyl cyclase (54 ± 5.4% and 25.4 ± 6.7% respectively). The effects on PGE1 and GTPγS stimulation support our suggestion that there is probably downstream desensitization. Similar effects of epinephrine-induced desensitization on these two activities were observed in the HA-βAR-6HIS. Forskolin (20 μM) stimulation was reduced only 8 to 14% in these cell lines, which is perhaps not surprising given that forskolin activates all adenylyl cyclases except type 9 and is unlikely to activate only that adenylyl cyclase coupled to the βAR. In fact, maximal forskolin activation is about double that of the maximal epinephrine stimulation.
Another possibility we evaluated was whether stimulation of Gi contributed to the epinephrine-induced decrease in V max. To test this, HA-βAR-6HIS and the PKA− cells were treated overnight in the presence or absence of pertussis toxin (100 ng/ml) and then desensitized by a 5-min treatment with epinephrine. Although pertussis toxin predictably increased theV max value for epinephrine stimulation of adenylyl cyclase in both controls and epinephrine-treated cells, it did not alter either the extent of the desensitization-induced decrease in the V max values for epinephrine stimulation of adenylyl cyclase or the overall extent of the desensitization.
Internalization and Recycling of the Mutant βARs
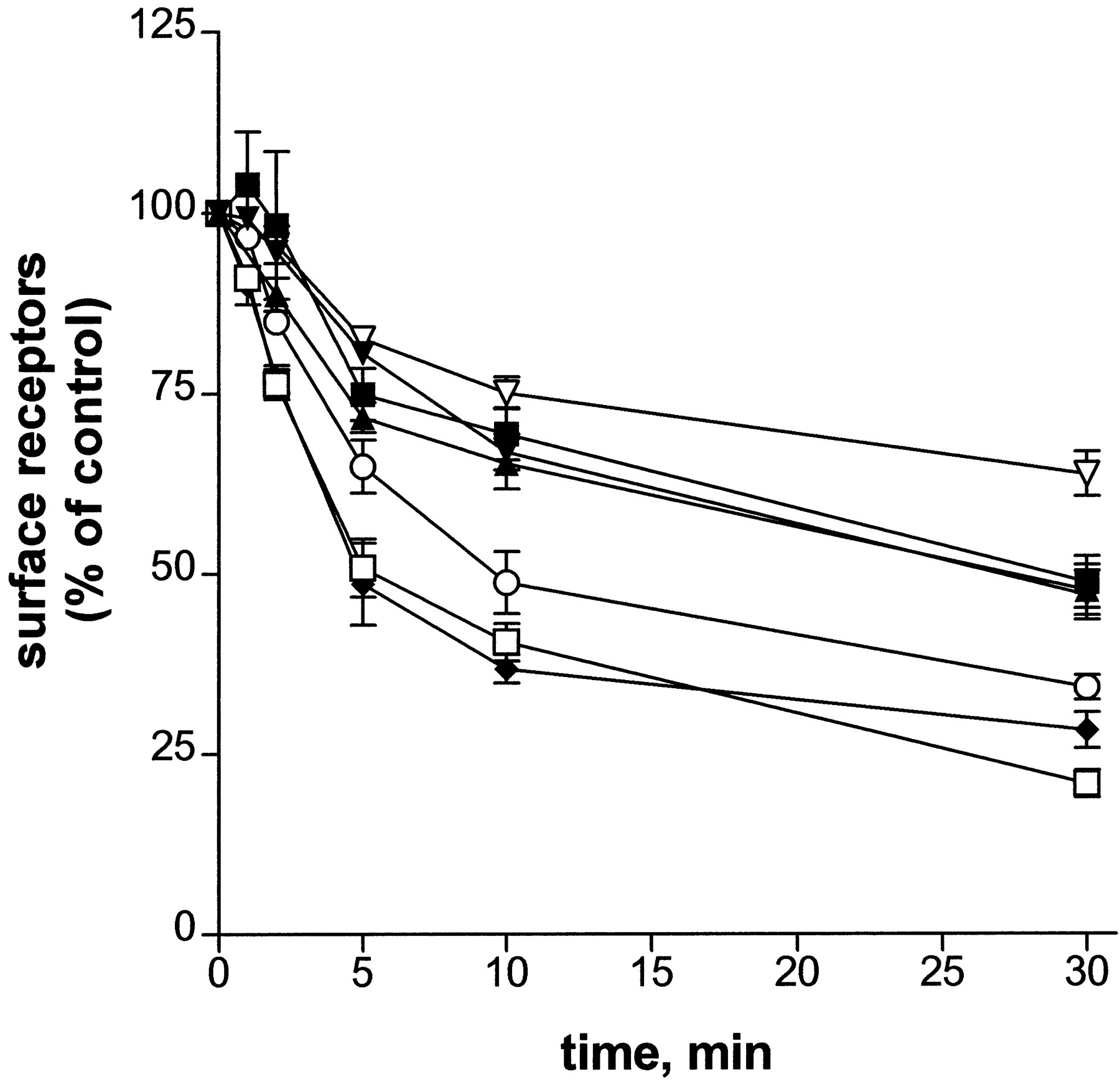
Cells expressing the various βARs were exposed to 10 μM epinephrine for various times, and receptor internalization was measured by [3H]CGP-12177 binding to intact cells. The results are shown in Fig.5 as the loss of surface receptor with time of epinephrine treatment. After 30 min of 10 μM epinephrine pretreatment, the percentage of surface receptors internalized was 79% (±2, n = 8), 66% (±2, n = 4), and 72% (±2, n = 4), for the HA-βAR-6HIS, PKA−, and S356A, respectively. The extent of internalization measured for the double mutants S355,356A and S356,364A and for the S364A mutant was reduced, with values of 52% (±3,n = 6), 53% (±3, n = 5), and 51% (±4, n = 5), respectively, a decrease of about 20% compared with PKA−. The triple-serine mutation caused the greatest decrease in the extent of internalization, giving a value of 36% (±3, n = 3). Fit of the averaged data shown in Fig. 5 to the curve for monoexponential decay showed that the observed rate of internalization was also reduced for the double, triple, and S364A mutants. The k obs determined for the HA-βAR-6HIS, PKA−, and S356A was 0.19 min−1, whereas that determined for the triple and double mutants and S364A was 0.11 min−1, a reduction of 42%. The correlation coefficients for the first-order decay curves were 0.996 or better for the different mutants. [3H]CGP-12177 binding performed in the presence of digitonin showed no loss of total βAR number during the internalization time course.

Time course of epinephrine-induced βAR internalization. Cells expressing the HA-βAR-6HIS (■), PKA− (○), S355,356A (▴), S356,364A (▾), S356A (♦), S364A (▪), or S355,356,364A (▿) were pretreated with carrier or 10 μM epinephrine for 1 to 30 min. After rinsing cells to remove epinephrine, the surface receptor number was measured using [3H]CGP-12177 binding as described under Materials and Methods. The surface receptor number measured for the carrier-treated control is set to 100%. The surface receptor number for the epinephrine treated samples is expressed as a percentage of control. Each point represents the mean ± S.E.M. of three to six assays, each performed in triplicate.
The rate of internalization is a composite of the rates of endocytosis and recycling (Morrison et al., 1996; Clark et al., 1999). Therefore, the rate of endocytosis can be calculated from the rate of recycling and the ratio of surface receptors to the total receptor number at steady state (eq. 6 under Materials and Methods). Determination of the recycling rate was necessary before concluding that the reduced observed rate of internalization for the double mutants and for S364A resulted from a reduced rate of endocytosis. To measure the rate of recycling, cells expressing the HA-βAR-6HIS, the PKA−, or the double and triple mutant βARs were pretreated with 1 μM epinephrine for 20 min, then rinsed to remove the hormone, and incubated for various times to allow the recycling of the βARs to the cell surface. The return of the βARs to the cell surface was measured by [3H]CGP-12177 binding (Fig.6). The HA-βAR-6HIS and mutant βARs all recycled to the cell surface after washout of epinephrine with similar rate constants, estimated from fit of the data to a monoexponential decay curve. From the average value of the rate constants, a recycling t1/2 of 10 min (krecycle= 0.07) was determined. Because the HA-βAR-6HIS and mutant βARs were found to recycle at the same rate, the differences in the rate and extent of internalization indicate different rates of endocytosis. The rate of endocytosis calculated for the double mutants was 0.08 min−1, a value 38% less than the rate of 0.13 min−1 calculated for PKA−. We were not able to obtain recycling rates for the triple mutant because the internalization was so small. However, it seems that its rate of recycling is not altered relative to the PKA− or the double mutants (0.07 min−1). Using this value, it can be calculated that the rate of endocytosis of the triple mutant is reduced about 70% relative to PKA−.
It was recently reported that the addition of various amino acids to the βAR carboxyl terminus inhibited receptor recycling to the plasma membrane (Cao et al., 1999). We measured internalization and recycling of the untagged WTβAR and found the data to be similar to those obtained for the HA-βAR-6HIS. In response to a 20-min pretreatment with 1 μM epinephrine, 66.2% (±2.77, n = 5) of the HA-βAR-6HIS, and 68.5% (±1.85, n = 2) of the untagged WTβAR were internalized. After removal of epinephrine and a 60 min recycling incubation, all but 16.7% (±3.09, n= 5) of the HA-βAR-6HIS, and 19.9% (±1.70, n = 2) of the untagged WTβAR had returned to the plasma membrane.
Observation of βAR Internalization Using Deconvolution Microscopy.
Cells expressing the PKA- or the triple and double mutants were incubated with antibody directed against the amino-terminal HA epitope tag, followed by treatment with either carrier or 10 μM isoproterenol for 5 or 30 min. The cellular location of the receptors in response to isoproterenol treatment was determined using immunofluorescent deconvolution microscopy. In the absence of agonist, all of the antibody-labeled mutant receptors remained at the cell surface. In the presence of agonist, there was rapid and substantial receptor internalization of the double mutants into peripheral endocytic vesicles (Fig. 7). The triple mutant seemed to internalize somewhat more slowly. Similar results were obtained using 10 μM epinephrine (data not shown). Although the immunofluorescent studies are not easily quantified, they are consistent with the results of CGP binding and indicate that internalization of the double and triple mutants and the PKA− results in a similar localization to endocytic vesicles.

Observation of mutant βAR internalization by immunofluorescence. Cells expressing the PKA−, the S355,356A, the S356,364A, or the S355,356,364A mutant receptors were incubated with antibody directed against the HA epitope and then exposed to either carrier or 10 μM isoproterenol for 5 or 30 min. Images were obtained using deconvolution immunofluorescence as described under Materials and Methods. The panels show representative fields. Magnification, 630×.
Phosphorylation of the Mutant βARs in Response to 10 μM Epinephrine.
Cells expressing the PKA−, S355,356A or the S356,364A mutant βARs, were labeled with [32P]orthophosphate and then treated with either 10 μM epinephrine or carrier for 2 min. The cells were solubilized and the βAR protein purified by procedure 1 as described under Materials and Methods. The purified βAR was subjected to SDS-PAGE and transferred to a PVDF membrane. The PhosphorImager analysis of a representative experiment is shown in Fig.8A. To estimate relative loading, a Western blot was performed on the same membrane, using antibody directed against the βAR carboxyl terminus (Fig. 8B). The data are representative of 4 independent experiments and show that the double mutants are rapidly phosphorylated in response to epinephrine pretreatment. Phosphorylation of the PKA−increased 7-fold in response to a 2-min pretreatment with 10 μM epinephrine, whereas phosphorylation of the double mutants S355,356A and S356,364A increased only 3.3- to 4-fold (Fig.9). The time courses of phosphorylation of the double mutants and PKA− were similar (data not shown), showing a maximum level at 2 to 5 min, after which phosphorylation declined.

Phosphorylation of the PKA− and double mutant βARs in response to a 2 min pretreatment with 10 μM epinephrine. Cells expressing the PKA−, the S355,356A or the S356,364A mutant, were labeled with 32P orthophosphate for 3 h and then exposed to either 10 μM epinephrine (epi) or carrier (c) for 2 min. The cells were solubilized and the receptor protein purified using Ni column chromatography and immunoprecipitation, as described by procedure 1 in “Materials and Methods”. The receptor protein was resolved by SDS-PAGE and transferred to a PVDF membrane. A, PhosphorImage of the PVDF membrane. Western blot analysis (B) was performed on the same membrane using antibody directed against the βAR carboxyl terminus. The data are representative of four similar experiments.

Phosphorylation of the PKA− and double mutant βARs measured as fold increase. Cells expressing the PKA−, S355,356A, or S356,364A mutant βARs were pretreated with 10 μM epinephrine or carrier for 2 min. The cells were solubilized and the βAR purified as described by procedure 1 under Materials and Methods. The fold increase in phosphorylation was determined by dividing the cpm obtained after epinephrine treatment with that measured in the basal, carrier-treated state. The data represent the mean ± S.E.M. of at least four experiments for each receptor type. * indicates significantly different from PKA− by an unpaired t test atP < .05.
To measure the phosphorylation of the double and triple serine mutant relative to the PKA−, [32P]labeled cells were pretreated with epinephrine for 2 min and the βARs purified by procedure 2 as discussed under Materials and Methods. The PhosphorImager scan and accompanying Western blots of a typical experiment (including comparison with S356,364A) are shown in Fig.10. In this purification procedure, the glycosyl residues are removed by treatment withN-glycosidase F (between the antibody affinity and the Talon affinity steps); this results in the migration of the βAR to a molecular mass of ≅ 48 to 50 kDa. The Western blot using the anti-HA antibody shows that similar levels of βAR were purified from the three cells lines (each antibody column was loaded with 375 fmol of solubilized βAR). The 2-min epinephrine treatment of the triple serine mutant and PKA− caused 2- and 15-fold increases in phosphorylation over basal, respectively. Phosphorylation of the double mutant was 6.4-fold over basal (i.e., 39% of PKA−). From three independent experiments, including the one in Fig. 10, we found the fold stimulation of the triple mutant over basal was 1.86 ± 0.18-fold compared with 16.6 ± 3.8 for PKA−. Thus the epinephrine-stimulated phosphorylation of the triple mutant is only ∼5% that of the PKA−. Nevertheless it is important to emphasize that phosphorylation was not eliminated.

Phosphorylation of PKA−, S356,364A, and S355,356,364A in response to a 2-min pretreatment with 10 μM epinephrine. Cells expressing the PKA−, the S356,364A, or the S355,356,364A mutant, were labeled with [32P]orthophosphate for 3 h and then exposed to either 10 μM epinephrine (epi) or carrier (c) for 2 min. The cells were solubilized and the receptor protein purified using antibody and Talon chromatography and digested with PNGase F, as described by procedure 2 under Materials and Methods. The receptor protein was resolved by SDS-PAGE and transferred to a nitrocellulose membrane. A, PhosphorImage of the nitrocellulose membrane. B, Western blot analysis was performed on the same membrane using antibody directed against the HA epitope in the βAR amino terminus. The data are representative of three experiments performed with the PKA− and the S355,356,364A mutant, and two experiments performed with the S356,364A mutant.
It can be seen that procedure 2 focuses the βAR to a tight band, greatly improving our ability to quantify 32P relative to the glycosylated βAR that spreads over a 15- to 20-kDa region of the gels. Additionally we have found that the background is reduced resulting in larger fold-stimulations relative to procedure 1.
Discussion
Our data show that mutations of either two or three of the serines in the βAR 355–364 domain caused a striking reduction in epinephrine-induced receptor-level desensitization of the βAR (EC50-shift) without altering the decrease inV max that seems to be downstream. These mutations were made in a receptor in which both PKA consensus sites were ablated to eliminate any contribution of PKA desensitization of the βAR. The complete elimination of the EC50shift in the S355,356,364A mutant coupled with the 90 to 95% loss of phosphorylation and considerably reduced extent of internalization (45%) relative to PKA− are consistent with the proposal that the crucial sites for homologous GRK-mediated desensitization of the βAR lie in the 355 to 364 amino acid region with the caveat that phosphorylation was not eliminated. Our data are consistent as well with the studies of rhodopsin phosphorylation by GRK1 (rhodopsin kinase). Two serines in the carboxyl tail of mouse rhodopsin are phosphorylated in vivo in response to light (Ohguro et al., 1995). These two serines lie in the region of rhodopsin most homologous to the 355 to 364 domain of the βAR [see Collins et al. (1991) for amino acid alignment]. In addition, in vitro studies have shown that rhodopsin kinase can phosphorylate the βAR (Benovic et al., 1986), and GRK2 (βARK1) can phosphorylate rhodopsin (Benovic et al., 1987). The evidence suggests that the βAR and rhodopsin share similar sites for recognition by their respective kinases.
Table 3 and Fig. 4 show further that the decreased desensitization of the triple and double mutants is almost completely attributable to the lack of change of the EC50 for epinephrine stimulation. What little desensitization is observed with S355,356,364A and S356,364A is caused for the most part by the decrease in theV max value. This is not significantly different from the decrease in V max values observed with the PKA− or the HA-βAR-6HIS (Table 3), demonstrating that the disruption of desensitization found in serine substitution mutants of the βAR 355–364 region results from changes in values of EC50 rather than those of V max. Loss of βAR/Gs coupling with receptor-level desensitization is predicted (by eqs. 4 and 5 under Materials and Methods) to be represented primarily by changes in the value of EC50 rather than changes in that ofV max at the high receptor density reported here (Whaley et al., 1994). The epinephrine-induced decreases in PGE1 and GTPγS stimulation of adenylyl cyclase support the idea that there is significant heterologous downstream desensitization. Unfortunately, we were unable to detect significant decreases in forskolin stimulation with desensitization; however, as noted previously, this could be explained by the fact that forskolin activates all adenylyl cyclases except type 9, and this may obscure the contribution of the subtype altered by the epinephrine pretreatment. The contribution of Gi to the 40% decrease inV max was also explored through the use of pertussis toxin and found not to contribute either to theV max or to the overall desensitization. At present therefore, the cause of the decrease inV max in these cells remains unexplained; however, the cells expressing the triple mutant will be ideal for examining this phenomenon in future studies because theV max effect is not complicated by receptor-level changes.
Although receptor-level desensitization of the double mutants was greatly impaired relative to the HA-βAR-6HIS and PKA−, internalization and phosphorylation were not comparably reduced. We considered how our results could be reconciled with the currently accepted scheme for the βAR desensitization process which proposes that GRK-phosphorylation of the βAR is followed by β-arrestin binding, β-arrestin-promoted endocytosis, and subsequent recycling (Krupnick and Benovic, 1998;Lefkowitz et al., 1998; Clark et al., 1999). Each step in this scheme of βAR regulation, including GRK phosphorylation, β-arrestin binding, and internalization, is dependent on the previous step. However, as we have shown previously in HEK 293 cells, the inter-relationships between these processes during the time of agonist stimulation are complex (January et al., 1997). Phosphorylation of the βAR shows a rapid rise, reaches a maximum by 2 to 5 min, and then declines, whereas levels of endocytosed βAR continue to increase until a steady state of endocytosis and recycling is achieved.
The most straightforward conclusion is that our double mutants have ablated two of the three crucial GRK sites that are required for β-arrestin-mediated desensitization, but that the additional GRK site must be blocked to achieve near complete blockade of β-arrestin binding. The first evidence for multisite interaction between receptors and arrestin came from studies of rhodopsin and visual arrestin. Mutagenesis studies showed that at least three regions of visual arrestin interact with phosphorylated, light-activated rhodopsin to terminate signaling (Gurevich and Benovic, 1993; Gurevich, 1998;Vishnivetskiy et al., 1999). Similar work suggests that multisite binding mediates β-arrestin-βAR interaction (Kovoor et al., 1999). Although domains of arrestin and β-arrestin important for receptor interaction have been identified, the corresponding sites in the respective receptors were not known previously. Regions of the receptors likely to be important for arrestin interaction include GRK-phosphorylated residues, the third intracellular loop (a region important for contact with G proteins), and undefined domains that result from agonist binding-induced conformational change (Gurevich and Benovic, 1993; Krupnick et al., 1994; Gurevich, 1998; Kovoor et al., 1999; Vishnivetskiy et al., 1999). In support of this model, the recently determined crystal structure of visual arrestin provided a firm structural basis for the proposed conformational alterations of arrestin and multisite binding to receptor (Granzin et al., 1998;Hirsch et al., 1999).
Given the validity of the multisite model of β-arrestin binding to the βAR, there are several schemes consistent with our desensitization, phosphorylation, and internalization data. When agonist-bound, the double or triple mutants may retain sites for low-affinity β-arrestin interaction sufficient to promote internalization, albeit at a reduced rate, but not adequate to provide a stable, uncoupled state of the βAR at the cell surface. A mutagenesis study of the m2 muscarinic receptor provides precedent for this scheme. Although two regions of the third intracellular loop were phosphorylated in response to agonist, mutagenesis of either region alone did not reduce receptor phosphorylation or internalization, and were described as “redundant” for these functions (Pals-Rylaarsdam and Hosey, 1997). In contrast, desensitization was eliminated by mutagenesis of the more carboxyl terminal region. The m2 muscarinic receptor study and our results support the model of multisite receptor-β-arrestin interaction and demonstrate that selective mutagenesis of the receptor can identify a subset of GRK phosphorylation sites important for high-affinity β-arrestin binding and desensitization but possibly leave intact other weak β-arrestin interaction sites that contribute to endocytosis.
As an alternative to the model of multisite βAR-β-arrestin interaction, the data reported here may indicate the existence of a separate but minor internalization pathway that is not dependent on receptor phosphorylation by GRK or β-arrestin binding. Arrestin-independent internalization pathways have been reported for the m1, m3, and m4 muscarinic receptors (Lee et al., 1998) and the angiotensin II type 1A receptor (Zhang et al., 1996). An internalization pathway that is independent of β-arrestin and GRK phosphorylation, but dependent on PKA, was reported for the secretin receptor (Walker et al., 1999). In addition, dominant negative β-arrestin does not completely block βAR internalization (Ferguson et al., 1996), perhaps because of the contribution of a β-arrestin-independent pathway. These studies suggest that the lack of correlation between the extent of βAR desensitization and internalization reported here may result from the use of an alternate internalization pathway that depends on agonist binding but not GRK phosphorylation or β-arrestin binding. Based on this model, the absence of crucial GRK phosphorylation sites in the serine mutants of the 355 to 364 domain would not block internalization through the alternate pathway.
Still another possibility that must be considered is based on our observation of a 2-fold phosphorylation in the triple mutant, even while receptor-level desensitization was eliminated. This result leaves open the possibility that there is an as-yet-unidentified phosphorylation site that could be involved in endocytosis of the βAR because the inhibitory effect of the triple mutant on endocytosis was far from complete. Further studies will be required to evaluate the location of this site and its possible contribution to internalization.
Regardless of which model best explains our results, the data described here resolve previous conflicting reports on the function of the βAR carboxyl tail serines and threonines (Bouvier et al., 1988; Hausdorff et al., 1989; Hausdorff et al., 1991; Yu et al., 1993). Although Hausdorff et al. reported global, nonspecific effects by substitution of four amino acids in the 355- to-364 region, we show that substitution of only two residues in this domain significantly reduces agonist-induced desensitization while only partially inhibiting internalization and phosphorylation. The selective mutagenesis strategy employed here more specifically impaired desensitization. The report that glycine substitution of serines 356 and 364 blocked internalization and resensitization without affecting desensitization (Yu et al., 1993) is very difficult to reconcile with our results. We found that alanine substitution of these same sites (S356,364A) resulted in only a partial inhibition of internalization, and recycling was unaffected. It is possible to rationalize the conflicting results if glycine and alanine substitution cause significantly different effects. In addition, their work was carried out in Chinese hamster ovary (CHO) cells, rather than the HEK 293 cells used here, and βAR internalization is greatly reduced in CHO cells compared with HEK 293 (Menard et al., 1997). Therefore a small effect of the mutations on the extent of internalization in HEK 293 cells may be a major effect in CHO cells.
We found that mutations of the βAR 355–364 domain did not affect recycling, because all of the mutants recycled with kinetics indistinguishable from those of HA-βAR-6HIS. However, we considered whether the carboxyl terminal HIS6 tag, included in the HA-βAR-6HIS and all the mutant βARs described here, might inhibit receptor recycling relative to the untagged WTβAR. It was recently reported that the addition of various amino acids to the βAR carboxyl terminus inhibited recycling to the cell surface (Cao et al., 1999). The study proposed that additions to the carboxyl terminus block the receptor PDZ-binding domain, composed of the last three amino acids, from mediating the protein-protein interactions required for recycling. We tested the effect of the HIS6tag by comparing recycling of the HA-βAR-6HIS and the untagged WTβAR. As we report here, the recycling of the HA-βAR-6HIS and the untagged WTβAR were indistinguishable. Our data are similar to those of Kallal et al. (1998), who found similar internalization and recycling for both the untagged WTβAR and the βAR labeled at the carboxyl terminus with green fluorescent protein. The kinetic data we obtained were similar to those of Morrison et al. (1996), who described the internalization and recycling of the HA-βAR, which has an untagged carboxyl tail. In addition, we found that the HIS6 tag did not affect βAR desensitization. Comparison of the HA-βAR-6HIS with the untagged WTβAR showed that their desensitization was similar, in agreement with previous work (January et al., 1997). After 30-min pretreatment with 10 μM epinephrine, the fraction activity remaining for the HA-βAR-6HIS, HA-βAR, and untagged WTβAR was 0.03 (±0.003, n = 9), 0.06 (±0.01, n = 6), and 0.07 (±0.01,n = 6), respectively. We show that agonist-induced desensitization, internalization, and recycling are clearly not impaired by addition of the HIS6 epitope tag to the receptor carboxyl terminus. However, we must be open to the possibility that there may be important cell-specific differences in the effects of C-terminal epitope tags that clearly block interactions with PDZ domain-containing proteins such as HNERF (Hall et al., 1998).
Previous work showed that both PKA- and GRK-dependent mechanisms contributed to receptor level desensitization of the βAR (Hausdorff et al., 1989; Yuan et al., 1994). The functional importance of the PKA consensus sites found in the third intracellular loop and carboxyl terminus of the βAR has been supported by mutagenesis studies (Hausdorff et al., 1989; Yuan et al., 1994) and by the use of S49 lymphoma cells lacking either Gs or PKA activity (Green and Clark, 1981; Green et al., 1981; Clark et al., 1988). Identification of the sites required for GRK-dependent, homologous desensitization has been more difficult. This is understandable in retrospect given that it seems that GRK-mediated phosphorylation, β-arrestin binding, internalization, and recycling may be tightly linked (Krupnick and Benovic, 1998; Lefkowitz et al., 1998; Clark et al., 1999) and, in addition, further confounded by PKA-mediated desensitization when concentrations of epinephrine in the pretreatment are high. In this article, isolation of GRK-mediated receptor desensitization and thereby simplification of its analysis was achieved by substitution of the serines in the two PKA consensus sites. Our work provides strong evidence that the serines in the βAR 355 to 364 play a key role in GRK-dependent desensitization.
Footnotes
- Received January 21, 2000.
- Accepted August 14, 2000.
-
Send reprint requests to: Dr. Richard B. Clark, University of Texas-Houston Medical School, Department of Integrative Biology and Pharmacology, P.O. Box 20708, Houston, Texas 77225. E-mail:richard.b.clark{at}uth.tmc.edu
-
This study was supported by National Institutes of Health Grants GM31208 (R.B.C.) and HL57445, HL50047 (B.J.K.) and HL03463 (R.H.M.)
Abbreviations
- βAR
- human β2-adrenergic receptor
- PKA
- cAMP-dependent protein kinase
- GRK
- G protein-coupled receptor kinase
- WT
- wild-type
- HEK
- human embryonic kidney
- HA
- hemagglutinin
- DMEM
- Dulbecco's modified Eagle's medium
- 125ICYP
- [125I]iodocyanopindolol
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- AT
- ascorbic acid/thiourea
- CGP
- [3H]CGP-12177
- PBSS
- PBS containing 0.12% sucrose
- DBM
- n-dodecyl-β-d-maltoside
- WGA
- wheat germ agglutinin
- PVDF
- polyvinylidene difluoride
- PAGE
- polyacrylamide gel electrophoresis
- PGE1
- prostaglandin E1
- CHO
- Chinese hamster ovary
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}