


Abstract
Sequence-specific small interfering RNA (siRNA) duplexes can be used for gene silencing in mammalian cells and as mechanistic probes for determining gene function. Transfection of siRNAs for the aryl hydrocarbon receptor (AhR) and AhR nuclear translocator (ARNT) mRNAs in MCF-7 breast cancer cells resulted in a 60 to 80% decrease in levels of AhR and ARNT proteins in whole-cell extracts and decreased binding of nuclear extracts to 32P-labeled dioxin-responsive element. siRNA for the AhR also decreased 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced CYP1A1 protein, CYP1A1-dependent activity, and luciferase activity in cells transfected with an Ah-responsive construct. 17β-Estradiol (E2) induces proliferation of MCF-7 cells through enhanced G0/G1 → S phase progression, and this response is inhibited in cells cotreated with E2 plus TCDD. The effects of TCDD on E2-induced cell-cycle progress were partially blocked in MCF-7 cells transfected with siRNA for AhR. The results also indicated that siRNA-dependent decreases in AhR protein in MCF-7 cells were accompanied by increased G0/G1 → S phase progression, suggesting a growth-inhibitory role for the “endogenous” AhR. Surprisingly, TCDD alone induced G0/G1 → S phase progression and exhibited estrogenic activity in MCF-7 cells transfected with siRNA for the AhR. In contrast, degradation of the AhR in HepG2 liver cancer cells resulted in decreased G0/G1 → S phase progression, and this was accompanied by decreased expression of cyclin D1, cyclin E, cyclin-dependent kinase 2 (cdk2), and cdk4. In the absence of ligand, the AhR exhibits growth-inhibitory (MCF-7) and growth-promoting (HepG2) activity that is cell context-dependent.
The aryl hydrocarbon receptor (AhR) is a ligand-activated nuclear transcription factor that is a member of the periodicity/ARNT/single-minded and basic helix-loop-helix protein families (Wilson and Safe, 1998; Gu et al., 2000; Massari and Murre, 2000). The transcriptionally active nuclear AhR complex is a heterodimer of the AhR and AhR nuclear translocator (ARNT) proteins, which interact with genomic cis-acting dioxin-responsive elements (DREs) in the CYP1A1 and other Ah-responsive genes (Swanson and Bradfield, 1993; Whitlock Jr., 1993; Whitlock, 1999). The ARNT protein, which was initially identified as a partner for the AhR (Reyes et al., 1992), is also called hypoxia-inducible factor 1β (HIF-1β), and many hypoxia-induced genes are regulated by the HIF-1α:HIF-1β complex interacting with hypoxia-responsive elements (Semenza, 1998). The AhR was initially identified as the intracellular receptor for the environmental toxicant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), which binds with high affinity to the AhR (Poland et al., 1976). Interactions of TCDD and related halogenated aromatic compounds with the AhR mediate their diverse species-/strain-, tissue-, and age-specific biochemical, toxic, carcinogenic, and anticarcinogenic responses (Poland et al., 1979; Poland and Knutson, 1982; Safe, 1990).
The physiological role of the AhR-ARNT heterodimer has been investigated in transgenic knockout mice that do not express the AhR protein (Fernandez-Salguero et al., 1995; Schmidt et al., 1996; Mimura et al., 1997). Not surprisingly, these knockout animals do not respond to the prototypical TCDD-induced biochemical (e.g., CYP1A1 induction) and toxic responses (Fernandez-Salguero et al., 1996); however, the three strains of mice deficient in the AhR exhibit both common and different phenotypes. These mice typically have problems in liver development, poor fecundity, and weight loss, suggesting that the AhR-ARNT complex regulates constitutive functions in the absence of exogenous ligand. These results are consistent with reports showing that AhR-ARNT alone may act as a transcription factor; however, this would not exclude a role for an unknown endogenous ligand (Ma and Whitlock Jr., 1996; Weiss et al., 1996; Chang and Puga, 1998; Wang et al., 1999).
The AhR also binds with moderate-to-low affinity to chemoprotective phytochemicals such as indole-3-carbinol, flavonoids, and carotenoids, which exhibit both AhR agonist and antagonist activities (Bjeldanes et al., 1991; Gradelet et al., 1997; Denison et al., 1998; Ashida et al., 2000). Research in our laboratory has identified selective AhR modulators that exhibit minimal AhR-mediated toxicities but inhibit 17β-estradiol (E2)-induced gene expression and mammary tumor growth in rodent models (Safe et al., 1999, 2000, 2001; McDougal et al., 2001). The molecular mechanisms of inhibitory AhR-estrogen receptor (ER) cross talk may be complex and dependent on multiple factors, including cell context. This study investigates AhR-ERα cross talk and other AhR-mediated pathways using small interfering RNA (siRNA) for the AhR that selectively degrades AhR mRNA and decreases AhR protein expression and function in breast cancer cells. Decreased expression of the AhR in MCF-7 breast cancer cells resulted in an increase in the percentage of cells in S phase and a decrease in G0/G1, suggesting that in the absence of exogenous ligand, the AhR suppresses the growth of this cell line. In contrast, degradation of the AhR in HepG2 liver cancer cells decreases G0/G1 → S phase progression, indicating a role for the AhR in enhancing the growth of this cell line, and this is associated with decreased expression of cyclin D1, cyclin E, cdk2, and cdk4. We also observed that in MCF-7 cells transfected with siRNA for the AhR, TCDD exhibited estrogenic activity, and this complements results of a previous study in AhR-deficient MCF-7 cells (Moore et al., 1994). Thus, selective gene silencing of the AhR in breast and liver cancer cells illustrates the usefulness of this approach for investigating cellular mechanisms and function of the gene targeted for degradation.
Materials and Methods
Chemical and Biochemical Cell Lines. MCF-7 and HepG2 cells were obtained from the American Type Culture Collection (Manassas, VA). Dulbecco's modified Eagle's medium plus Ham's F12 medium with and without phenol red, 100× antibiotic/antimycotic solution, propidium idodide, and E2 were purchased from Sigma Chemical Co. (St. Louis, MO). Fetal bovine serum was purchased from Intergen (Purchase, NY). [γ-32P]ATP (300 Ci/mmol) was obtained from PerkinElmer Life Sciences (Boston, MA). T4-polynucleotide kinase was purchased from Roche Diagnostics (Indianapolis, IN). Antibodies for Sp1, lamin A, AhR, ARNT, and CYP1A1 proteins were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Dr. Ming-Jer Tsai (Baylor College of Medicine, Houston, TX) kindly provided human ERα expression plasmid. The pDRE3 and pERE3 constructs contain three consensus DRE and ERE motifs, respectively; oligonucleotides containing these motifs were linked to the bacterial luciferase gene and cloned into BamHI-HindIII cut XP-2 plasmid obtained from the American Type Culture Collection. Lysis buffer, luciferase reagent, and RNase were obtained from Promega (Madision, WI). All other chemicals and biochemicals were the highest quality available from commercials sources. siRNA duplexes were prepared by Dharmacon Research (Lafayette, CO) and targeted coding regions of the AhR (1416 to 1434), ARNT (445 to 463), lamin A/C (608 to 626), and luciferase (GL2) (153 to 171). The siRNA duplexes used in this study are indicated below. Scrambled siRNA was derived from a message transcribed from the chloroplast genome of Euglena gracilis (accession number 70810, position 24750 to 24768). The design of individual siRNA duplexes is summarized in Table 1, and the approaches used for selecting specific sequences were comparable with those described by Harborth and coworkers (2001).
Summary of siRNAs used in this study
Transfection of MCF-7 and HepG2 Cells and Preparation of Nuclear Extracts. Cells were cultured in 6-well plates in 2 ml of Dulbecco's modified Eagle's medium plus Ham's F12 medium supplemented with 5% fetal bovine serum. When cells were approximately 50 to 60% confluent, siRNA duplexes and/or reporter gene constructs were transfected using Oligofectamine Reagent (Invitrogen, Carlsbad, CA). Taken from results of preliminary studies, 7 μl of 20 μM stock solution of siRNAs was transfected in each well to give a final concentration of 140 nM. Cells were harvested 48 to 56 h after transfection by manual scraping in 1× lysis buffer (Promega). Whole-cell extracts were frozen in liquid nitrogen for 30 s, vortexed for 30 s, and centrifuged at 12,000g for 1 min to yield lysates that were assayed for luciferase activity using luciferase assay reagent (Promega). β-Galactosidase activity was determined using Tropix Galacto-Light Plus assay system (Tropix, Bedford, MA) in a Lumi-count Micro-well plate reader (PerkinElmer Life Sciences). For preparing nuclear extracts, cells were washed in PBS (2×), scraped in 1 ml of 1× lysis buffer, incubated at 4°C for 15 min, and centrifuged at 14,000g for 1 min at 20°C. Cell pellets were initially washed in 1 ml of lysis buffer (3×), and lysis buffer supplemented with 500 mM KCl was then added to the cell pellet and incubated with frequent vortexing for 45 min at 4°C. Nuclei were pelleted by centrifugation at 14,000g for 1 min at 4°C, and aliquots of supernatant were stored at -80°C and used for gel shift assays.
Western Immunoblot. Forty-eight hours after transfection, cells were washed once with PBS and collected by scraping in 200 μl of lysis buffer [50 mM HEPES, 0.5 M sodium chloride, 1.5 mM magnesium chloride, 1 mM EGTA, 10% (v/v) glycerol, 1% Triton X-100, and 5 μl/ml of Protease Inhibitor Cocktail (Sigma)]. The lysates were incubated on ice for 1 h with intermittent vortexing followed by centrifugation at 40,000g for 10 min at 4°C. Equal amounts of protein from each treatment group were diluted with loading buffer, boiled, and loaded onto 7.5% SDS-polyacrylamide gel. Samples were electrophoresed at 150 to 180 V for 3 to 4 h, and separated proteins were transferred to polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA) in buffer containing 48 mM Tris-HCl, 29 mM glycine, and 0.025% SDS. Proteins were detected by incubation with polyclonal primary antibodies Sp1 (PEP2), lamin A/C (N-18), AhR (N-19), ARNT1 (C-19), CYP1A1 (G-18), cyclin D1 (M-20), cyclin E (C-19), cdk2 (M-2), cdk4 (C-22), Rb (C-15), and p27 (C-19) followed by blotting with horseradish peroxidase-conjugated anti-rabbit (for Sp1, cyclin D1, cyclin E, cdk2, cdk4, and p27), anti-goat (for lamin A, CYP1A1, AhR, and ARNT) or anti-mouse (for Rb) secondary antibody. Blots were then exposed to chemiluminescent substrate (PerkinElmer Life Sciences) and placed in Kodak X-Omat AR autoradiography film (Eastman Kodak, Rochester, NY). Band intensities were determined by a scanning laser densitometer (Sharp Electronics Corporation, Mahwah, NJ) using Zero-D Scanalytics software (Scanalytics Corporation, Billerica, MA).
FACS Analysis. Cells were transfected with siRNAs for AhR or scramble RNA, and after 36 h, cells were synchronized in serum-free media for 24 h and treated with Me2SO or 20 nM E2, 20 nM TCDD, or TCDD plus E2 for 18 to 20 h in serum-free medium. Cells were then trypsinized, and approximately 2 × 106 cells were centrifuged and resuspended after removal of trypsin in 1 ml of staining solution containing 50 μg/ml propidium iodide, 4 mM sodium citrate, 30 units/ml RNase, and 0.1% Triton X-100, pH 7.8. Cells were incubated at 37°C for 10 min, and then before FACS analysis, sodium chloride was added to give a final concentration of 0.15 M. Cells were analyzed on a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA) using CellQuest (BD Biosciences) acquisition software. Propidium iodide fluorescence was collected through a 585/42-nm band-pass filter, and list mode data were acquired on a minimum of 12,000 single cells defined by a dot plot of PI-width versus PI-area. Data analysis was performed with the use of ModFit LT software (Verity Software House, Topsham, ME) using PI-width versus PI-area to exclude cell aggregates. FlowJo (Treestar, Inc., Palo Alto, CA) was used to generate plots shown in the figures.
Electrophoretic Mobility Shift Assay. Consensus DRE oligonucleotide was synthesized and annealed, and 5-pmol aliquots were 5′-end–labeled using T4 kinase and [γ-32P]ATP (Moore et al., 1994). A 30-μl EMSA reaction mixture contained approximately 100 mM KCl, 3 μg of nuclear protein, 500 ng of salmon sperm DNA (Invitrogen) with or without unlabeled competitor oligonucleotide, and 10 fmol radiolabeled probe. After incubation for 20 min on ice, antibodies against AhR protein were added and incubated another 20 min on ice. Protein/DNA complexes were resolved by 5% polyacrylamide gel electrophoresis in 1× Tris-borate/EDTA (0.09 M Tris base, 0.09 M boric acid, and 2 mM EDTA, pH 8.3) at 120 V at 4°C for 2 to 3 h. Specific DNA/protein and antibody supershifted complexes were observed as retarded bands in the gel.
EROD Activity. EROD activity was determined as described previously (Willett et al., 1997). Trypsinized cells were seeded in 48-well plates and grown to 50% confluence. Thirty-six h after transfection with siRNAs, cells were treated with Me2SO or 10 nM TCDD for 18 to 20 h. Cells were then washed with PBS; 200 μl of PBS was added to each well, and cells were incubated at 37°C for 2 min. Ethoxyresorufin (1.25 μg) was added to each well and incubated for 10 min at 37°C, and the reaction was stopped by adding 100 μl of fluorescamine. EROD activity and protein concentration were determined on a CytoFluor 2350 plate reader as described previously (Willett et al., 1997). Each treatment was carried out in triplicate, and results are presented as means ± S.D.
Statistical Analysis. Statistical significance was determined by analysis of variance and Scheffe's test, and the levels of probability are noted. The results are expressed as means ± S.D. for at least three separate (replicate) experiments for each treatment.
Results
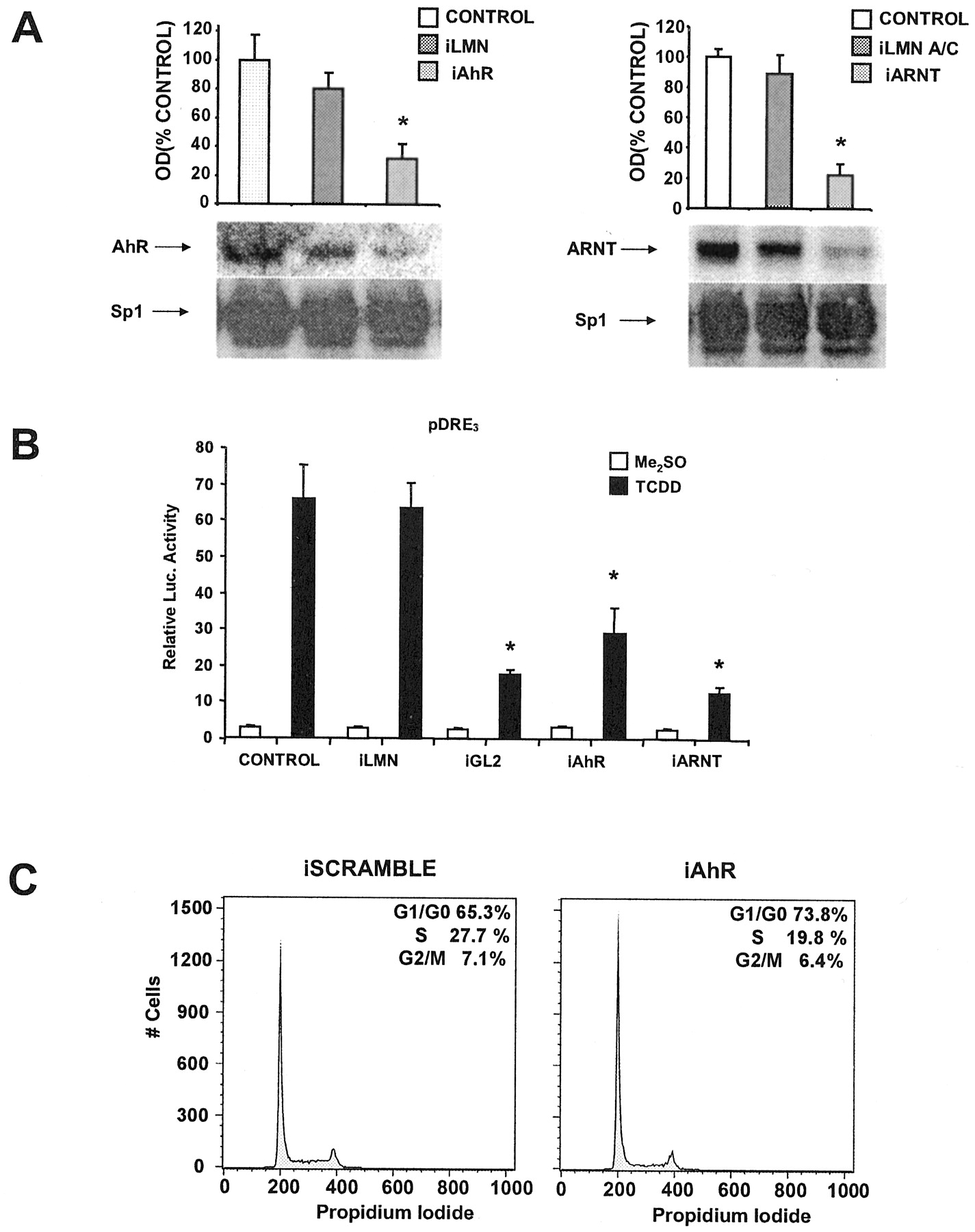
MCF-7 breast cancer cells are ERα-positive and express the AhR and ARNT proteins (Harris et al., 1989; Wormke et al., 2000). Results summarized in Fig. 1A demonstrate that AhR levels are decreased in MCF-7 cells transfected with siRNA for the AhR, whereas levels were unchanged in control cells and cells treated with siRNA for lamin A. Sp1 protein is used as a loading control for these experiments because it is highly expressed and unaffected by treatment with E2 or AhR agonists (Wormke et al., 2000). This experiment has been replicated several times, and AhR protein levels are typically decreased by 60 to 80% in whole-cell extracts depending on the transfection efficiency in the individual experiments. Results shown in Fig. 1B demonstrate the specificity of the siRNAs and show that treatment with siRNA for lamin A decreases lamin A protein levels by approximately 65%, whereas siRNA for the AhR did not affect lamin protein. Using a similar approach, we also showed that siRNA for ARNT specifically decreases ARNT protein expression in MCF-7 cells, whereas siRNA for lamin A did not affect levels of ARNT protein (Fig. 1C).
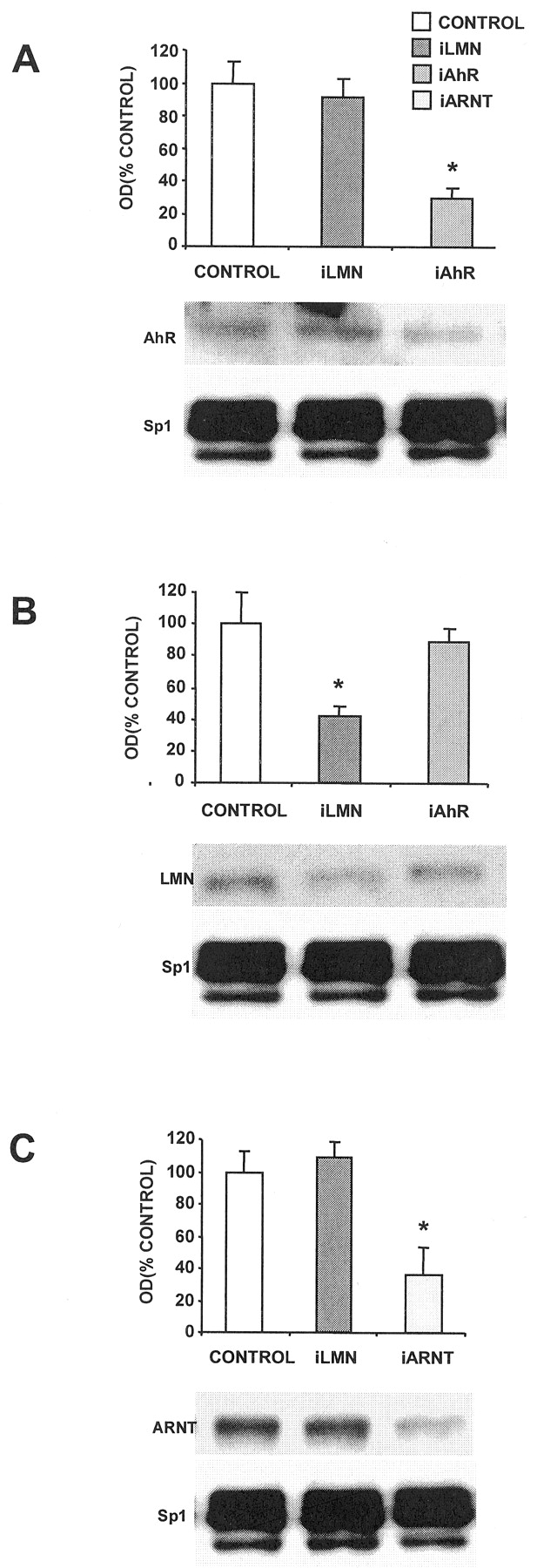
siRNAs for AhR (iAhR) and ARNT (iARNT) decrease their corresponding proteins in MCF-7 cells. A, effects on AhR protein in MCF-7 cells. Cells were transfected with siRNA for lamin A (iLMN) and iAhR, and whole-cell extracts were analyzed for AhR and Sp1 proteins by Western blot analysis as described under Materials and Methods. Results are means ± S.D. for three replicate determinations for each treatment group, and a significant (p < 0.05) decrease in AhR protein levels was observed only in cells treated with iAhR. B, effects of siRNAs on lamin A in MCF-7 cells. Cells were treated as described in A, and lamin A and Sp1 proteins were detected by Western blot analysis. Treatment with iLMN significantly (p < 0.05) decreased lamin A protein. C, effects of siRNAs on ARNT protein. Experiments were carried out as described in A, and iARNT significantly (p < 0.05) decreased ARNT protein in MCF-7 cells, whereas iLMN did not affect ARNT protein levels. Sp1 protein serves as a loading and reference control protein that is not affected by the siRNAs used in this study.
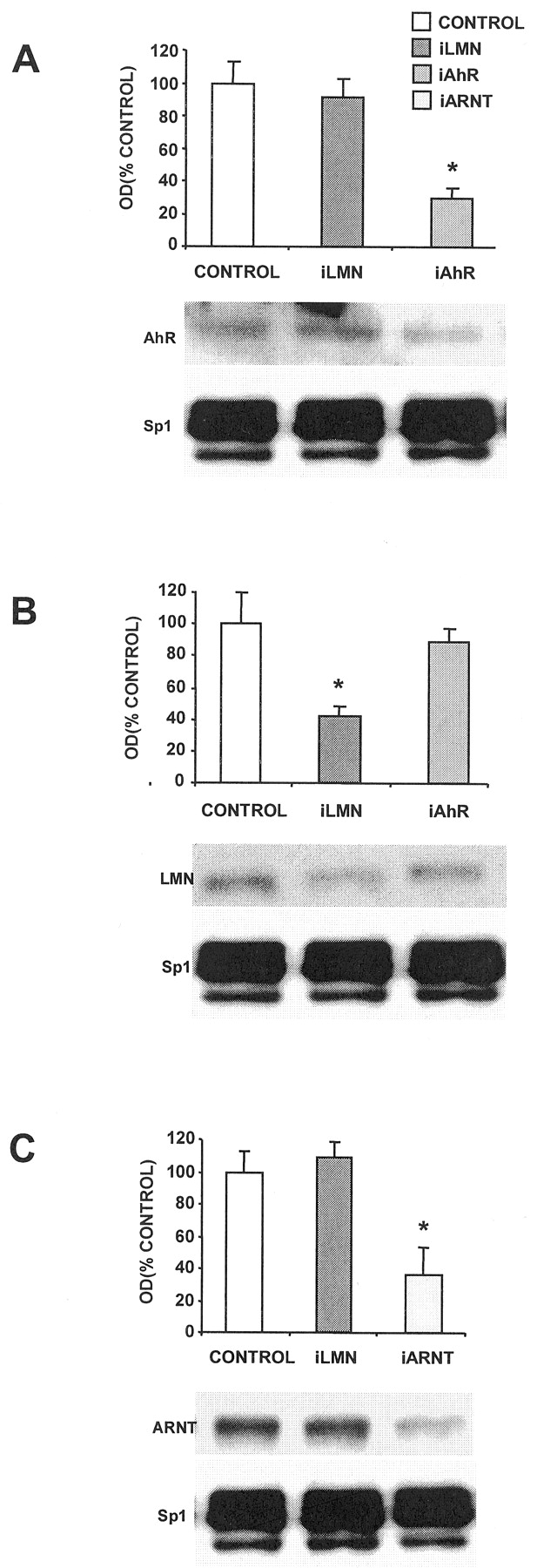
MCF-7 cells were treated with siRNA for lamin A, AhR, and ARNT, and nuclear extracts were incubated with 32P-labeled DRE and analyzed by gel mobility shift assays (Fig. 2). In control cells and cells treated with siRNA for lamin A, a weak complex was observed after treatment with Me2SO (lanes 2 and 4), and an intense retarded band was observed using nuclear extracts from cells treated with 10 nM TCDD (lanes 3, 5, and 9). In extracts from cells treated with siRNAs for AhR or ARNT, there was a marked decrease in retarded band intensities in extracts from Me2SO- (lanes 6 and 10) and TCDD- (lanes 7 and 11) treated cells. The specifically bound complex was decreased after incubation with unlabeled DRE (lane 8) and supershifted with AhR antibodies (lane 9). These results complement Western blot analyses of whole-cell lysates (Fig. 1, A to C) showing that siRNAs for AhR and ARNT decrease expression of their corresponding proteins in MCF-7 cells.

Binding of [32P]DRE with nuclear extracts from breast cancer cells treated with iLMN, iAhR, or iARNT. MCF-7 cells were treated with Me2SO (D) or TCDD (T) and transfected with iAhR, iARNT, or iLMN or not transfected (CTL), and binding of nuclear extracts to [32P]DRE was determined in gel mobility shift assays as described under Materials and Methods. Only iAhR or iARNT decreased the intensity of the specifically bound AhR/ARNT:DRE complex (arrow). Similar results were observed in duplicate experiments.
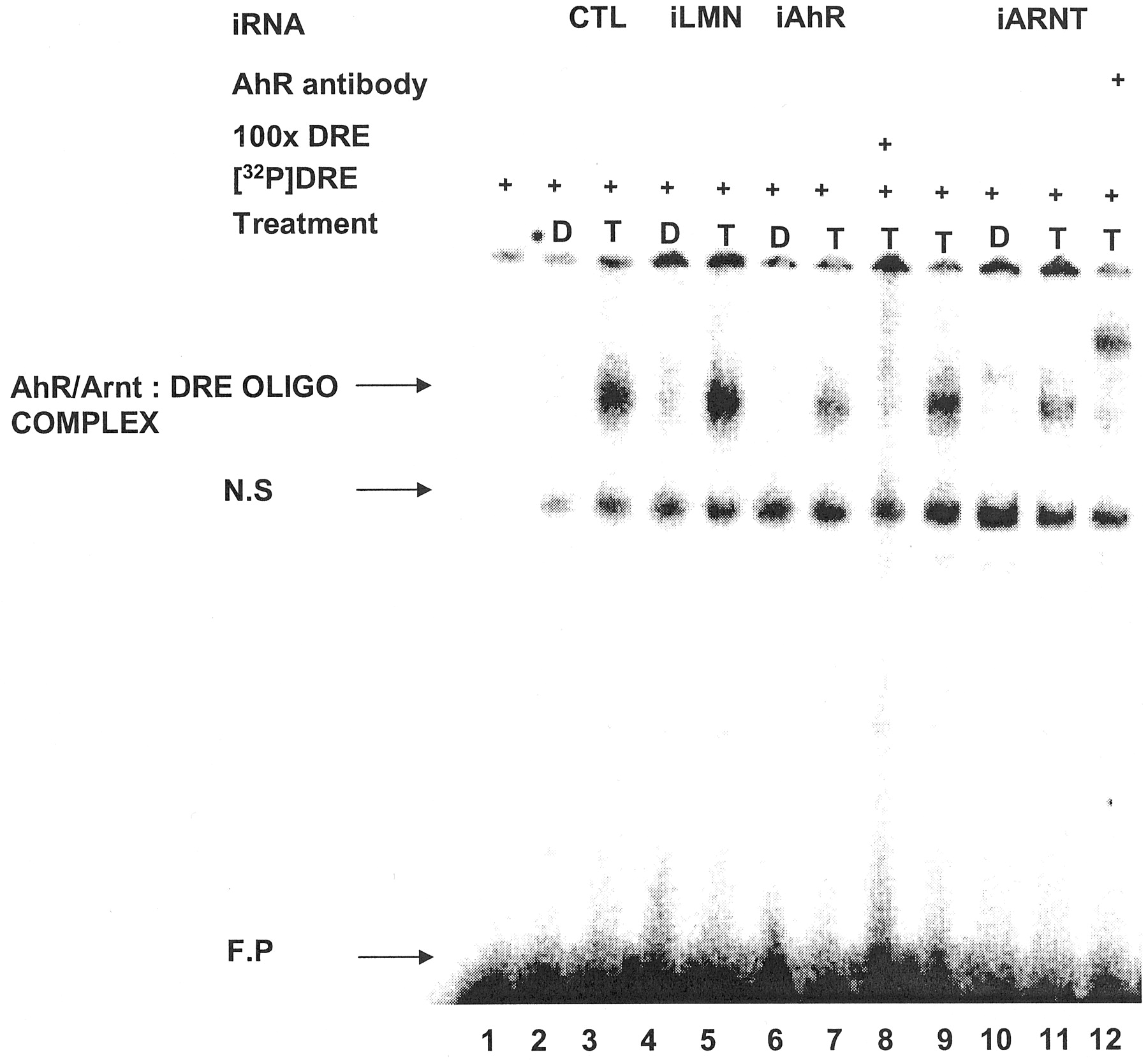
TCDD induces CYP1A1 mRNA and protein levels in multiple cells/tissues (Whitlock, 1999), including MCF-7 cells as indicated in Fig. 3A. In cells treated with siRNA for lamin A, there was a slight decrease in CYP1A1 protein levels in the control and TCDD-induced response; however, after treatment with siRNA for AhR, CYP1A1 protein levels induced by TCDD were decreased by >65%. In a separate experiment, the effects of siRNAs for lamin A and AhR on induction of CYP1A1-dependent EROD activity also showed that only siRNA for the AhR decreased induction of EROD activity by TCDD (Fig. 3B). A complementary study used an Ah-responsive construct containing three tandem consensus DREs linked to a luciferase reporter gene. The results (Fig. 3C) showed that siRNAs for AhR and ARNT inhibited the induction of luciferase activity by TCDD (compared with control cells), whereas siRNA for lamin A and scrambled siRNA (data not shown) did not affect Ah-responsiveness. As a positive control, siRNA for GL2 inhibited luciferase activity in cells treated with solvent or TCDD.

iAhR inhibits TCDD-induced transactivation. A, CYP1A1 protein. Cells were transfected with iLMN or iAhR, treated with Me2SO or 20 nM TCDD, and CYP1A1 protein was determined by Western blot analysis as described under Materials and Methods. **, significant (p < 0.05) decreases in activity compared with control cells. B, EROD activity. The treatment groups were comparable with those outlined in A, and EROD activity was determined as described under Materials and Methods. *, significant (p < 0.05) decreases in activity compared with control cells. C, luciferase activity. The treatment groups included those described in A except that scrambled siRNA was used as control and iGL2 was also transfected. Cells were transfected with pDRE3, and luciferase activity was determined as described under Materials and Methods. *, significantly (p < 0.05) decreased activity compared with control cells. Results are expressed as means ± S.D. for at least three replicate experiments for each treatment group.
Previous studies have demonstrated that TCDD and related AhR agonists inhibit expression of E2-induced genes and proliferation of ER-positive breast cancer cells (Safe et al., 2000). The results in Table 2 summarize FACS analysis of the effects of Me2SO (solvent control), E2, TCDD, and their combination on MCF-7 cell-cycle progression in which interactions between the AhR- and ERα-mediated pathways are primarily directed at changes in the percentage of cells in G0/G1 and S phases. The control for this experiment with MCF-7 (and HepG2) cells was the scrambled siRNA, and previous studies in MCF-7 cells also showed that transfection alone did not affect cell-cycle progression (Abdelrahim et al., 2002). E2 induced a >6.1 to 6.6% increase in MCF-7 cells in S phase (compared with Me2SO), whereas TCDD alone decreased the percentage of cells in S phase and inhibited E2-induced G0/G1 → S phase progression. In solvent (Me2SO)-treated MCF-7 cells transfected with siRNA for AhR, there was a >2.7 to 3.3% increase in cells in S phase compared with cells treated scrambled siRNA and with Me2SO alone (Fig. 3A). These results suggest that in the absence of exogenous ligand, the AhR inhibits G0/G1 → S phase progression of MCF-7 cells. E2 induced an 8.9% increase in cells in S phase transfected with siRNA for the AhR, and this was only slightly decreased in cells cotreated with E2 plus TCDD. These results confirm that the AhR is required for activation of growth-inhibitory AhR-ERα cross talk by TCDD (Safe et al., 2000). A comparison of the effects of TCDD alone in MCF-7 cells and in AhR-depleted cells treated with siRNA for the AhR indicates that TCDD induces AhR-independent G0/G1 → S phase progression and exhibits estrogen-like mitogenic activity (observed in at least three separate experiments). Therefore, the estrogenic activity of TCDD was further investigated in MCF-7 cells cotransfected with a construct containing three tandem EREs (pERE3) and siRNAs for lamin A, luciferase, and the AhR (Fig. 4). E2 induced luciferase activity in cells transfected with siRNA for lamin A or the AhR, and minimal activity was observed in cells transfected with siRNA for luciferase (iGL2). In wild-type Ah-responsive MCF-7 cells transfected with siRNA for lamin A, TCDD slightly decreased luciferase activity as previously observed using other E2-responsive constructs in MCF-7 cells (Moore et al., 1994; Safe et al., 2000). In contrast, TCDD significantly increased luciferase activity in cells cotransfected with pERE3 and siRNA for the AhR, and this complemented the mitogenic activity of TCDD in these same AhR-deficient cells (Table 2). The estrogenic activity of TCDD (and E2) in AhR-deficient cells was inhibited after cotreatment with the antiestrogen ICI 182,780 (Fig. 4B); minimal interactions (TCDD plus ICI 182,780) were observed in cells transfected with siRNA for lamin A. Moore and coworkers (1994) previously developed AhR-defective MCF-7 cells that express ARNT but low-to-nondetectable AhR protein. Inhibitory AhR-ERα cross talk was also not observed in this cell line, and treatment of these cells with TCDD caused an increase in cell growth and significantly induced reporter gene activity in cells transfected with an E2-responsive construct containing the ERE from the vitellogenin A2 gene promoter.
Cell-cycle distribution of MCF-7 cells treated with E2, TCDD, and TCDD plus E2, and small inhibitory RNA for AhR

Effects of iLMN, iAhR, and iGL2 on luciferase activity in MCF-7 cells transfected with pERE3 and treated with Me2SO, 30 nM E2, 30 nM TCDD, 1 μM ICI 182,780, or their combination. A, effects of iAhR on induced luciferase activity. MCF-7 cells were cotransfected with pERE3 along with iLMN, iGL2, or iAhR and treated with Me2SO, TCDD, or E2. Luciferase activity was determined as described under Materials and Methods. *, significant (p < 0.05) induction. B, antiestrogen inhibition of TCDD- and E2-induced transactivation. Cells were transfected with pERE3 and iLMN or iAhR and treated with Me2SO, E2, TCDD, ICI 182,780, or combinations, and luciferase activity was then determined as described under Materials and Methods. *, significant (p < 0.05) induction; **, significant inhibition by ICI 182,780. Results summarized in A and B are means ± S.D. for three replicate determinations for each treatment group.
The growth-inhibitory role of the endogenous AhR was in contrast to findings in studies in AhR-deficient rodent liver cancer cells in which AhR expression was associated with enhanced cell proliferation (Ma and Whitlock Jr., 1996). The results in Fig. 5A demonstrate that siRNAs for AhR and ARNT decrease expression of their respective proteins in Ah-responsive human HepG2 liver cancer cells, and induction of luciferase by TCDD in cells transfected with pDRE3 was also decreased in cells cotransfected with the same siR-NAs (Fig. 5B). siRNA for lamin A served as a control for these transfection studies, and this oligonucleotide did not affect levels of AhR/ARNT protein or luciferase inducibility by TCDD. Similar results were observed using scrambled siRNA (data not shown). FACS analysis of HepG2 cells transfected with scrambled siRNA or siRNA for the AhR (Fig. 5C) indicated that in AhR-deficient HepG2 cells, there was an 8% decrease in cells in S phase and a comparable increase in G0/G1. These results suggest that in HepG2 cells, the endogenous AhR enhances cell-cycle progression as previously reported in rodent cancer cell lines (Ma and Whitlock Jr., 1996; Weiss et al., 1996). In contrast, in breast cancer cells (Table 2), the AhR is growth-inhibitory, and this demonstrates the importance of cell context on the role for the endogenous AhR in Ah-responsive breast and liver cancer cell lines.

siRNAs for the AhR or ARNT decrease protein expression, TCDD-induced transactivation, and affect cell-cycle progression in human HepG2 cells. A, decreased protein expression. HepG2 cells were transfected with iAhR, iLMN, or iARNT, and AhR, ARNT, or Sp1 proteins were determined in the various treatment groups by Western blot analysis. B, decreased Ah-responsiveness. Cells were cotransfected with pDRE3 and scrambled RNA (control), iLMN, iGL2, iAhR, or iARNT and treated with Me2SO or 20 nM TCDD, and luciferase activity was determined as described under Materials and Methods. Results summarized in A and B are means ± S.D. for three replicate determinations for each treatment group. *, significant (p < 0.05) decreases in activity. C, effects of siRNA for the AhR on cell-cycle progression of HepG2 cells. HepG2 cells were transfected with scrambled RNA or iAhR, and the percentage of distribution of cells in G0/G1, S, and G2/M was determined by FACS analysis as described under Materials and Methods. In a second study, the percentage of cells in G0/G1:S:G2/M were 69.1:24.12:6.79 (plus scrambled siRNA) and 74.98:18.04:6.98 (plus siRNA for the AhR).
Because decreased AhR expression in MCF-7 and HepG2 cells affected G1 → S phase progression in both cell lines, we further investigated the modulation of several key cell-cycle regulatory proteins that are important in this phase of the cell cycle. The results in Fig. 6 show that in HepG2 cells transfected with siRNA for the AhR, there were significant decreases in cyclin D1, cyclin E, cdk2, and cdk4 protein expression, whereas no significant changes in Rb or p27 proteins were observed. Immunoblot analysis showed low-to-nondetectable levels of p21 protein in HepG2 (and MCF-7) cells. Thus, decreased proliferation of HepG2 cells transfected with siRNA AhR is consistent with decreased expression of several proteins required for G1 → S phase progression. In contrast, expression of these same proteins was unchanged in MCF-7 cells transfected with siRNA for the AhR. This suggests that other genes/proteins associated with increased proliferation of AhR-deficient MCF-7 cells must be affected, and these are currently being investigated.

Effects of AhR gene silencing on cell-cycle enzymes in MCF-7 and HepG2 cells. MCF-7 or HepG2 cells were transfected with iLMN or iAhR as described for the FACS analysis experiments (Figs. 4 and 6C), and levels of cyclin D1, cyclin E, cdk2, cdk4, Rb, and p27 proteins were determined in whole-cell lysates by Western blot analysis as described under Materials and Methods. Determinations were carried out in triplicate, and in HepG2 cells, relative protein levels in AhR-depleted cells compared with cells treated with iLMN are presented as means ± S.D. *, significant (p < 0.05) decreases in protein levels. Minimal-to-nondetectable p21 protein was detected in all groups (data not shown).
Discussion
The development of transgenic animal models in which specific gene(s) have been ablated, overexpressed, or conditionally expressed has provided unique insights into their physiological significance and roles in various diseases including cancer. Analogous approaches have been used for studies in mammalian cells using transiently or stably transfected expression plasmids for specific genes or their antisense/dominant-negative counterparts. RNA interference associated with double-stranded RNA that is rapidly processed into siRNAs has been identified in many eukaryotes (Sharp, 2001). Recent studies have demonstrated that siRNA oligonucleotides can be successfully used for gene silencing in mammalian cells (Elbashir et al., 2001; Harborth et al., 2001; Abdelrahim et al., 2002; Tuschl and Borkhardt, 2002). Initial applications of this technique by Elbashir and coworkers (2001) in HeLa, NIH3T3, COS-7, and 293 cells and subsequent studies in several different mammalian cell lines have demonstrated that gene silencing can target multiple genes, and this approach has numerous applications (Tuschl and Borkhardt, 2002). For example, research in our laboratory (Abdelrahim et al., 2002) has shown that siRNA for Sp1 decreases Sp1 protein expression in MCF-7 cells and also blocks E2-dependent transactivation of a GC-rich construct through interactions of ERα/Sp1. Moreover, silencing of Sp1 inhibited hormone-induced cell-cycle progression of MCF-7 cells, showing that ERα/Sp1-mediated genes play an important role in the growth of breast cancer cells.
In this study, we successfully used siRNA for AhR to decrease AhR protein expression in MCF-7 cells, and siRNAs for lamin A and ARNT also silence their corresponding genes, resulting in 60 to 80% decreased expression of their corresponding proteins (Figs. 1 and 2). AhR-mediated induction of CYP1A1 has been extensively investigated as a model for understanding the molecular mechanisms of AhR action (Whitlock, 1999), and the results in Fig. 3 demonstrate that siRNA for the AhR blocks induction of CYP1A1 protein, EROD activity, and DRE-dependent reporter gene activity, and siRNA for ARNT gave similar results in some of the assays. These data confirm the role of AhR/ARNT in mediating the induction of CYP1A1 and also illustrate that the siRNA approach can be used for targeting the AhR and ARNT.
Several studies report that TCDD inhibits E2-induced gene/reporter gene activity in breast cancer cells, and inhibitory AhR-ERα cross talk is also observed for cell proliferation and cell-cycle progression (Wang et al., 1998). Treatment of MCF-7 cells with E2 significantly enhances G0/G1 → S phase progression of ER-positive breast cancer cells, and this response is inhibited after cotreatment with TCDD (Wang et al., 1998). The results summarized in Table 2 also show that E2 and E2 plus TCDD primarily act on the G0/G1 → S phase of the cell cycle and that TCDD alone is growth-inhibitory as reported previously (Wang et al., 1998). Not unexpectedly, in cells transfected with siRNA for the AhR, the inhibitory effects of TCDD on E2-induced G0/G1 → S phase progression were dramatically decreased as demonstrated by FACS analysis of the whole cells (transfected plus untransfected). Moreover, in cells transfected with siRNA for the AhR and treated with Me2SO (solvent), there was an increase in the percentage of cells in S or G2/M phase, and this was also observed in all the treatment groups in the AhR-depleted cells. These results show that in the absence of TCDD, the AhR is growth-inhibitory in MCF-7 cells, and this constitutes a function for the endogenous AhR in this cell line. Expression of several proteins required for G1 → S phase progression was investigated in AhR-deficient MCF-7 cells (Fig. 6); significant changes in levels of cyclin D1, cyclin E, cdk2, cdk4, Rb, or p27 (or p21) proteins were not observed. Currently, we are using microarrays to identify specific AhR-regulated genes that play a role in inhibiting breast cancer cell growth.
Phenotypic changes observed in AhR knockout mice suggest that the AhR complex exhibits exogenous ligand-independent activity as a transcription factor, and this is supported by other reports, including studies in cell lines with defective or mutated AhR expression (Ma and Whitlock Jr., 1996). Ma and Whitlock (1996) showed that AhR-defected mouse Hepa1 cells exhibited a different morphology, longer doubling times, and a higher percentage of cells in G0/G1 compared with wild-type (AhR-positive) cells. Similar results were reported for AhR-defective rat hepatoma 5L cells, which also exhibit an increased percentage of cells in G0/G1 compared with wild-type Ah-responsive cells (Weiss et al., 1996). The cell context-dependent differences in endogenous AhR function in human breast cancer (growth-inhibitory) versus rodent liver cancer (growth-promoting) cells was confirmed in this study using a human hepatoma cell line (HepG2) in which siRNA for the AhR decreased AhR protein expression, resulting in an increased percentage of cells in G0/G1 and a decreased percentage of cells in S phase (Fig. 5). The ligand-independent effects of the AhR in HepG2 cells was further investigated by determining the expression of several proteins required for G1 → S phase progression in HepG2 cells transfected with siRNA for the AhR (Fig. 6). In AhR-depleted HepG2 cells, there was significantly decreased expression of cyclin D1, cyclin E, cdk2, and cdk4 proteins, and this was consistent with the higher percentage of HepG2 cells in G0/G1 compared with cells expressing the AhR. These results suggest that these four genes/proteins may be regulated by the endogenous AhR in liver cancer cells, and the molecular mechanisms of ligand-independent AhR gene regulation are currently being investigated.
FACS analysis of AhR-deficient MCF-7 cells suggests that TCDD exhibits mitogenic activity (Table 2). The estrogen-like activity of TCDD was surprising; however, previous studies in AhR-deficient benzo[a]pyrene-resistant MCF-7 cells also showed that TCDD induced a small but insignificant increase in cell proliferation and reporter gene activity in cells transfected with an E2-responsive construct containing a cathepsin D gene promoter insert (Moore et al., 1994). TCDD (1 nM) significantly increased secretion of procathepsin D protein in these AhR-deficient cells. The ER agonist activity of TCDD was confirmed in MCF-7 cells transfected with pERE3 and siRNA for lamin (control) or AhR. In AhR-deficient cells, both TCDD and E2 induced luciferase activity, and these responses were inhibited by the antiestrogen ICI 182,780. Thus, TCDD activates both the AhR and ER in breast cancer cells; however, because of the high affinity of TCDD for the AhR, the ER agonist response is only observed in AhR-deficient cells. A previous report showed that indolo[3,2-b]carbazole, an acid-catalyzed condensation product of the phytochemical indole-3-carbinol, was also an AhR and ER agonist in MCF-7 cells (Liu et al., 1994). Unlike TCDD, indolo[3,2-b]carbazole activated both pathways in Ah-responsive MCF-7 cells, and this may be caused by the lower affinity of this compound for the AhR (Bjeldanes et al., 1991). TCDD and related compounds may activate the ER through other pathways, such as the modulation of kinase activities (Matsumura, 1994), and this is currently being investigated.
In summary, results of this study demonstrate that ligand-independent actions of the AhR on cell proliferation are dependent on cell context, and both growth-inhibitory (breast) and growth-promoting (liver) functions can be observed in cancer cell lines. The results also demonstrate that TCDD exhibits estrogenic activity in AhR-deficient MCF-7 cells, and it is possible that activation of ER signaling by TCDD may be the predominant response in cells with high ER/AhR protein ratios. Ongoing studies are focused on developing cancer cell lines that stably express specific siRNAs that can be used for investigating the function of other transcription factors.
Footnotes
-
This study was supported by the financial assistance of the National Institutes of Health (ES04176 and ES09106), the Texas Agricultural Experiment Station, and the Sid Kyle endowment.
-
ABBREVIATIONS: AhR, aryl hydrocarbon receptor; ARNT, aryl hydrocarbon receptor nuclear translocator; DRE, dioxin-responsive element; ERE, estrogen response element; E2, 17β-estradiol; ER, estrogen receptor; HIF, hypoxia-inducible factor; siRNA, small interfering RNA; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; LMN, lamin A; cdk, cyclin-dependent kinase; PBS, phosphate-buffered saline; FACS, fluorescence-activated cell sorting; PI, phosphatidylinositol; EROD, ethoxyresorufin O-dealkylase; iAhR, small interfering RNA for aryl hydrocarbon receptor; iLMN, small interfering RNA for lamin A; iARNT, small interfering RNA for aryl hydrocarbon receptor nuclear translocator; iGL2, small interfering RNA for luciferase; ICI 182,780, 7α-[9-(4,4,5,5,5-pentafluoropentylsulfinyl)nonyl]estra-1,3,5(10)-triene-3,17-β-diol.
- Received November 8, 2002.
- Accepted March 7, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}