


Abstract
Steroid hormones modulate activity of the nuclear receptor constitutive active receptor (CAR, or constitutive androstane receptor) in mouse liver. Progesterone and testosterone repress the constitutive activity of mouse CAR (mCAR) in cell-mediated transfection assays, whereas estrogens activate the repressed receptor. This repression and activation is not observed with human CAR. To define the structural basis that confers the hormone responsiveness to mCAR, we constructed various chimeric and mutated receptors and examined their response to steroid hormones. The hormone responsiveness resided near or within AF-2 domain of mCAR. Moreover, a single mutation of threonine at position 350 to the corresponding methionine in the human counterpart abolished the repression of mCAR by steroid hormones. Coactivation by steroid receptor coactivator 1 (SRC-1) of mCAR did not depend on the threonine 350. However, overexpression of SRC-1 counteracted progesterone to repress mCAR activity. Thus, threonine 350 seems to regulate hormone responsiveness of mCAR by interfering indirectly an interaction of the receptor with a coactivator.
The nuclear receptor CAR plays a key role in activating the transcription of genes encoding xenochemical/steroid–metabolizing enzymes in response to phenobarbital (PB) and other PB-type inducers (Wei et al., 2000;Zelko and Negishi, 2000; Sueyoshi and Negishi, 2001; Ueda et al., 2002). The receptor is retained in the cytoplasm of noninduced hepatocytes. Upon PB induction, CAR translocates to the nucleus, forms a heterodimer with retinoid X receptor, and activates a phenobarbital-responsive enhancer module found in PB-inducible genes (Trottier et al., 1995; Park et al., 1996; Honkakoski et al., 1998a,b;Ramsden et al., 1999; Sueyoshi et al., 1999; Sugatani et al., 2001). Induction of these enzymes confers a higher metabolic capability to organisms for their defense mechanism against the xenochemical toxicity and/or carcinogenicity. In addition to such xenochemicals as PB and TCPOBOP, steroid hormones can also be regulatory factors that modulate the CAR-mediated activation of gene transcription. Estrogens activate mouse CAR (mCAR) and induce the Cyp2b10 gene in mouse liver, whereas androgens and progesterone repress estrogen-activated mCAR (Kawamoto et al., 2000). The receptor is promiscuous in that it is activated by many structurally diverse endogenous as well as exogenous chemicals; paradoxically, CAR also exhibits strong specificity, such as species specificity in its response to chemicals. For example, unlike mCAR, human CAR (hCAR) does not respond to steroid hormones (Kawamoto et al., 2000). The structural basis of this paradoxical nature (i.e., chemical promiscuity with species specificity) remains elusive.
In a cell-based transfection assay, CAR was characterized as a constitutive active receptor, indicating that CAR is activated in the absence of agonistic chemicals. Pharmacological doses of estrogens can activate mCAR in cell-based transfection assays as well as in mouse liver in vivo (Kawamoto et al., 2000). To acquire the ability to be activated by estrogen, mCAR needs to be first repressed by progesterone or testosterone. Thus, the hormone responsiveness of mCAR depends entirely on the repression by progesterone and/or testosterone. To define the structural basis of the hormone responsiveness, we constructed chimeric receptors between the mouse and human receptors and measured their response to steroid hormones in cell-based transfection assays. Using site-directed mutants, we demonstrate that residue 350 (threonine in mCAR) confers hormone responsiveness to mCAR (repression by testosterone and progesterone and reactivation by estrogen). Although the Ca2+/calmodulin kinase inhibitor KN-62 is also known to repress mCAR (Kawamoto et al., 2000), the repression by KN-62 did not depend on threonine at position 350 in mCAR, suggesting that the structural basis for hormonal repression differs from that for repression by xenochemicals. Experiments were also performed to understand the role of Thr350 in the coactivation of mCAR by SRC-1. Because species specificity is a general characteristic observed with various nuclear orphan receptors, it is critical to delineate the mechanism of species specificity at the molecular level to understand the roles of the receptors in different species, thereby predicting the effect of a given chemical.
Materials and Methods
Plasmids.
Mouse SRC-1 cDNA, corresponding to amino acid residues from 633 to 1450, was amplified and cloned intoEcoRI and HindIII sites of pcDNA3.1/myc-His(−)B plasmid. mCAR and hCAR expression vectors were constructed by cloning the entire coding sequence into BamHI and XhoI sites of the pCR3 plasmid, as described previously (Sueyoshi et al., 1999). The reporter plasmid, (NR1)5-tk-luciferase, was described elsewhere (Kawamoto et al., 1999). For hCAR/mCAR chimera expression plasmids construction, analysis by polymerase chain reaction (PCR) was conducted using mCAR or hCAR cDNA in pCR3 as templates. The amplified fragments used for the chimeras constructions were nucleotides encoding amino acids 1 to 86 of mCAR and 77 to 348 of hCAR (mhh; see Fig. 1 legend for explanation of letter codes), 1 to 116 of mCAR and 107 to 348 of hCAR (mmh), 1 to 76 of hCAR and 87 to 358 of mCAR (hmm), 1 to 106 of hCAR and 117 to 358 of mCAR (hhm), 1 to 159 of mCAR and 150 to 348 of hCAR (mh159), 1 to 224 of mCAR and 215 to 348 of hCAR (mh224), 1 to 315 of mCAR and 306 to 348 of hCAR (mh315), and 1 to 345 of mCAR and 336 to 348 of hCAR (mh345). The PCR reactions for these fragments were produced using Pfu polymerase and enzymatically phosphorylated primers. The amplified fragments, in the combinations described above, were ligated, and a second PCR amplification was performed on the ligated DNA using the primers for the 5′ and 3′ end of the chimeric DNA. The second PCR products were cloned into pCR3 vector with newly createdBamHI and XhoI sites at the 5′ and the 3′ ends, respectively. Mutations were introduced by PCR using the QuickChange site-directed mutagenesis system (Stratagene, La Jolla, CA) according to the instruction manual. Using appropriate nucleotide primers, methionine 340 in hCAR was mutated to threonine, Thr350 to methionine, and glycine 354 to encode glutamine. All chimeras and mutations were confirmed by sequencing.
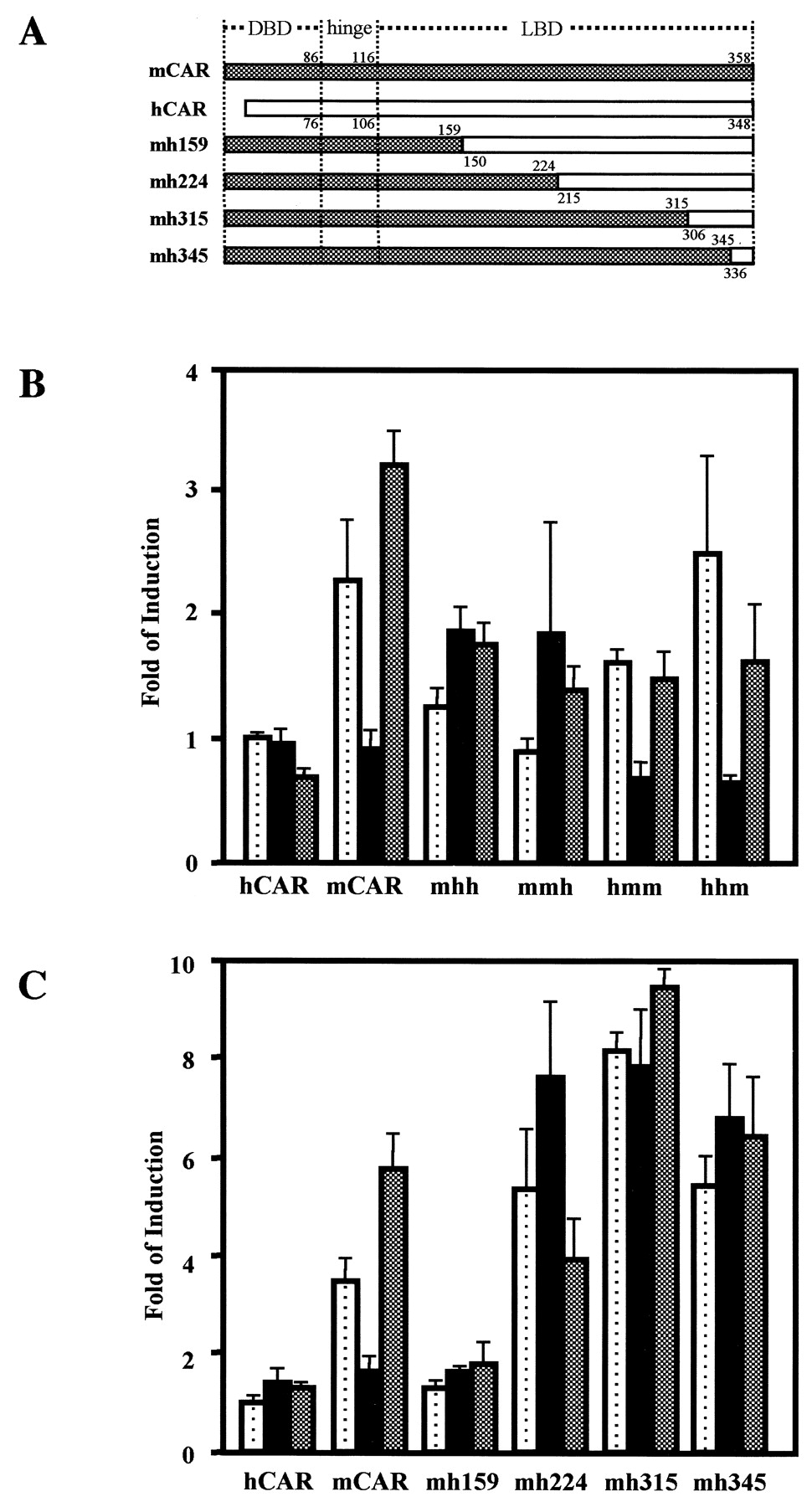
Domain-based chimera of the CAR proteins. A, putative domains are indicated on receptor sequences. Numbers indicate the positions of amino acid residues at which chimeric receptors were constructed. B, chimeric receptors were transfected into HepG2 cells, and NR1-luciferase reporter gene activity was measured as described under Materials and Methods. Each domain (DBD, hinge, or LBD) is denoted as either m for mCAR or h for hCAR (e.g., mhh contains mouse DBD and human hinge and LBD; hmm contains human DBD and mouse hinge and LBD). Dotted, filled, and shaded bars show luciferase activity of the HepG2 cells treated with DMSO, progesterone (10 μM), or progesterone plus estradiol (10 μM), respectively. These bars were generated from triplicate experiments with standard deviations. C, mCAR LBD was successively replaced with the corresponding regions of hCAR were constructed as indicated in A. These chimeric receptors were transfected into HepG2 cells, and NR1-luciferase reporter gene activity was measured. Dotted, filled, and shaded bars show luciferase activity of the HepG2 cells treated with DMSO, progesterone (10 μM), or progesterone plus estradiol (10 μM), respectively. These bars were generated from triplicate experiments with standard deviations.
Transfection.
HepG2 cells were cultured in minimal essential medium supplemented with 10% fetal bovine serum. HepG2 cells were plated in 24-well plates 1 day before transfection. (NR1)5-tk-luciferase plasmid (0.1 μg) was cotransfected with CAR expression plasmid (0.2 μg) and pRL-SV40 (0.2 μg) into HepG2 cells by calcium phosphate coprecipitation using CellPhect Transfection Kit (Amersham Biosciences, Piscataway, NJ). In separate experiments, pcDNA3.1-SRC-1 (0.1 μg) was also cotransfected into HepG2 cells. Sixteen hours later, the transfected cells were treated for another 24 h with KN-62, progesterone, TCPOBOP, PB, and/or estradiol (E2). Luciferase activity was measured using the Dual-Luciferase reporter assay system (Promega, Madison, WI).
Results
Chimera Receptors with hCAR and mCAR.
The putative DNA binding domain (DBD), ligand binding domain (LBD), and hinge region were defined from the amino acid sequences of mCAR and hCAR based on the reported multiple sequence alignments (Giguere, 1999) (Fig.1A). Subsequently, cDNAs encoding various chimeric receptors were constructed between mCAR and hCAR at the junctions of the domains and hinge region and were cloned into pCR3. By cotransfecting with the reporter plasmid (NR1)5-tk-luciferase into HepG2 cells, the function of chimeric receptors was examined with respect to their response to steroid hormones, such as progesterone and E2. In the cell-mediated transfection assays, mCAR activity was repressed by progesterone and reactivated by E2; however, these steroid hormones did not alter the activity of hCAR. The hormone-responsive repression and activation of mCAR were abolished only when the LBD was replaced with its human counterpart (Fig. 1B). Consistent with the role of mLBD (i.e., LBD of mCAR) in the hormone responsiveness, hCAR acquired this responsiveness by replacing its LBD with mLBD. Thus, the hormone response activity was associated with mLBD.
To delineate the hormone responsiveness to a region of mLBD, chimeric receptors within the mLBD were constructed by replacing their corresponding sequences in the hCAR: mh159, mh244, mh315, and mh345 (Fig. 1A). Then, their hormone responsiveness was examined in a cell-based transfection assay. None of these chimeric receptors was repressed by progesterone (Fig. 1C), indicating that the residues after position 345 in mCAR could be a determining factor for the hormone responsiveness. Given the sequence alignment, the carboxyl-terminal 13 residues (from positions 346 to 358 of mCAR) constitute the so-called AF2 domain. The carboxyl-terminal sequence of nuclear steroid receptors, the AF2 domain modulates a trans-activation function of the receptors. mCAR lost its constitutive activity after an AF2 mutation (Leu352Ala or Glu355Ala) or an AF2 deletion in a cell-based transfection assay (Choi et al., 1997). These Leu and Glu residues are conserved in both mCAR and hCAR and are not likely to determine species differences in the capability of responding to steroid hormones.
Site-Directed Mutagenesis on mCAR.
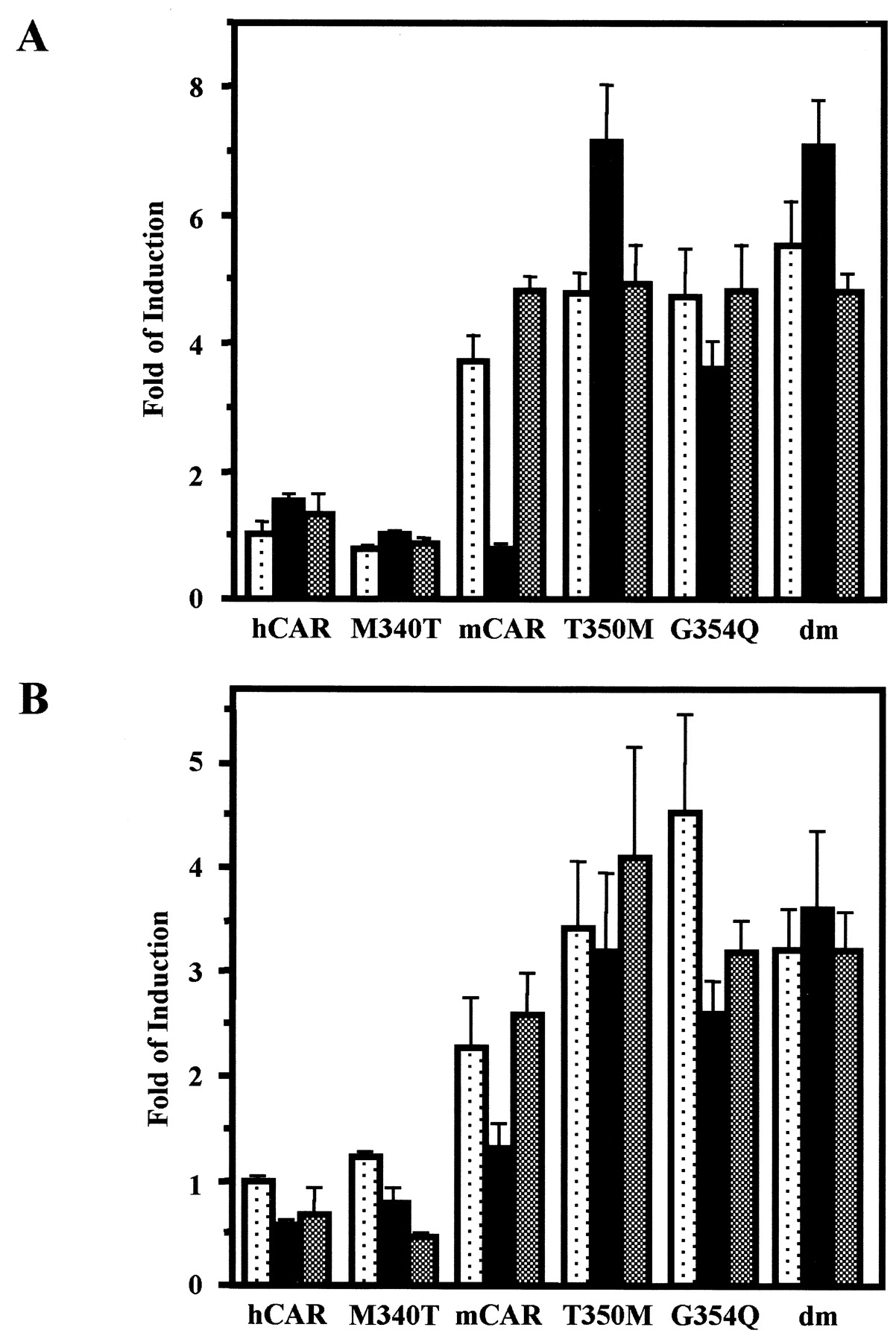
Alignment of 13 residues with the corresponding residues of hCAR (from positions 336 to 348) revealed two amino acid differences: Thr350 and Gly354 of mCAR were substituted in hCAR with Met340 and Gln344, respectively. Accordingly, Thr350 and Gly354 were mutated to methionine (mCART350M) and glutamine (mCARG354Q), respectively. The cDNAs of the mutated receptors were cloned into pCR3 vectors and were transfected into HepG2 cells. The mutant mCART350M lost its ability to show repressed activity by progesterone, whereas mCARG354Q retained a degree of repression by progesterone (Fig. 2A). The double mutation (mCART350M/G354Q) showed activity similar to that of the Thr350 mutant (mCART350M). In dose-dependent experiments, a low micromolar dose of progesterone was sufficient to repress the activity of wild-type mCAR, whereas the mutant mCART350M was not repressed by 10 μM progesterone (Fig. 3A). E2 fully activated wild-type mCAR in the presence of progesterone (10 μM), whereas the mutated receptor could not be activated by E2 (Fig. 3B). In addition to progesterone, testosterone repressed wild-type but not mutant mCAR (Fig. 2B). These results indicate that Thr350 of mCAR is a primary determinant conferring hormone responsiveness to mCAR. This, however, does not eliminate the possibility that other residues may also be involved in regulating hormone response activity of mCAR. Substituting Met340 with threonine (hCARM340T) did not alter the nonresponsiveness of the human receptor to steroid hormones (Fig. 2, A and B).

Mutational analysis of mCAR. There are two amino acid residues that differ between mCAR and hCAR within their C-terminal 13 residues. These residues, Thr350 and Gly354, in mCAR were singularly and doubly mutated to the corresponding residues in hCAR: T350M, G354Q, and dm, respectively. Met340 in hCAR was also mutated to Thr. The mutated mCARs were transfected into HepG2 cells, and NR1-luciferase reporter gene activity was measured as described under Materials and Methods. A, dotted, filled, and shaded bars show luciferase activity of the HepG2 cells treated with DMSO, progesterone (10 μM), or progesterone (10 μM) plus estradiol (10 μM). These bars were generated from triplicate experiments with standard deviations. B, all other experimental treatments were the same as those in A, except that testosterone (10 μM) was used instead of progesterone.

Dose-dependent responses to steroid hormones. A, repression of NR1-luciferase reporter gene activity by progesterone was examined for wild-type CAR(■) and mutant mCART350M (○) as described under Materials and Methods. The concentrations of progesterone used were 0.1, 0.3, 1, 3, and 10 μM. The percentage of control was calculated by considering the activity of DMSO-treated cells to be 100%. Standard deviations were calculated from triplicate experiments. B, activation by estradiol of NR1-luciferase reporter gene activity was measured in the presence of progesterone (10 μM) as described under Materials and Methods. The concentrations of estradiol used were 0.1, 0.3, 1, 3, and 10 μM. The percentage of control was calculated by considering the activity of DMSO-treated cells to be 100%. Standard deviations were calculated from triplicate experiments.
SRC-1 Effect on Wild-Type and T350M mCAR.
The AF2 domain regulates receptor activity through its direct interaction with coregulators (Glass and Rosenfeld, 2000). SRC-1, a coactivator, was shown previously to enhance CAR-mediated activity of phenobarbital-responsive enhancer module in a cell-based transfection assay (Muangmoonchai et al., 2001; Zelko et al., 2001). Thr350 resides within or near a predicted AF2 domain of mCAR. The role of Thr350 in regulating coactivation was examined by cotransfection of an SRC-1 expression plasmid. Coexpression of SRC-1 similarly increased thetrans-activation activity of both wild-type mCAR and mutant mCART350M 4-fold (Fig.4). Even at a concentration of 10 μM, progesterone did not repress the SRC-1–dependently increased activity of the mutant receptor. On the other hand, SRC-1 prevented the repression of the wild-type mCAR by progesterone at any concentration up to 10 μM. These results suggest that Thr350 plays no role in the coactivation by SRC-1 in the absence of progesterone under the experimental conditions used but it does regulate the coactivation in the presence of progesterone. In addition to the steroid hormones progesterone and testosterone, KN-62 (a Ca2+/calmodulin kinase inhibitor) is also known to repress the constitutive activity of mCAR but not that of hCAR (Kawamoto et al., 2000). The question arose as to whether Thr350 is involved in the repression by KN-62 as well as by steroid hormones. First, various domain-based chimeric receptors were used to localize a region of mRNA responsible for the repression by KN-62 (Fig.5A). Among them, KN-62 repressed the activity of chimeras hmm and hhm, and the repressed chimeric receptors were reactivated by TCPOBOP. These results indicate that mLBD dictates the KN-62 repression of mCAR as well as the reactivation by TCPOBOP. Next, the role of Thr350 in repression of KN-62 was examined (Fig. 5B). KN-62 similarly repressed the activities of both wild-type mCAR and mutant mCART350M, indicating that Thr350 plays no role in the repression by KN-62. The degree of repression was influenced by coexpression of SRC-1: 80% in the absence of SRC-1 and only 20 to 30% repression its presence. However, the repression was not affected by the mutation of Thr350. Thr350 does not seem to play a role either in the coregulation by SRC-1 or in the repression by KN-62. This is in sharp contrast to the critical role of Thr350 in the repression by steroid hormones.

Effect of SRC-1 on mCAR activity. The wild-type mCAR or the mutant mCART350M was cotransfected with or without SRC-1 expression plasmid in HepG2 cells in various concentrations of progesterone (0.1, 0.3, 1, 3, and 10 μM). Then, NR1-luciferase reporter gene activity was measured as described under Materials and Methods. The percentage of control was calculated by considering the corresponding activity of DMSO-treated HepG2 cells to be 100%: the wild-type receptor with and without SRC-1 (■ and ▪, respectively) and the mutant with and without SRC-1 (○ and ●, respectively). Standard deviations were calculated from triplicate experiments.
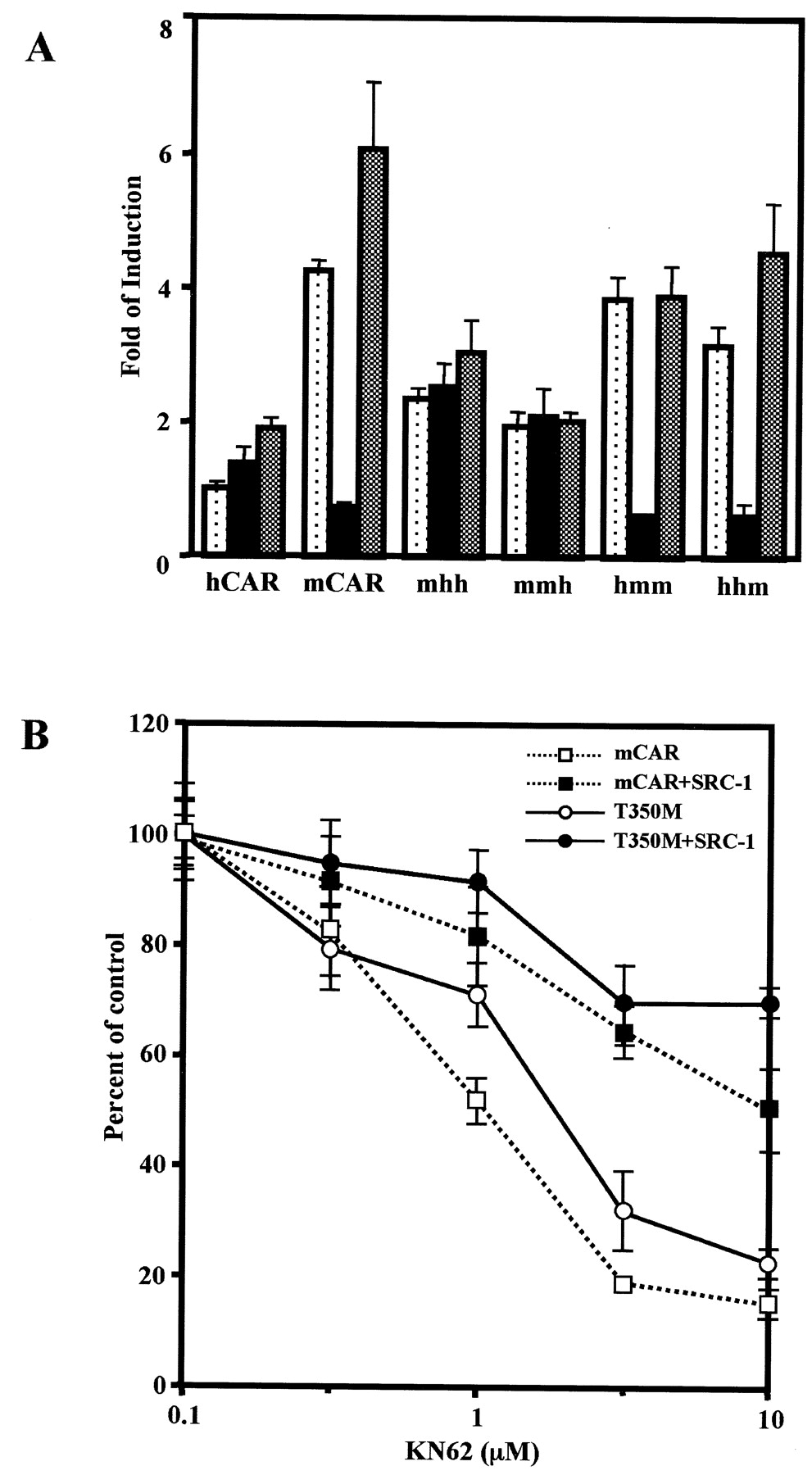
Repression by KN-62 of the mCART350Mactivity. A, the domain-based chimeric receptors, as indicated in Fig.1, were transfected into HepG2 cells, and NR1-luciferase reporter gene activity was measured as described under Materials and Methods. All abbreviations of the chimera are the same as those in Fig. 1. Dotted, filled, and shaded bars show luciferase activity of HepG2 cells treated with DMSO, KN-62 (10 μM), and KN-62 plus TCPOBOP (250 nM), respectively. These bars were generated from triplicate experiments with standard deviations. B, KN-62 repression was examined with and without the coexpression of SRC-1. With the exception that KN-62 at various concentrations (0.1, 0.3, 1, 3, and 10 μM) was used instead of progesterone, all other experimental treatments and representations are the same as those shown in Fig. 4. Standard deviations were calculated from triplicate experiments.
Discussion
Our study demonstrated that only the identity of a single residue at position 350 is sufficient to confer the steroid hormone responsiveness to mCAR, despite the fact that the receptor can be promiscuously activated by numerous endogenous and foreign chemicals. Other nuclear orphan receptors such as peroxisome proliferator-activated receptors, pregnane X receptor, and farnesoid X receptor share more or less the same promiscuity. This paradoxical characteristic of the receptor function is reminiscent of the drug/steroid-metabolizing enzymes, such as cytochrome P450. As a member of the large family, cytochrome P450 is an enzyme that generally metabolizes a broad range of substrates, yet a given cytochrome P450 can often be characterized by a unique substrate. Moreover, a single amino acid residue is sufficient to confer unique substrate specificity to a given cytochrome P450 (Lindberg and Negishi, 1989; Negishi et al., 1996). These paradoxical characteristics (i.e., coexistence of promiscuity and specificity) seem to be fundamental to proteins and enzymes that encounter practically unlimited numbers of foreign chemicals. However, the structural principles governing the paradox are not understood. Considering the large number of genes that are collectively regulated by the nuclear orphan receptors in response to diverse chemicals, finding the structural principles that govern activation and repression is critical to predict the pharmacological as well as the toxicological effects of a given chemical. As observed with P450 polymorphisms, the structural information will help us to understand genetic polymorphism and its biological implications in nuclear orphan receptors when it becomes evident.
Footnotes
- Received January 4, 2002.
- Accepted February 21, 2002.
-
S.K. is a Japan Society for the Promotion of Science Research fellow.
Abbreviations
- PB
- phenobarbital
- mCAR
- mouse nuclear constitutive active receptor
- hCAR
- human nuclear constitutive active receptor
- CAR
- nuclear constitutive active receptor or constitutive androstane receptor
- SRC-1
- steroid receptor coactivator 1
- AF2
- activation function 2
- PCR
- polymerase chain reaction
- tk
- thymidine kinase
- TCPOBOP
- 1,4-bis [2-(3,5-dichloropyridyloxy)]benzene
- E2
- estradiol
- KN-62
- (8)-5-isoquinolinesulfonic acid, 4-[2-(5-isoquinolinyl-sulfonyl)methylamino]-3-oxo-(4-phenyl-1-piperazinyl)-propyl]phenyl ester
- DBD
- DNA binding domain
- LBD
- ligand binding domain
- DMSO
- dimethyl sulfoxide
- U.S. Government




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}