


Abstract
Fusion proteins between the human 5-hydroxytryptamine (5-HT)1A receptor and either wild type or certain pertussis toxin-resistant forms of Go1α and Gi1α display constitutive GTPase activity that can be inhibited by the inverse agonist spiperone. Addition of recombinant regulator of G protein signaling (RGS) 1 or RGS16 to membranes expressing these fusion proteins resulted in elevation of this constitutive GTPase activity without significantly altering the binding affinity of antagonist/inverse agonist ligands. For a 5-HT1Areceptor-(Cys351Ile)Go1α fusion protein the increase in basal GTPase activity was greater than 4-fold. Enzyme kinetic analysis demonstrated that the effect of RGS1 was as a GTPase-activating protein for the fusion construct. In the presence of the RGS proteins, both agonists and inverse agonists produced much more robust regulation of high-affinity GTPase activity than in their absence. This allowed detection of the partial agonist nature of WAY100635, which has been described previously as a neutral antagonist at the 5-HT1A receptor. Of a range of ligands studied, only haloperidol functioned as a neutral ligand in the presence of RGS1. These studies show that addition of a recombinant RGS protein provides a simple and novel means to elevate the fraction of basal membrane GTPase activity contributed by the constitutive activity of a receptor. By so doing, it also greatly enhances the ability to detect and analyze the effects of inverse agonists and to discriminate between neutral ligands and those with low levels of positive intrinsic efficacy.
Constitutive activity of G protein-coupled receptors (GPCRs) has been one of the most highly studied topics in pharmacology in the recent past (Leurs et al., 1998; Pauwels and Wurch, 1998; de Ligt et al., 2000). Such studies have provided novel insights into the mechanisms of action of GPCRs and introduced the term inverse agonist for ligands able to suppress this activity (Milligan et al., 1995). So widespread have the studies been that compounds, generally now described as neutral antagonists, that bind receptors but fail to alter their activity are considered to be relatively uncommon.
Direct measures of regulation of the activation of a G protein by a GPCR can be provided by monitoring either exchange of a poorly hydrolyzed analog of GTP for GDP on the G protein α-subunit (Wieland and Jakobs, 1994) or the subsequent hydrolysis of authentic GTP by the GTPase activity of this subunit (Gierschik et al., 1994). This GTPase activity and its regulation can be analyzed using basic enzyme kinetics. However, when using both purified G proteins and membrane preparations, the rate of GTP hydrolysis has routinely been noted to be much slower than the rate of deactivation of a range of G protein-mediated events in vivo. Such discrepancies informed searches for GTPase-activating proteins (GAPs) capable of accelerating the turn-off reaction. The largest family of such GAPs for heterotrimeric G proteins in mammals are the regulators of G protein signaling (RGS) proteins (De Vries et al., 2000), comprising more than 20 polypeptides that contain a highly conserved RGS domain within their sequence. These act as GAPs for many G proteins and can be shown to alter the effectiveness of downstream signal transduction (Berman et al., 1996;Druey et al., 1996; Doupnik et al., 1997; Hepler et al., 1997; Saitoh et al., 1997). Wide-ranging experiments (Berman et al., 1996) and the crystal structure of the core RGS domain of RGS4 complexed with Gi1α (Tesmer et al., 1997) indicated that the mechanism of these proteins was via stabilization of the transition state required for GTP hydrolysis.
For many native GPCRs in cell membranes, their degree of constitutive activity is modest. Thus, when measuring inhibition of GTPase activity by potential inverse agonists, it may be difficult to obtain precise information. Because an RGS protein must be expected to function as a GAP for GTP loaded by the constitutive activity of a GPCR and after agonist activation, we reasoned that RGS proteins may be used to enhance the dynamic range of GTPase activity arising from the presence of constitutively activated GPCRs.
We demonstrate that this is the case, that the extent of this effect varies dependent upon the identity of the G protein studied, that it can provide a substantially more robust analysis of compounds with inverse agonist activity, and that this approach is well suited to the study of ligands with positive but low intrinsic activity.
Experimental Procedures
Materials.
All materials for tissue culture were supplied by Invitrogen (Paisley, Strathclyde, UK). The 5-HT1Areceptor antagonist [3H]MPFF (70.5 Ci/mmol) and [γ-32P]GTP (30 Ci/mmol) were obtained from PerkinElmer Life Sciences (Boston, MA). The 5-HT1A receptor antagonist [3H]WAY100635 (83.0 Ci/mmol) was from Amersham Biosciences (Piscataway, NJ). Pertussis toxin was purchased from Sigma (St. Louis). Oligonucleotides were purchased from Cruachem (Glasgow, Strathclyde, UK). All other chemicals were from Sigma and Roche Molecular Biochemicals (Summerville, NJ).
Construction of Plasmids Encoding 5HT1A-Gi1α and 5HT1A-Go1α Fusion Proteins.
The human 5-HT1A receptor clone in pSP64 (a gift from Glaxo-Wellcome, Stevenage, UK) was digested withXbaI/BamHI, and the resulting 1.5-kilobase pair fragment was ligated to pcDNA3. To obtain the open reading frame of 1.3 kilobase pairs, PCR was carried out using the following primers to introduce a HindIII restriction site at the 5′ end and to remove the stop codon and introduce a BamHI restriction site at the 3′ end, respectively: 5′-CTGAAGCTTATGGATGTGCTCAGCCCTGGTC-3′; 5′-CTGGGATCCCTGGCGGCAGAAGTTACACTTAATG-3′ (restriction enzyme sites underlined). The PCR fragment was digested withHindIII and BamHI and ligated into pcDNA3 to make the plasmid p5HT. To link the Gi1α wild-type (cys351)cDNA to the 5HT1Areceptor sequence, PCR was carried out on Gi1α to produce compatible restriction sites. The oligonucleotides used to do this were 5′-CTGGGATCCGGCTGCACACTGAGCGCTGAG-3′ at the 5′ end and 5′-GAGAATTCTTAGAAAGAGACCACAGTC-3′ for the 3′ end. The plasmid p5HT was digested with BamHI/EcoRI as was the Gi1α PCR fragment, and the two were ligated to give the plasmid p5HTGi1. To construct the 5-HT1A-(Gly351) and (Ile351)Gi1α fusion proteins, plasmid (Gly or Ile351)Gi1α in pBS was digested with SacII/EcoRI, and the 730-base pair fragment was used to replace the corresponding fragment in p5HTGi1. Equivalent strategies were used to produce the 5-HT1A-(Ile351)Go1α fusions. The constructs were then sequenced to verify the DNA sequence.
Cell Culture and Stable Expression.
HEK 293 cells were maintained in Dulbecco's modified Eagle's medium containing 10% (v/v) newborn calf serum and 2 mM l-glutamine. Cells were seeded into 100-mm culture dishes and grown to 60 to 80% confluence (18–24 h) before transfection with 5 μg of appropriate cDNAs using DOTAP reagent (Roche Molecular Biochemicals). Forty-eight hours after transfection, the cells were split 1:4 into medium containing 800 μg/ml G418 sulfate (Calbiochem, San Diego, CA). A 100-mm dish of untransfected HEK 293 cells was also split into the same medium as a control dish. About 1 week later, after all the cells in the control dish had died, G418-resistant cells in the transfected dishes were picked and transferred into 24-well plates using autoclaved pipette tips. About 20 clones of each cDNA were picked and grown in 1 ml/well G418 medium (400 μg/ml). Clones were amplified, membrane preparations were made, and their binding of [3H]4-(2′- methoxy)-phenyl-1-[2′-(N-2"-pyridinyl)-p-fluorobenzamido]ethyl-piperazine was determined.
Preparation of Membranes.
Plasma membrane-containing P2 particulate fractions were prepared from cell pastes that had been stored at −80°C after harvesting. Cell pellets were resuspended in Tris/EDTA buffer [10 mM Tris HCl, pH 7.5, and 0.1 mM EDTA], and rupture of the cells was achieved with 25 strokes of a hand-held Teflon-on-glass homogenizer. Unbroken cells and nuclei were removed by centrifugation at low speed (1600 rpm) in a refrigerated microcentrifuge. The supernatant fraction was then centrifuged at 50,000 rpm for 30 min in an Optima TLX ultracentrifuge with a TLA100.2 rotor (Beckman Coulter, Inc., Fullerton, CA). The pellets were resuspended in Tris/EDTA buffer to a final protein concentration of 1 mg/ml and stored at −80°C until required.
[3H]WAY100635 Binding Studies.
Binding assays were performed by adding 5 μg of membrane protein to an assay buffer (20 mM HEPES, 10 mM MgCl2, 0.1% ascorbic acid, and 10 μM pargyline, pH 7.4) containing [3H]WAY100635 (0.25–12 nM). Nonspecific binding was determined in parallel in the presence of 100 μM 5-HT. Samples were incubated at 30°C for 40 min and then terminated by rapid filtration through GF/C filters. The filters were washed three times with 5 ml of ice-cold wash buffer (20 mM HEPES, 10 mM MgCl2, and 0.1% ascorbic acid, pH 7.4) and then counted. In a number of experiments, recombinant RGS1 was also added to the binding assays. In competition binding assays, [3H]WAY100635 was present at 1 nM.
High-Affinity GTPase Assays.
High-affinity GTPase assays were performed essentially as described previously (Wise et al., 1997a,1997b; Wise and Milligan, 1997) adapted to a 96-well microtiter plate assay (Hoffmann et al., 2001). Nonspecific GTPase activity was assessed by parallel assays in the presence of 100 μM GTP. GTPase saturation data were analyzed by nonlinear regression using Prism version 2.01 (GraphPad Software, San Diego, CA).
Purification of GST-Tagged RGS1 and RGS16.
GST-RGS1 (Denecke et al., 1999) was kindly donated by Dr. A. Meyerdierks (Department of Medical Microbiology, Medizinischer Hochschule, Hannover, Germany). The full coding region of the human RGS1 gene except the initiation codon was cloned in-frame in the BamHI and SalI sites of the vector pGEX4T1 (Amersham Biosciences).
GST-RGS16 (Chen et al., 1997) was kindly donated by Dr. C. W. Fong (Institute of Molecular and Cell Biology, Singapore). In essence, the GST-RGS16 was constructed in a similar way as for RGS1. The coding region of the RGS16 gene was fused to the GST gene of the vector pGEX-2TK2 (derived from pGEX-2KT, Amersham Biosciences) with modifications in the multiple cloning region.
GST-RGS fusion proteins were isolated from transformedEscherichia coli BL21 cells. In brief, E. coliBL21 cells were transformed with the appropriate plasmids and plated on LB agar plates containing 100 μg/ml ampicillin. The next day, cells were washed from the plate and used to inoculate 400 ml of LB medium (supplemented with 100 μg/ml ampicillin) at an OD660 of 0.1. Cells were grown for 1 h before induction of the expression of the GST-RGS fusion proteins by addition of 1 mM isopropyl β-d-thiogalactoside. Cells were allowed to express the fusion protein for 4 h, after which the cells were harvested by centrifugation. Pellets were stored at −80°C or used immediately for GST affinity purification of the GST-tagged proteins. Pellets were resuspended in BugBuster solution (Novagen, Madison, WI) with 5 ml/g wet tissue and containing 10 μl of Benzonase (Novagen) to reduce viscosity. Cells were incubated with 1 mg/ml lysozyme for 30 min on ice followed by sonication to disrupt the cells (4 × 30-s pulses). Dithiothreitol (5 mM) was added to the lysed cells before the addition of 200 μl of 50% (w/v) slurry of glutathione-Sepharose 4B (Amersham Biosciences). Samples were incubated at room temperature on a rotary wheel for 30 min and the Sepharose harvested by centrifugation at 500g. The beads were washed three times with 2 ml of ice-cold phosphate-buffered saline and the GST fusion proteins eluted from the beads with 20 mM reduced glutathione in a Tris-HCl buffer, pH 7.4. Purity of the isolated protein was visually inspected after SDS-PAGE. Protein amounts were determined according to Bradford (1976) after precipitation of a small amount of protein with 6% trichloroacetic acid to remove the glutathione.
Miscellaneous.
All experiments were performed on a minimum of three occasions using cells or membrane preparations derived from different cell passages. Where appropriate, data are presented as means ± S.E.M.
Results
Membranes stably expressing a fusion protein between the human 5-HT1A receptor and a pertussis toxin-resistant form of Gi1α in which cysteine351 of the G protein was replaced by isoleucine have higher levels of basal high-affinity GTPase activity than those expressing a similar level of a fusion protein between the 5-HT1A receptor and a form of Gi1α in which this cysteine is replaced by glycine (Kellett et al., 1999). This difference reflects a constitutive capacity of the 5-HT1A receptor to activate (Cys351Ile)Gi1α because the 5-HT1A receptor inverse agonist spiperone (Barr and Manning, 1997; Newman-Tancredi et al.,1997a, 1997b; Kellett et al., 1999; Milligan et al., 2001) is able to reduce basal high-affinity GTPase activity in these membranes but not in those expressing the 5-HT1Areceptor-(Cys351Gly)Gi1α fusion protein (Kellett et al., 1999).
The 5-HT1Areceptor-(Cys351Ile)Gi1α fusion protein could be immunodetected as an 85-kDa polypeptide (Fig.1A). Addition of 1 μM recombinant RGS1 (Hoffmann et al., 2001) to membranes of pertussis toxin-pretreated cells stably expressing this construct resulted in an increase in basal high-affinity GTPase activity (Fig. 1A) and a greatly enhanced capacity to measure stimulation of GTPase activity upon addition of increasing concentrations of 5-HT (Fig. 1A). The 5-HT1Areceptor selective agonist 8-OH-DPAT was as effective as 5-HT (Fig.1B), and 1 μM recombinant RGS16 (Hoffmann et al., 2001) also enhanced the effects of the agonists (Fig. 1B). This was not observed upon equivalent additions of the RGS proteins to membranes expressing the 5-HT1Areceptor-(Cys351Gly)Gi1α fusion protein or to membranes of mock-transfected cells (data not shown). We also constructed and stably expressed fusion proteins between the human 5-HT1A receptor and both (Cys351Ile)Go1α and (Cys351Gly)Go1α. After pertussis toxin treatment and membrane preparation, addition of recombinant forms of either RGS1 (Fig. 2) or RGS16 (data not shown) also elevated basal high-affinity GTPase activity in the membranes expressing the 5-HT1Areceptor-(Cys351Ile)Go1α fusion protein (Fig. 2). This polypeptide could also be immunodetected as an 85-kDa polypeptide (Fig. 2). As before, the RGS proteins had little effect in membranes expressing the Gly351-containing version of this fusion protein (data not shown). Noticeably, however, compared with the effects (less than 2-fold) on basal GTPase activity in membranes expressing 5-HT1Areceptor-(Cys351Ile)Gi1α (Fig. 1A), 1 μM RGS1 increased basal activity some 4-fold in membranes expressing the 5-HT1Areceptor-(Cys351Ile)Go1α fusion protein (Fig. 2). The stimulatory effects of both 5-HT (Fig. 2) and 8-OH-DPAT (data not shown) on GTPase activity were again greatly enhanced compared with those observed in the absence of the RGS (Fig.2). Half-maximal effects of RGS1 required some 50 nM for 5-HT1Areceptor-(Cys351Ile)Go1α and some 90 nM for 5-HT1Areceptor-(Cys351Ile)Gi1α (Fig. 3). Enzyme kinetic analysis of the high-affinity GTPase activity indicated that increasing concentrations of RGS16 elevated the V max of both basal (Fig. 4A) and 100 μM 5-HT-stimulated (Fig. 4B) GTPase activity of the 5-HT1Areceptor-(Cys351Ile)Go1α fusion protein. Moreover, although in the absence of an RGS protein, 5-HT increased V max of membranes expressing the fusion protein (Table 1), it did so without producing an alteration in apparentK m for GTP (Table 1). By contrast, both in the absence and the presence of 5-HT, the effects of the RGS proteins reflected a combination of increased GTPaseV max and increases in the observedK m for GTP (Fig. 4; Table 1). To ensure that the effects of the RGS proteins were not restricted to the fusion proteins containing the Cys351Ile mutations similar experiments were performed on membranes expressing fusion proteins between the 5-HT1A receptor and wild-type forms of either Gi1α or Go1α. For both of these constructs RGS1 markedly enhanced basal GTPase activity and synergistically increased the effect of a maximally effective concentration of 5-HT in membranes of cells that had not been pretreated with pertussis toxin (Fig.5).

Agonist-mediated stimulation of the GTPase activity of a 5-HT1A receptor-(Cys351Ile) Gi1α fusion protein; effects of RGS proteins. A, HEK 293 cells stably expressing a 5-HT1Areceptor-(Cys351Ile) Gi1α fusion protein were pretreated with pertussis toxin (25 ng/ml, 24 h) before harvest and membrane preparation. The capacity of varying concentrations of 5-HT to regulate high-affinity GTPase activity was measured in the absence (open symbols) (absence of 5-HT = 28.9 ± 0.6 pmol/min/mg membrane of protein) and presence (filled symbols) (absence of 5-HT = 47.6 ± 0.1 pmol/min/mg membrane of protein) of 1 μM recombinant RGS1. GTPase activity was measured at 0.5 μM GTP. Inset, membranes from parental HEK 293 cell (A) and those expressing either 5-HT1A receptor-(Cys351Ile) Gi1α (B) or 5-HT1Areceptor-(Cys351Ile) Go1α (C) were resolved by SDS-PAGE and transferred to nitrocellulose. Immunodetection of 5-HT1A receptor-(Cys351Ile) Gi1α was achieved using an antiserum that identifies the extreme C terminus of Gi1α. B, the effects of 5-HT and 8-OH-DPAT (both 100 μM) to modulate the GTPase activity of the 5-HT1Areceptor-(Cys351Ile) Gi1α fusion protein in the absence or presence of RGS1 or RGS16 (both 1 μM) were measured.
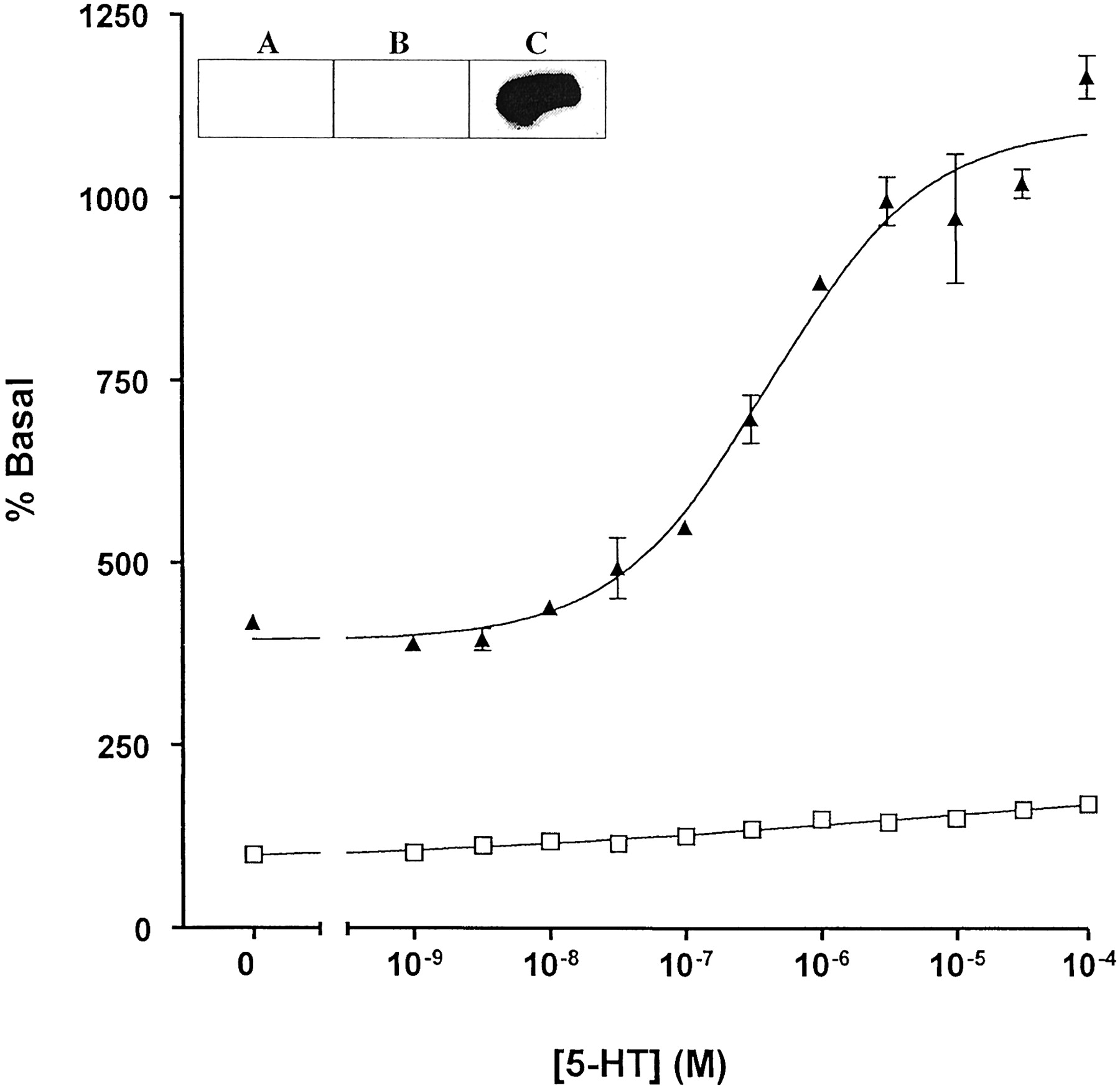
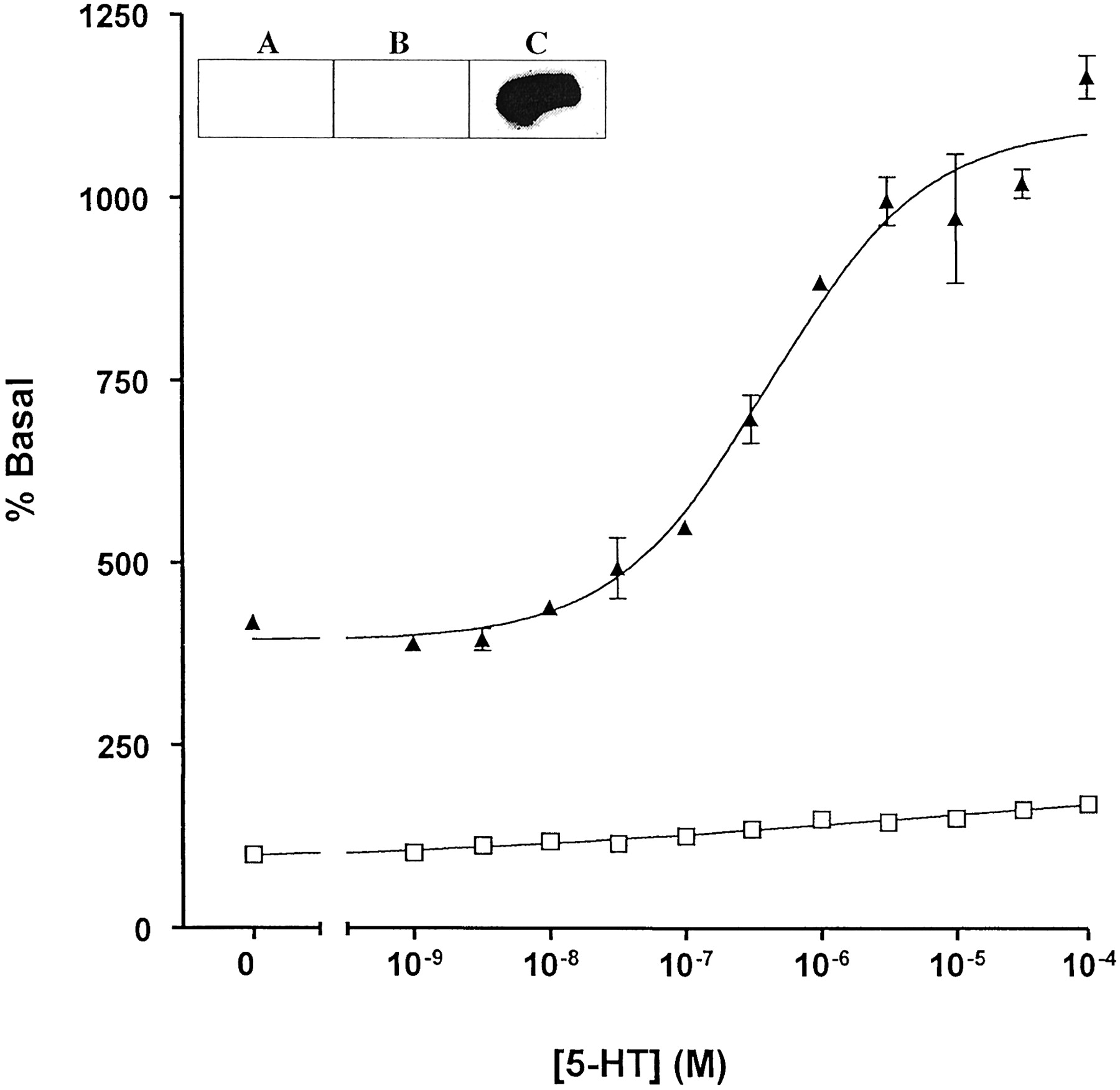
5-HT-mediated stimulation of the GTPase activity of a 5-HT1A receptor-(Cys351Ile) Go1α fusion protein: effects of RGS1. HEK 293 cells stably expressing a 5-HT1A receptor-(Cys351Ile) Go1α fusion protein were pretreated with pertussis toxin (25 ng/ml, 24 h) before harvest and membrane preparation. The capacity of varying concentrations of 5-HT to regulate high-affinity GTPase activity was measured in the absence (open symbols) (absence of 5-HT = 23.9 ± 0.4 pmol/min/mg membrane of protein) and presence (filled symbols) (absence of 5-HT = 99.2 ± 2.3 pmol/min/mg membrane of protein) of recombinant RGS1. GTPase activity was measured at 0.5 μM GTP. Inset, membranes from parental HEK 293 cell (A) and those expressing either 5-HT1A receptor-(Cys351Ile) Gi1α (B) or a 5-HT1Areceptor-(Cys351Ile) Go1α (C) were resolved by SDS-PAGE and transferred to nitrocellulose. Immunodetection of 5-HT1A receptor-(Cys351Ile) Go1α was achieved using an antiserum that identifies the extreme C terminus of Go1α.

The potency of RGS1 to regulate basal GTPase activity of 5-HT1A receptor-containing fusion proteins. Varying concentrations of recombinant RGS1 were added to membranes from pertussis toxin-pretreated cells expressing either the 5-HT1A receptor-(Cys351Ile) Go1α (squares) or the 5-HT1A receptor-(Cys351Ile) Gi1α fusion protein (triangles). High-affinity GTPase activity was then measured at 0.5 μM GTP.

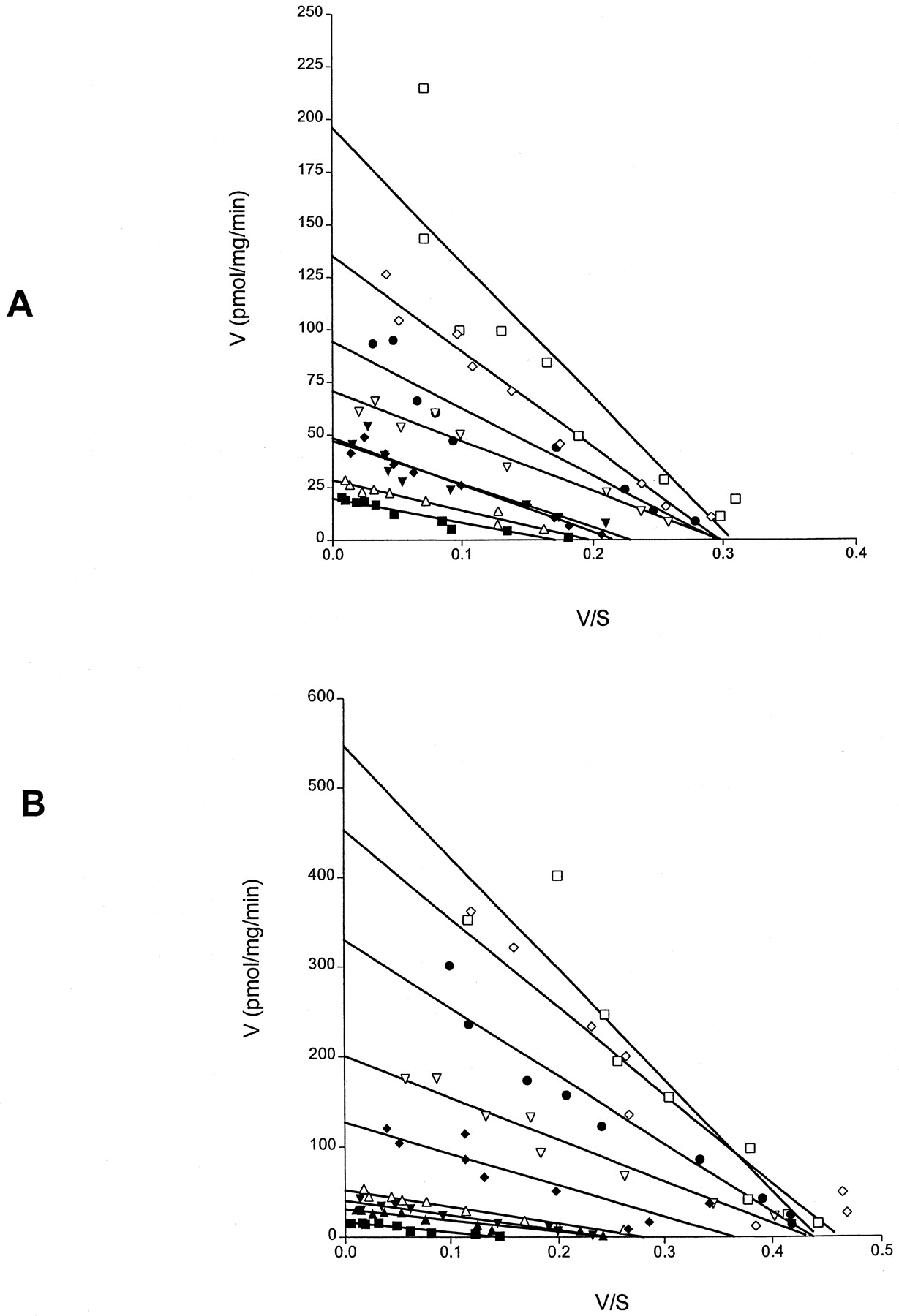
Effects of varying concentrations of RGS16 on basal and agonist-stimulated GTPase activity of a 5-HT1Areceptor-(Cys351Ile) Go1α fusion protein. Enzyme kinetic analysis. GTPase activity of membranes expressing the 5-HT1A receptor-(Cys351Ile)Go1α fusion protein was measured at a wide range of concentrations of GTP in the absence (A) or presence (B) of 100 μM 5-HT. Assays also contained 0 (filled squares), 1 nM (filled triangles), 10 nM (filled diamonds), 50 nM (open triangles), 100 nM (filled circles), 500 nM (open diamonds) or 1 μM (open squares) RGS16.
WAY100635 is a partial agonist at the 5-HT1A receptor

Agonist and RGS1 regulation of the GTPase activity of 5-HT1A receptor fusion proteins containing wild-type G proteins. GTPase activity was measured at varying concentrations of GTP in cell membranes expressing either the 5-HT1Areceptor-Gi1α fusion protein (A) or the 5-HT1A receptor-Go1α fusion protein (B). Basal activity (squares) and the effects of 100 μM 5-HT (triangles), 1 μM RGS1 (inverted triangles), or both 5-HT and RGS1 (diamonds) were assessed. Data are shown from a representative experiment.
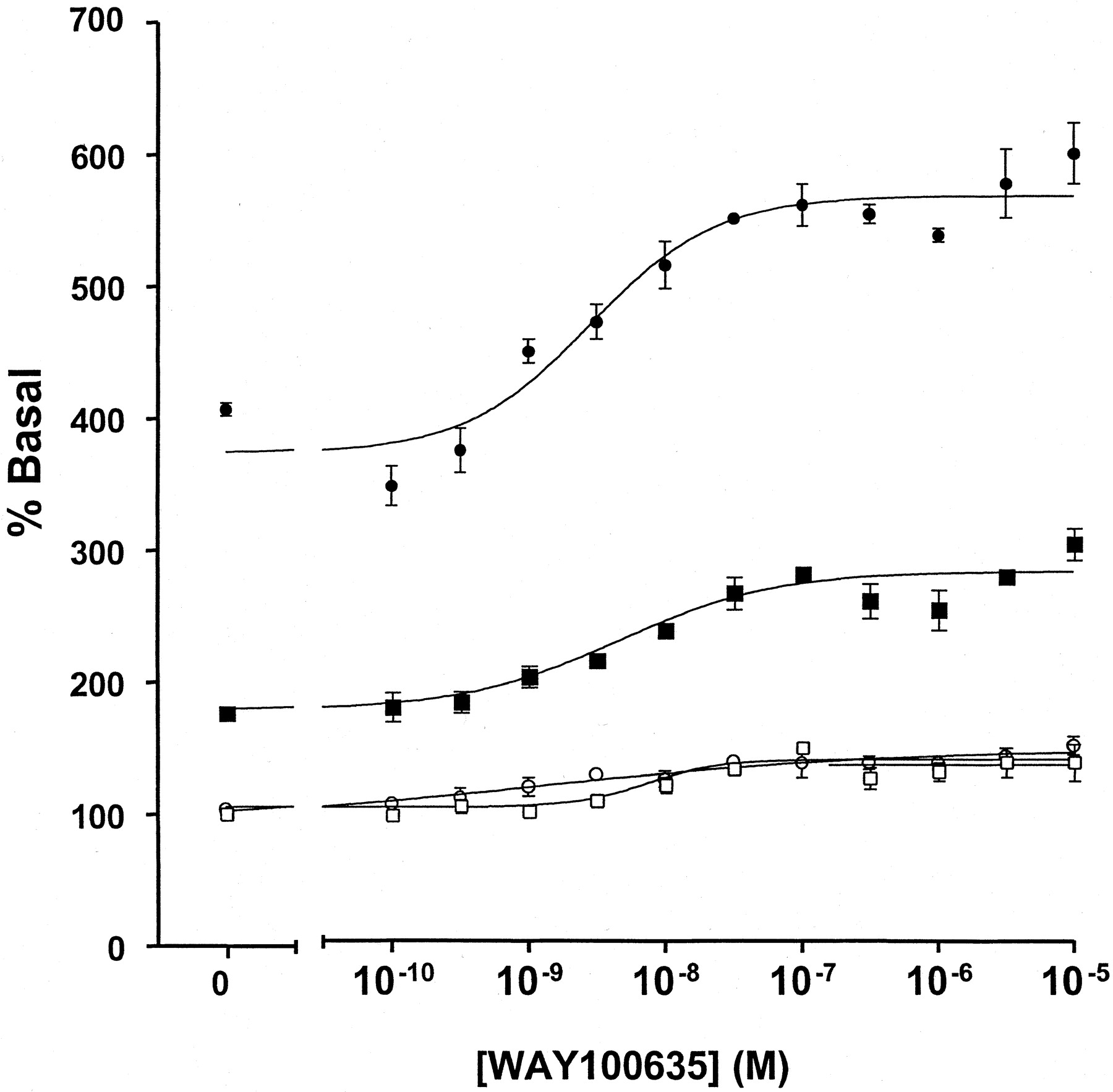
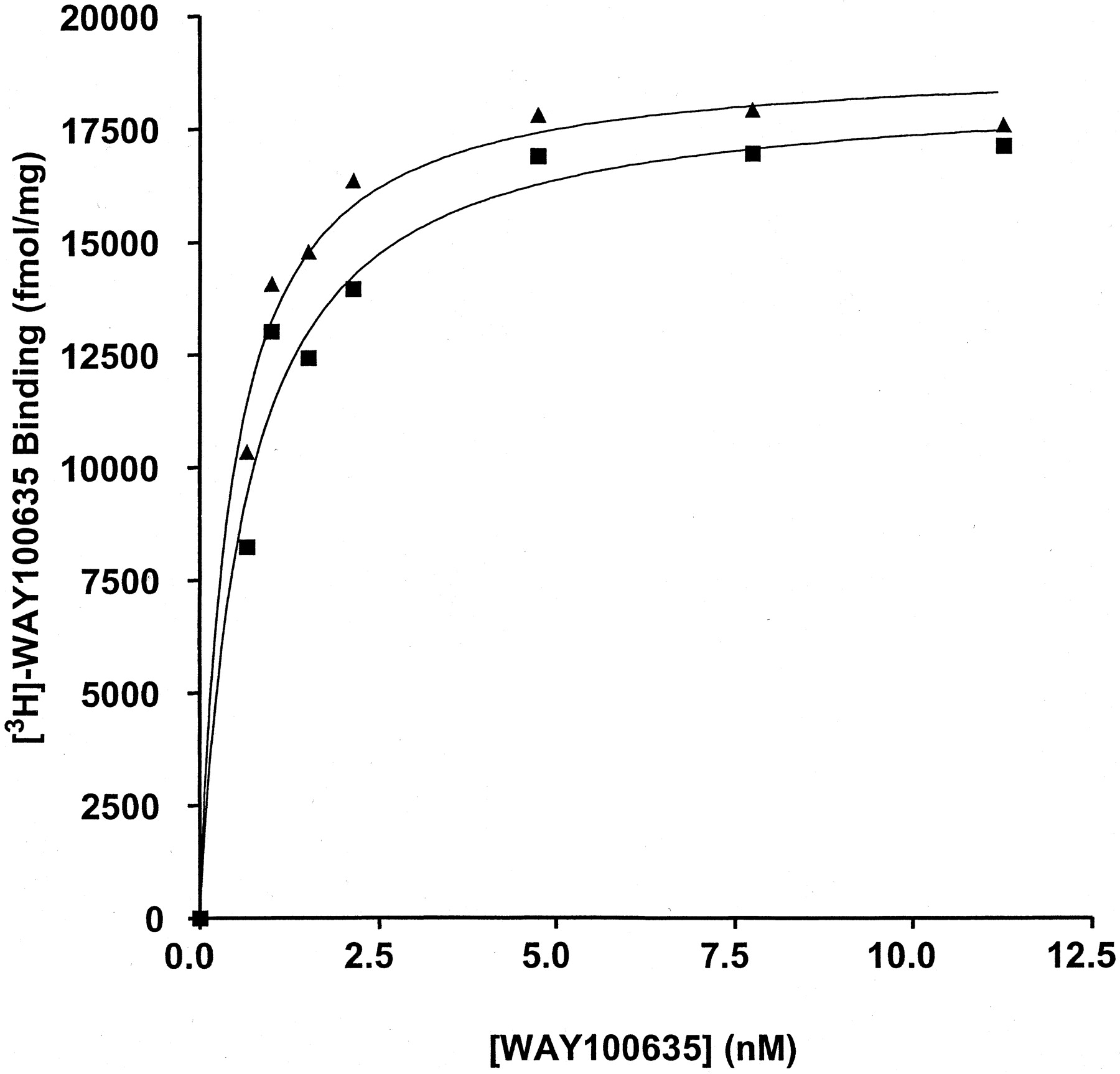
This effect of the RGS proteins was extremely useful in demonstrating weak agonism of WAY100635. In membranes expressing either the 5-HT1Areceptor-(Cys351Ile)Go1α or the 5-HT1Areceptor-(Cys351Ile)Gi1α fusion proteins, this compound acted as either a neutral antagonist or a very weak partial agonist (Fig. 6) with little ability to alter basal GTPase activity. However, in the additional presence of 1 μM RGS1 WAY100635 clearly functioned as a low-efficacy partial agonist (Fig. 6; Table 1). Furthermore, the enhanced sensitivity imbued to detection of efficacy in the presence of an RGS protein allowed good estimates to be obtained for EC50 for WAY100635 (2.8–4.6 × 10−9 M) (Fig. 6). This was not possible without addition of the RGS. In the absence of RGS1, enzyme kinetic analysis showed that the effects of 1 μM WAY 100635 were, like 5-HT, achieved by an increase in V max without alteration in K m for GTP (Table 1). In the presence of RGS1, WAY100635 further increased enzyme activity over that achieved by RGS1 alone but had little or no further effect on theK m for GTP (Table 1). Importantly, addition of RGS1 to membranes expressing these fusion proteins had little effect of the affinity or maximal capacity of [3H]WAY100635 to bind to the receptor (Fig.7; data not shown).

WAY100635 is a weak partial agonist at the 5-HT1A receptor. The effects of varying concentrations of WAY100635 to regulate basal high-affinity GTPase activity were measured in membranes of pertussis toxin-pretreated cells expressing either the 5-HT1A receptor-(Cys351Ile) Go1α fusion protein (circles) or the 5-HT1Areceptor-(Cys351Ile) Gi1α fusion protein (squares). Experiments were performed in the absence (open symbols) or presence (filled symbols) of 1 μM RGS1.

RGS1 does not alter the binding characteristics of [3H]WAY100635. The specific binding of varying concentrations of [3H]WAY100635 was measured in membranes of pertussis toxin-pretreated HEK 293 cells expressing the 5-HT1A receptor-(Cys351Ile) Gi1α fusion protein in the absence (squares) or presence (triangles) of 1 μM RGS1.
Because the majority of wild-type receptors display relatively low levels of constitutive activity (Lefkowitz et al., 1993; Rossier et al., 1999), it is often difficult to monitor functional differences between inverse agonists and neutral antagonists. Mutational alteration of the receptor is often required to boost the level of constitutive activity for such studies (Lefkowitz et al., 1993; Samama et al., 1993;Scheer and Cotecchia, 1997). However, as RGS proteins function to catalyze the GTP hydrolysis rate (De Vries et al., 2000), the elevated GTPase activity of the fusion proteins in the absence of ligands but the presence of RGS reflects a response to constitutive loading of GTP onto the fusion protein. Dependent on their relative intrinsic activity, therefore, inverse agonists would be expected to reduce or eliminate the effect of the RGS proteins. If this is so, then the capacity to detect inverse agonism should be markedly improved in the presence of an RGS. To test this hypothesis, we measured the ability of varying concentrations of the previously characterized 5-HT1A receptor inverse agonist spiperone to reduce basal high-affinity GTPase activity in membranes expressing either the 5-HT1Areceptor-(Cys351Ile)Gi1α or the 5-HT1Areceptor-(Cys351Ile)Go1α fusion proteins in the absence and presence of RGS1. As anticipated from previous studies (Kellett et al., 1999; Milligan et al., 2001), in the absence of RGS1, spiperone inhibited basal GTPase activity in membranes expressing the 5-HT1Areceptor-(Cys351Ile)Gi1α fusion protein by some 50% with an EC50 value of 6.7 × 10−8 M (Fig.8A). In the presence of 1 μM RGS1, spiperone displayed a similar EC50 value (4.6 × 10−8 M). However, with the elevation in constitutive, receptor-mediated GTPase activity, this was substantially easier to measure (Fig. 8A). The precision of measurement was even more pronounced when the experiments were repeated using the 5-HT1Areceptor-(Cys351Ile)Go1α fusion protein. Although spiperone could be shown to be an inverse agonist at this construct, in the absence of the RGS, this was difficult to quantitate with precision (Fig. 8B). Because the 4-fold elevated basal activity in these membranes observed on addition of RGS1 was attenuated almost completely by spiperone, this was easy to measure and provided an EC50 value of 3.2 × 10−8 M for the ligand (Fig. 8B). Importantly, the ability and potency of spiperone to compete with [3H]WAY100635 for binding to the fusion constructs was also unaltered by the presence of the RGS (Fig. 8C; data not shown).
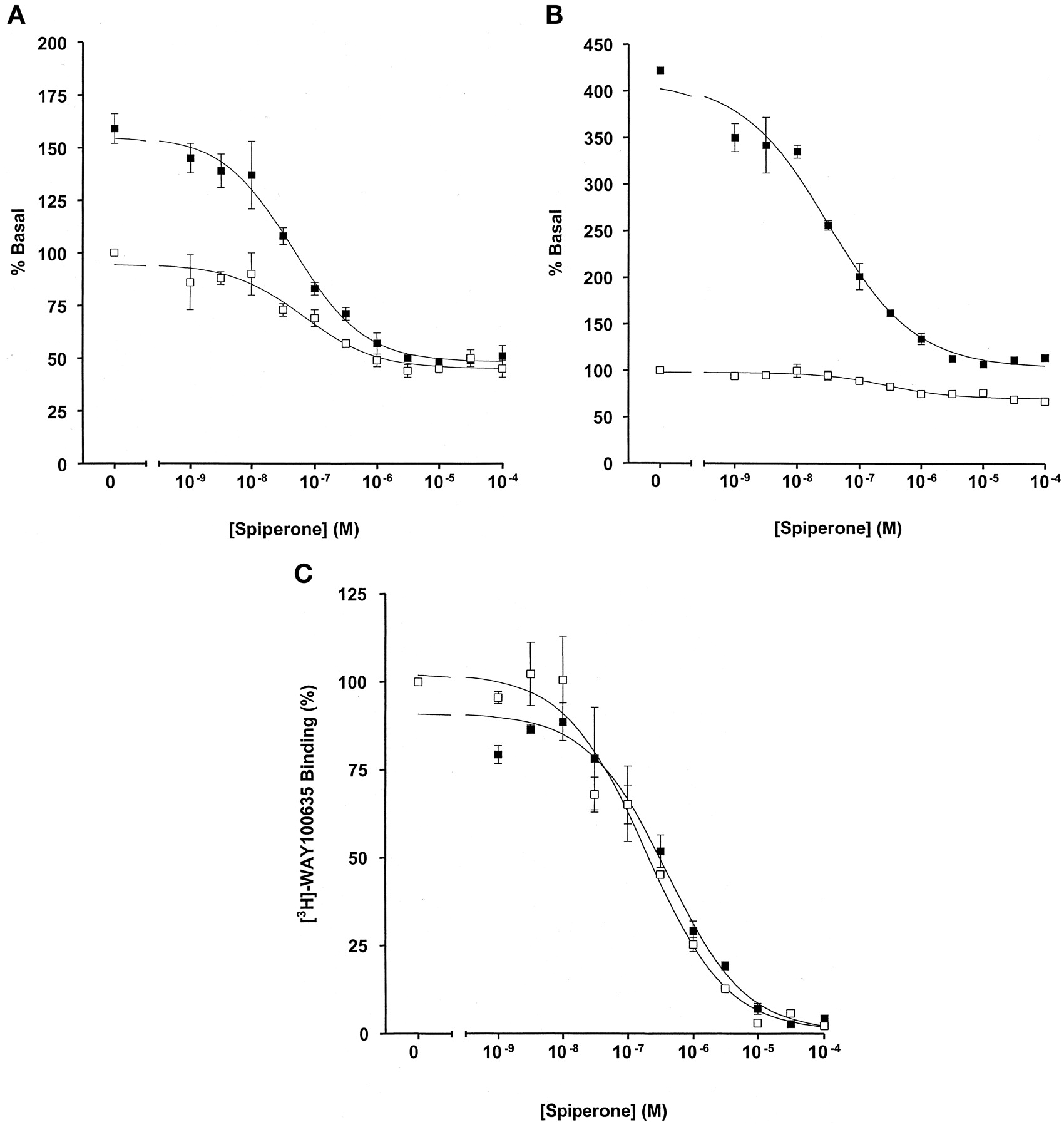
RGS1 enhances detection of the inverse agonist properties of spiperone at the 5-HT1A receptor. Membranes of pertussis toxin-treated HEK 293 cells expressing either the 5-HT1A receptor-(Cys351Ile) Gi1α fusion protein (A) or the 5-HT1Areceptor-(Cys351Ile) Go1α fusion protein (B) in the absence (open symbols) or presence (filled symbols) of 1 μM RGS1 were exposed to varying concentrations of spiperone and high-affinity GTPase activity measured at 0.5 μM GTP. C, the ability of spiperone to compete with [3H]WAY100635 for binding to the 5-HT1A receptor-(Cys351Ile) Gi1α fusion protein was assessed in the absence (open symbols) and presence (filled symbols) of 1 μM RGS1.
To explore the basis of the effect of the RGS proteins, GTPase assays were again performed at a wide range of concentrations of GTP. Basal activity in membranes expressing the 5-HT1Areceptor-(Cys351Ile)Go1α fusion protein was adequately described by a single function withK m for GTP in the region of 100 nM. In the presence of 1 μM RGS1, the increase in basal activity was shown to represent both an increase in GTPaseV max and an increase in the observedK m for GTP (Fig.9). Spiperone, at a maximally effective concentration, reduced V max and returned the observed K m for GTP to a value close to that of the basal state (Fig. 9).

Mechanism of action of RGS1 on the basal GTPase activity of the 5-HT1A receptor-(Cys351Ile) Go1α fusion protein; enzyme kinetic analysis and the effects of spiperone. The ability of 1 μM RGS1 (squares) to regulate the basal (circles) GTPase activity of membranes expressing the 5-HT1A receptor-(Cys351Ile) Go1α fusion protein was measured at a wide range of concentrations of GTP in the absence (open symbols) or presence (filled symbols) of 100 μM spiperone. In the example displayed, GTPaseV max in the basal state was 23.4 ± 0.9 pmol/min/mg membrane of protein, and the measuredK m for GTP was 145 ± 11 nM. RGS1 alone increased the GTPase V max to 146 ± 11.0 pmol/min/mg membrane of protein and increased markedly theK m for GTP (492 ± 61 nM). Spiperone reduced basal GTPase (12.7 ± 1.5 pmol/min/mg membrane of protein) without altering the K m for GTP (100 ± 25 nM). In the presence of both RGS1 and spiperone, the values wereV max 23.6 ± 2.7 pmol/min/mg membrane of protein and K m 143 ± 33 nM.
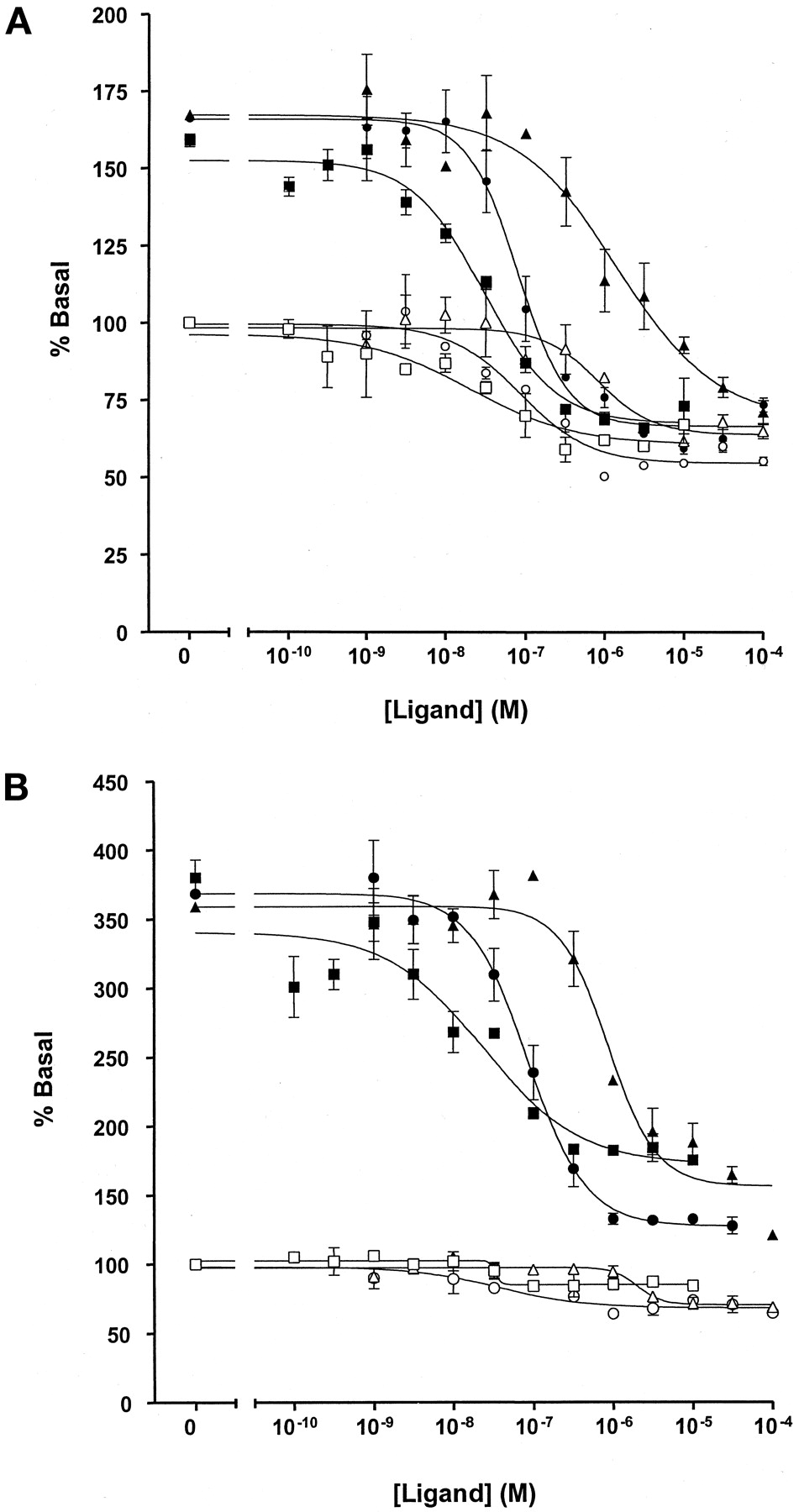
A series of other ligands with affinity at the 5-HT1A receptor was also examined. For methiothepin, (+)-butaclamol, and chlorpromazine, inverse agonist activity could be easily detected at both the 5-HT1Areceptor-(Cys351Ile)Gi1α and the 5-HT1Areceptor-(Cys351Ile)Go1α fusion proteins when the assays were performed in the presence of RGS1 (Fig. 10). These compounds were without effect on basal GTPase activity in membranes of parental HEK 293 cells (data not shown). Data on inverse agonism of these compounds was either impossible to quantitate effectively [5-HT1Areceptor-(Cys351Ile)Go1α] (Fig. 10A) or was substantially less convincing [the 5-HT1Areceptor-(Cys351Ile)Gi1α] (Fig. 10B) when the assays were performed in the absence of an RGS. Only in the case of haloperidol were conclusions that it functioned as an essentially neutral ligand at the 5-HT1A receptor-fusion proteins confirmed when the assays were also performed in the presence of RGS1 (Fig.11A). This was not a reflection of a lack of binding of haloperidol to the fusion proteins (Fig. 11B). A small inhibition of GTPase activity was noted for the highest concentrations of haloperidol tested (Fig. 11A), but this is probably a nonspecific effect because these concentrations are not consistent with the affinity of haloperidol at the 5-HT1Areceptor (Fig. 11B). High concentrations of haloperidol were also able to reverse the inhibition of GTPase activity produced by spiperone (Fig. 11C), confirming that spiperone was acting as an inverse agonist at the 5-HT1A receptor-fusion proteins.
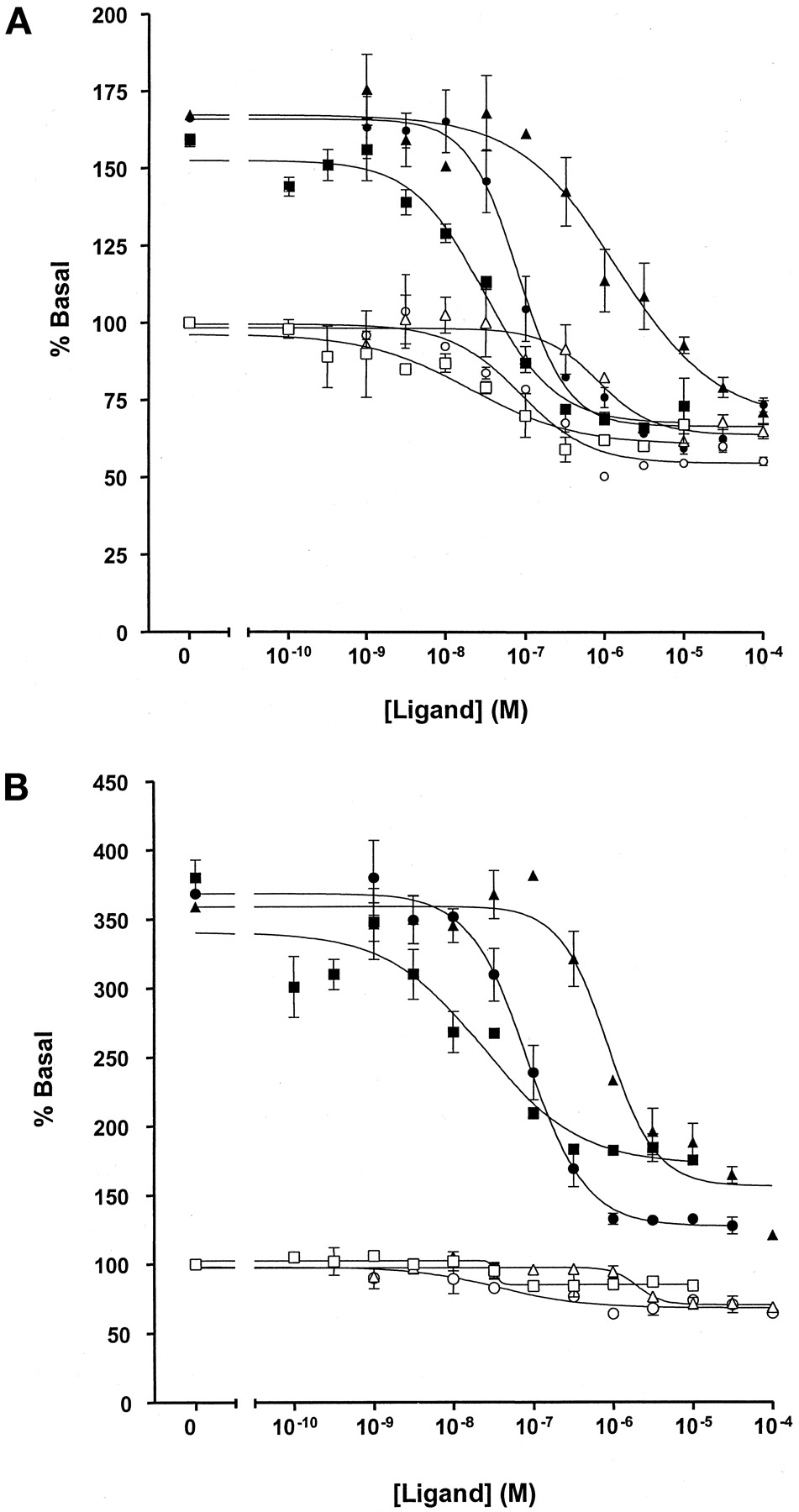
A range of ligands act as inverse agonists at the 5-HT1A receptor. Enhanced detection in the presence of RGS1. Basal GTPase activity in membranes expressing the 5-HT1A receptor-(Cys351Ile) Gi1α (A) or to the 5-HT1A receptor-(Cys351Ile) Go1α (B) fusion proteins was measured in the absence (open symbols) or presence (filled symbols) or 1 μM RGS1. The ability of varying concentrations of methiothepin (squares), (+)-butaclamol (circles), and chlorpromazine (triangles) to regulate GTPase activity was then measured at 0.5 μM GTP.

Haloperidol is a neutral ligand at the 5-HT1A receptor. A, The effect of varying concentrations of haloperidol on GTPase activity in membranes expressing the 5-HT1A receptor-(Cys351Ile)Gi1α (squares) or the 5-HT1Areceptor-(Cys351Ile)Go1α (diamonds) fusion proteins was assessed in the absence (open symbols) or presence (filled symbols) of 1 μM RGS1. B, competition by haloperidol for binding of [3H]WAY100635 to the 5-HT1Areceptor-(Cys351Ile)Gi1α(filled symbols) or the 5-HT1Areceptor-(Cys351Ile)Go1α (open symbols) fusion proteins. C, The basal GTPase activity of the 5-HT1Areceptor-(Cys351Ile)Go1α fusion protein was measured in the presence of 1 μM RGS1 and regulation of this activity by 0.1 μM spiperone, 10 μM haloperidol, or a combination of the two ligands assessed. ∗, significantly different from basal,p < 0.01.
Discussion
5-HT is a key neurotransmitter, and the 5-HT1A receptor is an important target for drug action. This reflects that presynaptic 5-HT1Areceptors in the raphe nuclei control 5-HT release throughout the brain and that postsynaptic 5-HT1A receptors are important in functions that include memory and stress modulation. Indeed, gene knockout studies have confirmed key roles for the 5-HT1A receptor in anxiety and anti-depressive actions (Heisler et al., 1998; Ramboz et al., 1998).
The capacity of recombinant RGS proteins to elevate basal high-affinity GTPase activity in membranes expressing fusion proteins between the 5-HT1A receptor and each of wild type and pertussis toxin resistant (Cys351Ile) forms of both Gi1α and Go1α, but not in membranes from mock transfected cells, provides clear evidence for constitutive information transfer between this receptor and these G proteins. It is noteworthy that we have previously recorded constitutive activity of the 5-HT1A receptor- (Cys351Ile) Gi1α fusion protein but not for a similar construct containing (Cys351Gly)Gi1α (Kellett et al., 1999) because we have also demonstrated that the interactions between GPCRs and Gi-family G proteins are of higher affinity and more conducive to productive information transfer when a hydrophobic amino acid is used to replace the pertussis toxin-sensitive cysteine (Waldhoer et al., 1999; Moon et al., 2001a). RGS1 was half-maximally effective at some 50 nM, a concentration akin to that recently noted for its regulation of an agonist activated α2A-adrenoceptor-Go1α fusion protein (Hoffmann et al., 2001). Interestingly, this effect of RGS1 was much more pronounced in membranes expressing the Go1α rather than the Gi1α-containing fusion protein. Again, this was previously noted for agonist stimulation of α2A-adrenoceptor-containing fusion proteins (Cavalli et al., 2000) and may indicate that in a membrane environment, it is a more efficient GAP for Go1α than Gi1α.
Constitutive activity of GPCRs and the capacity of this to be suppressed by inverse agonists has been very actively studied in recent years. However, because the level of constitutive activity of many wild-type receptors is relatively low, many of these studies have used mutationally modified receptors with enhanced agonist-independent activity to allow amplification of the constitutive signal. There is, however, clear evidence that at least certain GPCRs do display significant constitutive activity in vivo and that regulation of this activity may have pathophysiological significance (Morisset et al., 2001). An attractive feature of the use of recombinant RGS proteins to boost the constitutive GTPase activity of the 5-HT1A receptor fusion proteins is that their accepted mechanism of action is to promote the rate of hydrolysis of GTP. By contrast, receptor ligands regulate the rate of guanine nucleotide exchange of the G protein (Gilman, 1987). Because nucleotide exchange is the rate-limiting step in the activation/deactivation cycle (Gilman, 1987), then the effect of the RGS is simply to boost the capacity of the fusion protein to load GTP in response to either constitutive or agonist-induced activity. Proof of this mechanism of action of RGS1 and RGS16 on the constitutive activity of the 5-HT1A receptor-(Cys351Ile) Go1α fusion protein was obtained by enzyme kinetic analysis of the GTPase activity. Addition of these RGS proteins increased both GTPase V max and theK m for GTP. We have previously shown that acceleration of the GTP hydrolytic activity must be accompanied by both of these features (Cavalli et al., 2000; Hoffmann et al., 2001). It was, thus, anticipated that an inverse agonist would restore both of these features and this was observed when a maximally effective concentration of spiperone was added in concert with RGS1 (Fig. 9). We selected spiperone for these studies because it has previously been well characterized as an inverse agonist at the 5-HT1A receptor. These observations and the related effects of added RGS proteins on 5-HT stimulated GTPase activity raise an interesting issue. When 5-HT is added to membranes expressing either the 5-HT1Areceptor-(Cys351Ile) Go1α or the 5-HT1Areceptor-(Cys351Ile) Gi1α fusion proteins the effect is to increase GTPaseV max, but it has little effect on the observed K m for GTP (Table 1; Kellett et al., 1999). However, with addition of the RGS proteins, as with the situation with basal activity, both the GTPase activity and the apparent K m for GTP are markedly increased. These observations imply that the membrane preparations themselves have little functionally relevant RGS activity. It is currently unclear whether this reflects low levels of cellular expression of RGS proteins or whether cell homogenization and membrane preparation results in removal of much of the endogenous RGS. This will be explored in future studies, but clearly RGS proteins are, at best, peripheral membrane proteins with anchorage to the membrane being provided by combinations of post-translational acylation, cysteine string motifs, N-terminal amphipathic α-helices and possibly other means (Chen et al., 1999; Druey et al., 1999; De Vries et al., 2000). It also seems that a number of RGS proteins are not confined to the plasma membrane but are present in cellular compartments including the cytosol, Golgi, and nucleus (Wylie et al., 1999; Chatterjee and Fisher, 2000).
In the recent past, fusion proteins between receptors and G protein α-subunits (Seifert et al., 1999; Milligan, 2000; Guo et al., 2001;Wurch and Pauwels, 2001) have been employed to explore the details of topics as diverse as the relative intrinsic activity of different agonist ligands (Jackson et al., 1999), the selectivity of receptors for closely related G proteins (Moon et al., 2001b), and the regulation of post-translational acylation of both receptors and G proteins (Loisel et al., 1999; Stevens et al., 2001). Herein, the use of such fusion proteins containing the human 5-HT1Areceptor and pertussis toxin-resistant forms of Gi-family G proteins allowed prior pertussis toxin treatment of the cells to define that ligand and RGS regulation of the high-affinity GTPase activity was a direct monitor only of effects on the fusion protein-linked G proteins. Because antagonist binding is generally insensitive to the G protein interaction state of receptors it is hardly surprising that the affinity for antagonist ligands was highly similar for each of the fusion constructs employed in these studies and that this was not altered by addition of the recombinant RGS proteins (Figs. 7 and 8C).
Furthermore, because ligand occupancy of the 5-HT1A receptor is required to suppress the effects of the RGS proteins on constitutive GTPase activity of the fusion proteins, it was also anticipated that the pEC50 value for the ligands as inverse agonists should be in good agreement with measures of ligand affinity. Measures of pEC50 were difficult to obtain for such ligands at the 5-HT1Areceptor-(Cys351Ile)Go1α construct in the absence of an RGS because the measurable effects of the ligands were small. Affinity estimates were, thus, of low precision. However, for the 5-HT1Areceptor-(Cys351Ile)Gi1α construct, such estimates were not altered significantly by the presence of the RGS. This is a more complex issue for agonist ligands because we have previously noted that the presence of an RGS reduces the potency of high-efficacy agonists at the α2A-adrenoceptor, although it does not alter their binding affinity (Hoffmann et al., 2001). When examining a range of ligands with inverse agonist activity at the 5-HT1A receptor fusion proteins, it was clear that the rank order of potency was the same for their capacity to inhibit basal GTPase activity and their affinity to bind to the receptor with methiothepin >+ butaclamol > chlorpromazine. Furthermore, because the only effects of haloperidol on GTPase activity occurred at substantially higher concentrations than are consistent with ligand occupancy of the receptor (Fig. 11), this indicates that these effects are not specific. Thus, over the pharmacologically relevant concentration range, haloperidol was shown as a true neutral ligand for the 5-HT1A receptor.
The general strategy adopted in these studies should be suitable for any Gi/Go-coupled receptor that possesses a degree of constitutive activity. Furthermore, although Gs seems to be resistant to the effects of traditional RGS proteins, it is possible to employ chimeric G proteins to overcome this limitation. For example, the effects of vasopressin V2 receptor agonists became sensitive to RGS regulation when the receptor was constructed into a fusion protein with a Gi/Gs chimera that is activated by the receptor and retains high affinity to bind RGS1 (Feng et al., 2002).
These studies provide an entirely novel means of enhancing the sensitivity of studies on regulation of high-affinity GTPase activity and, thus, of examining the detailed characteristics of weak partial agonists and inverse agonists.
Footnotes
- Received October 15, 2001.
- Accepted January 25, 2002.
-
Financial support for this work was provided by the Medical Research Council and the Wellcome Trust. P.W. is a recipient of a “CASE” studentship from the Biotechnology and Biosciences Research Council.
Abbreviations
- 5-HT
- 5-hydroxytryptamine
- GPCR
- G protein-coupled receptor
- GAP
- GTPase-activating protein
- HEK
- human embryonic kidney
- RGS
- regulator of G protein signaling
- 8-OH-DPAT
- 8-hydroxy-2-dipropylaminotetralin
- PAGE
- polyacrylamide gel electrophoresis
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}