


Abstract
The nuclear constitutive active receptor (CAR) is a key transcription factor regulating phenobarbital (PB)-inducible transcription of various hepatic genes that encode xenobiotic/steroid-metabolizing enzymes. CAR is retained in the cytoplasm of noninduced livers and translocates into the nucleus after PB induction (Mol Cell Biol19:6318-6322, 1999). HepG2 cells lack the capability of retaining CAR in the cytoplasm; thus, the receptor spontaneously accumulates in the nucleus. We have now cloned and characterized a tetratricopeptide repeat (TPR) protein, designated cytoplasmic CAR retention protein (CCRP), for its ability to accumulate the receptor in the cytoplasm of cotransfected HepG2 cells. CCRP directly interacts with the ligand-binding domain of CAR and mediates the formation of a cytoplasmic CAR-CCRP-90-kDa heat shock protein (hsp90) ternary complex. Simultaneous expression of fluorescent protein-tagged CAR and CCRP reveals their colocalization with tubulin in mouse liver in vivo. Thus, these results indicate that CCRP may be a component of the CAR-hsp90 complex and involved in retaining the receptor in the cytoplasm of both HepG2 cells and probably in vivo liver cells.
As a coordinate regulator of hepatic gene expression in defense against xenobiotic toxicity, the nuclear receptor CAR plays a key role in regulating hepatic genes that encode xenobiotic/steroid-metabolizing enzymes such as cytochrome P450 and UDP-glucuronosyltransferase (Honkakoski et al., 1998; Sueyoshi et al., 1999; Wei et al., 2000; Sueyoshi and Negishi, 2001; Sugatani et al., 2001; Ueda et al., 2002; Yamamoto et al., 2003). CAR is compartmentalized in the cytoplasm of noninduced mouse liver (Kawamoto et al., 1999; Zelko et al., 2001). After PB induction, CAR translocates into the nucleus to form an active transcriptional complex with the retinoid X receptor. Thus, the nuclear translocation becomes an initial step in the process of CAR-mediated transcriptional activation in liver in vivo. However, the molecular and cellular mechanisms of the CAR nuclear translocation remain poorly understood.
One of the major difficulties in investigating the nuclear translocation mechanism is the lack of a cell system in which CAR is properly sequestered in the cytoplasm and translocates into nucleus in response to inducers. Expression of CAR in transformed cells, such as HepG2, resulted in spontaneous accumulation of the receptor in the nucleus (Kawamoto et al., 1999). Receptors such as glucocorticoid receptor (GR), vitamin D3 receptors, and aryl hydrocarbon receptor (AhR) are retained in the cytoplasm of transfected cells and translocate into the nucleus in response to their agonists (Pratt, 1993; Pollenz et al., 1994; Pratt and Toft, 1997). GR and AhR are present as complexes with hsp90, in which there is an additional co-chaperone specific to each receptor. These specific co-chaperones play an important role in the cytoplasmic retention of the receptors (Kazlauskas et al., 2000, 2001; Davies et al., 2002). For example, the GR-hsp90 complex contains immunophilin FK-binding proteins (FKBPs) (Tai et al., 1992; Nair et al., 1997). Immunophilin-like proteins called hepatitis B virus protein X-associated protein 2 (XAP2), AhR-associated protein 9, and AhR-interacting protein are specific constituents of the AhR-hsp90 complex (Carver and Bradfield, 1997; Ma and Whitlock, 1997; Meyer et al., 1998). These FKBP and XAP2 proteins are characterized by their multiple TPR motifs (Blatch and Lässle, 1999). In our recent work, CAR was partially purified from mouse liver cytosol by column chromatography and was chemically cross-linked. Western blot analysis using anti-CAR and anti-hsp90 antibodies demonstrated comigration of CAR and hsp90, thus indicating that CAR also forms a complex with hsp90 in mouse liver cytosol (Yoshinari et al., 2003). However, a unique co-chaperone such as FKBP or XAP2 has not yet been found in the CAR-hsp90 complex. Identification and coexpression of such a chaperone may retain CAR in the cytoplasm of HepG2 cells, providing a model for studying nuclear translocation of the receptor.
In attempting to identify such a chaperone, we performed yeast two-hybrid screening of a mouse liver cDNA library using CAR as bait and have cloned a TPR protein. For the first time, we have produced an in vitro cell system in which CAR accumulates in the cytoplasm. A TPR protein, previously known as mDj11 in mouse, TRP-2 in human, and dTPR-2 in Drosophila (Murthy et al., 1996; Kazemi-Esfarjani and Benzer, 2000; Ohtsuka and Hata, 2000) has now been identified as a chaperone that binds to CAR, forming a ternary complex with Hsp90, and is here designated CCRP. Coexpression and immunoprecipitation assays reveal that CCRP retains CAR in the cytoplasm of HepG2 cells by mediating the binding of the receptor to hsp90. Gene delivery through tail-vein injection of plasmid DNA was performed to directly express fluorescent protein-tagged CAR and CCRP in mouse liver in vivo. These coexpression studies have shown the colocalization of CAR and CCRP with tubulin, thus suggesting their association in the liver in vivo. Identification of CCRP and conversion of HepG2 cells to a phenotype capable of accumulating CAR in the cytoplasm will help in further investigation of the molecular mechanisms retaining CAR in the cytoplasm and translocating it into the nucleus after treatment with inducers.
Materials and Methods
Antibodies. Anti-CCRP antibody was raised against bacterially expressed recombinant mDj11 in rabbit. An anti-mCAR peptide antibody was produced in rabbit using the 20 N-terminal residues conjugated with keyhole limpet hemocyanin as the antigen and was purified using the peptide and a SulfoLink kit (Pierce, Rockford, IL). Anti-hsp90 monoclonal antibody was purchased from BD Transduction Laboratories (San Diego, CA) and Affinity Bioreagents (Golden, Co); anti-V5 and anti-V5-HRP antibodies were purchased from Invitrogen (Carlsbad, CA); and ImmunoPure Immobilized Protein A and ImmunoPure Immobilized Protein L were purchased from Pierce.
Plasmids. cDNAs encoding full-length mCAR and CCRP were amplified using proper primers and cloned into pcDNA3.1/V5-His-Topo plasmids (Invitrogen, Carlsbad, CA): pcDNA3.1/V5-His-CAR and pcDNA3.1/V5-His-CCRP. The following deletion mutants were also constructed: pcDNA3.1/V5-His-LBD (mCAR, Asn117 to Ser358); pcDNA3.1/V5-His-DBD (mCAR, Met1 to Met86); pcDNA3.1/V5-His-ΔAF2 (mCAR, Met1 to Thr350); pcDNA3.1/V5-His-tprAB (CCRP, Met1 to Agr379); pcDNA3.1/V5-His-J (CCRP, Lys380 to Gly494); pcDNA3.1/V5-His-ΔtprB (CCRP, Met1 to Asn209 plus Lys380 to Gly494). YFP-tagged CCRP expression vector was constructed by cloning the respective cDNAs into pEYFP-c1 (BD Biosciences Clontech, Palo Alto, CA). All plasmids were verified by nucleotide sequencing. CFP-tagged hCAR expression vector, hCAR/pCFP-c1, was reported previously (Kawamoto et al., 1999). Expression vectors for subcellular localization markers; pEYFP-Tub for tubulin, pECFP-ER for endoplasmic reticulum, pECFP-Mito for mitochondria, and pECFP-Golgi for Golgi apparatus were obtained from BD Biosciences Clontech (Palo Alto, CA).
Yeast Two-Hybrid Screening. Matchmaker Gal4 Two Hybrid System cDNA library from mouse liver (CLONTECH) in pGAD10 was screened using protocol provided from the manufacturer. To construct the hCAR L342A/pAS2-1 vector for bait, hCAR cDNA was cloned into pAS2-1 and Leu342 was mutated to Ala using the QuikChange site directed mutagenesis kit (Stratagene, La Jolla, CA). Yeast cells (Y190) were sequentially transformed with the bait construct and the mouse liver library DNA using Yeastmaker Yeast Transformation System (BD Biosciences Clontech). Fifty-three positive colonies were isolated from 1.6 × 107 primary transformants. Among them, three matched their sequences with mDj11 (Ohtsuka and Hata, 2000), and its full-length cDNA (now called CCRP) was amplified.
Expression of Fluorescent Protein-Tagged CAR in Liver in Vivo. Expression of CCRP, hCAR, and subcellular localization markers in liver in vivo and their microscopic detection were performed as reported previously (Zelko et al., 2001; Sueyoshi et al., 2002). Mouse liver sections of 30-μm thickness were analyzed by confocal laser scanning microscopy using a Zeiss LSM510 microscopy system for YFP and CFP images. A 100× oil immersion objective (1.4 numeric aperture) was used for scanning with a pixel size of 0.06 μm. Samples on the slides were excited at 514 nm (YFP) or 458 nm (CFP). YFP emission was detected using a 530-nm long-pass filter, and CFP fluorescence was detected using a 470- to 500-nm band pass filter. Pinhole size was set for 0.6 μm confocal slices.
Cell Culture and Fraction. HepG2 cells were cultured in minimal essential medium supplemented with 10% fetal bovine serum and antibiotics (100 unit/ml penicillin and 100 μg/ml streptomycin) on plastic dishes (10-cm diameter) at 37°C for 24 h before transfection. The CellPhect calcium phosphate coprecipitation method (Amersham Biosciences, Piscataway, NJ) was used to transfect plasmids. Cells were washed twice with phosphate-buffered saline, collected by centrifugation, and homogenized in 10 mM HEPES buffer, pH 7.6, containing 10 mM KCl, 1.5 mM MgCl2, 20 mM Na2MoO4, 0.3% Nonidet P-40, 0.2 mM phenylmethylsulfonyl fluoride, 2 μg/ml pepstatin A, 2 μg/ml leupeptin, and 1 mM dithiothreitol (buffer A). The homogenate was centrifuged at 4,000g for 10 min and the resulting supernatant was again centrifuged at 17,800g for 10 min to obtain the cytoslic fraction. After being washed three times with buffer A, the 4,000g pellet was re-suspended in 10 mM HEPES buffer, pH 7.6, containing 0.1 M KCl, 3 mM MgCl2, 0.1 mM EDTA, 1 mM Na3VO4, 2 μg/ml pepstatin A, 2 μg/ml leupeptin, 0.2 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol. The resulting suspension was mixed at 4°C for 1 h in the presence of 0.4 M NaCl and centrifuged at 50,000 rpm for 15 min to obtain nuclear extracts. All procedures were carried out at 4°C.
Immunoprecipitation. Anti-mCAR N20 (5 μg), anti-CCRP (5 μg), or anti-hsp90 (5 μl; ABR-Affinity BioReagents, Golden, CO) antibodies were added to 1 to 2 mg of cytosolic protein, and the mixture was incubated for 3 to 24 h at 4°C. Twenty microliters of a 50% slurry of protein A-agarose (anti-mCAR N20 or anti-CCRP) or protein l-agarose (anti-hsp90) was added and incubated for an additional hour. After rapid centrifugation, the resulting pellets were washed five times with 1 ml of 50 mM Tris-HCl buffer, pH 7.5, containing 0.15 M NaCl, 20 mM Na2MoO4, and 0.2% of Nonidet P-40, and the remaining slurry was boiled for 10 min in NuPAGE LDS sample buffer (Invitrogen) before immunoblotting.
GST Pull Down Assay. The cDNA of CCRP was cloned into pGEX4T-3. The GST-CCRP fusion protein was expressed in Escherichia coli BL21 cells and purified using glutathione-Sepharose (Amersham Biosciences). V5 peptide-tagged CARs (pcDNA3.1/hCAR and pcDNA3.1/mCAR) were expressed using the TnT coupled reticulocyte lysate system (Promega, Madison, WI). In vitro-translated CAR was incubated with GST-CCRP fusion protein on a glutathione resin at room temperature for 30 min. The resin was washed four times with HEPES-NaOH buffer, pH 7.6, containing 0.1 M NaCl and 0.1% Triton X-100. Subsequently, proteins were extracted from the resin with SDS-PAGE sample buffer, separated on a 10% Tris-glycine SDS-PAGE gel, transferred to Immobilon-P membrane (Millipore, Bedford, MA), and immuno-stained with anti-V5 antibody.
Western Blot and Northern Hybridization. Proteins were separated on a NuPAGE Novex 4-12% Bis-Tris Gel (Invitrogen) or a 10% SDS-polyacrylamide gel, and transferred to Immobilon-P membrane. Subsequently, these membranes were incubated for 1 h at 25°C with primary rabbit anti-CCRP (1:500 dilution), anti-V5 (1: 5000 dilution), anti-V5-HRP (1:5000 dilution), or anti-hsp90 (1:5000 dilution) antibodies in 5% nonfat milk in Tris-buffered saline/0.2% Tween 20 and were incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:5000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA). Finally, protein bands were visualized using ECL Western blotting detection reagent (Amersham Biosciences). For Northern analysis, mouse Multiple Tissue Northern Blot membrane was purchased from BD Biosciences Clontech. The membrane was hybridized with 32P-labeled CCRP cDNA under highstringency conditions.
Results
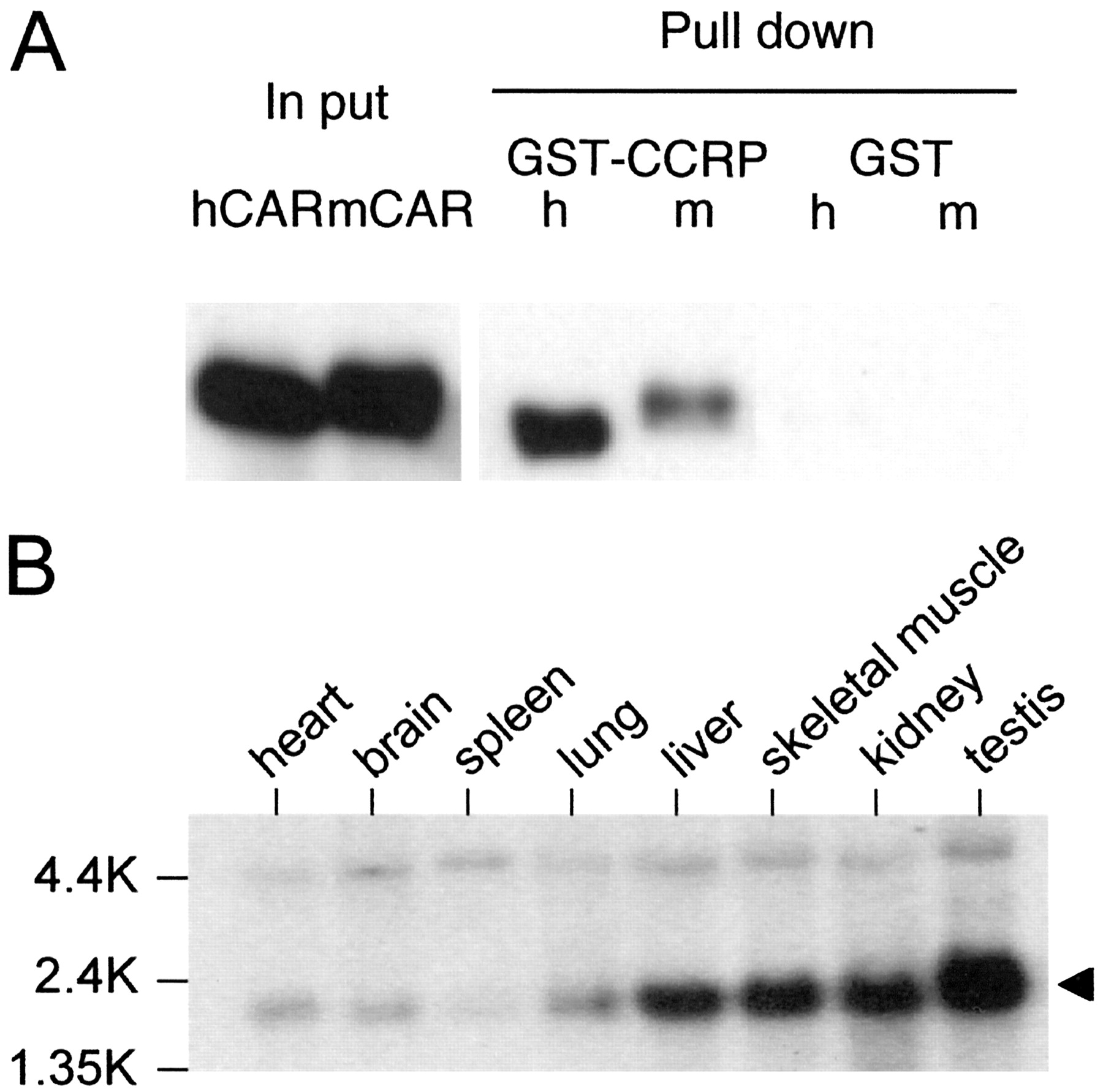
Cloning of CAR-Binding Protein. Using yeast two-hybrid and CAR as bait, mouse liver cDNA library was screened to isolate cDNAs that encode probable binding proteins to the receptor. The deduced amino acid sequence of such a cDNA matched with that of the mouse homolog of bacterial DnaJ (Ohtsuka and Hata, 2000). Subsequently, a GST pull-down assay provided direct evidence that both mouse and human CARs bound to the mouse homolog, now called CCRP (GenBank accession number AY323828; chromosome 11) (Fig. 1A). Northern hybridization analysis showed ubiquitous expression of CCRP mRNA in mouse tissues (Fig. 1B). Liver was one of the tissues (including skeletal muscle, kidney, and testis) in which the mRNA was expressed at high levels. Given this experimental evidence, we investigated further whether CCRP plays a role in the intracellular localization of CAR.

Binding of CCRP to CAR and expression of CCRP mRNA. A, GST pull down assay. In vitro-translated V5 peptide-tagged hCAR (h) or V5 peptide-tagged mCAR (m) was incubated with GST-CCRP fusion protein (GST-CCRP) or GST on a glutathione resin. Proteins extracted from the resin were separated by 10% SDS-PAGE, blotted, and stained with anti-V5 antibody. “In put” indicates the in vitro translated hCAR and mCAR that was run for comparison (5% of protein used for GST pull-down assay). B, Northern hybridization. Multiple-tissue Northern blot membrane (BD Biosciences Clontech) was hybridized with 32P-labeled CCRP cDNA. Arrow indicates the most prominent transcript of approximately 1.8 kilobases.
Cytosolic Accumulation of CAR by CCRP. mCAR was expressed alone or coexpressed with CCRP in HepG2 cells and subjected to Western blot analysis (Fig. 2). When only the receptor was expressed, it was equally distributed in cytosolic and nuclear fractions from the HepG2 cells (Fig. 2A). CCRP was exclusively recovered in the cytosolic fraction. Coexpression of CCRP resulted in a dramatic increase of the receptor in the cytosolic fraction (Fig. 2A). In contrast, no such increase was observed for CAR in the nuclear extracts. The mCAR levels increased as a function of CCRP, peaking when 3 μg of CCRP-expression plasmid was cotransfected (Fig. 2B). Unlike that in the cytosolic fraction, mCAR did not increase its levels in the nuclear extracts even at the highest expression of CCRP, although they varied arbitrarily. The expression of CCRP did not alter levels of hsp90 in the cytosolic fractions. Thus, presumably via its interaction with CAR, CCRP was capable of retaining the receptor in the cytoplasm of HepG2 cells. Despite its cytoplasmic accumulation capability, CCRP did not deplete mCAR from the nuclear fraction (Fig. 2A). Because CAR is a small molecule of ∼40 kDa, the receptor could have diffused passively into the nucleus before a complex with CCRP was formed.

Cytosolic accumulation of CAR by coexpression of CCRP in HepG2 cell. A, HepG2 cells were cotransfected with pcDNA3.1/V5-His-mCAR (3 μg) and pcDNA3.1/V5-His-CCRP (3 μg) for 21 h. Then the medium was replaced and the cells were continued in culture for additional 24 h before harvesting. Cytosols (CYT) and nuclear extracts (NE) were prepared from the HepG2 cells. The cytosolic and nuclear proteins (40 and 8 μg, respectively) were prepared from the HepG2 cells, separated by10% SDS-PAGE, blotted, and stained for CAR with anti-V5, for CCPR with anti-CCRP, and for hsp90 with anti-hsp90 antibodies. Minor bands appeared with the anti-CCRP antibody, which may be a human homolog of CCRP or an immunoreactive endogenous protein. Exposure time was 1 min for mCAR and CCRP and 15 s for hsp90. B, HepG2 cells were cotransfected with pcDNA3.1/V5-His-mCAR (3 μg) and pcDNA3.1/V5-His-CCRP (various amounts up to 6 μg) for 16 h. Then, the medium was replaced and the cells were continued cultured for an additional 24 h before harvesting. Cytosol (CYT) and nuclear fraction (NE) were prepared and the cytosolic and nuclear proteins (40 and 8 μg, respectively) were analyzed by Western blot using anti-V5 antibody, as described under Materials and Methods. Exposure time was 15 min for mCAR and 1 min for CCRP. C, pcDNA3.1/V5-His-mCAR was transfected alone or with pcDNA3.1/V5-His-CCRP into HepG2 cells for 18 h. Then the medium was replaced and the cells were continued in culture for an additional 24 h. The cells were treated with 250 nM TCPOBOP or DMSO for an additional hour.
Treatment with a potent mCAR activator 1, 4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) decreased the cytosolic level of mCAR in the absence of coexpressed CCRP (Fig. 2C). More importantly, the decrease of the cytosolic receptor occurred in accordance with its increase in the nuclear fraction. The HepG2 cells seemed to retain a degree of ability to actively translocate mCAR into the nucleus in response to TCPOBOP. When CCRP was coexpressed, TCPOBOP treatment neither decreased the accumulated cytosolic mCAR nor increased nuclear mCAR. Thus, overexpression of CCRP seemed to inhibit the TCPOBOP-elicited nuclear translocation of mCAR in HepG2 cells.
Formation of a CAR-CCRP-hsp90 Complex. To examine the direct interaction of mCAR with CCRP, anti-CAR immunoprecipitates from the cytosolic fractions were subjected to Western blot analysis. When both mCAR and CCRP were coexpressed, the latter was coprecipitated by anti-CAR antibody (Fig. 3A). Conversely, mCAR was coprecipitated with anti-CCRP antibody (Fig. 3B). These results indicate that CCRP forms a complex with mCAR, which then accumulates in the cytoplasm of HepG2 cells. Nuclear receptors generally form a complex with hsp90, and mCAR has been shown to do this in mouse liver cytoplasm (Yoshinari et al., 2003), thus an immunoprecipitation assay was designed to determine whether mCAR and CCRP bound to hsp90 in the cytosolic fraction of the coexpressed HepG2 cells. Western blot analysis using an anti-V5 antibody could simultaneously detect both mCAR and CCRP, which were tagged with V5 peptide, and allowed estimation of the relative ratio of mCAR and CCRP in a given sample. Such analysis revealed that the anti-hsp90 antibody coprecipitated mCAR and CCRP in an approximately one-to-one molecular ratio, even though the level of CCRP at input far exceeded that of mCAR (Fig. 3C). Figure 3C shows an mCAR signal after precipitation that is 70 times that of the input signal; this is caused by 70 times the amount of input material being used for precipitation than loaded in well “inp”. Thus, the amount of mCAR precipitated by anti-hsp90 antibody indicated complete precipitation from the cytosolic fraction. This was confirmed by increasing the concentration of the anti-hsp90 antibody, which did not increase the amount of mCAR precipitated. However, only approximately 2% of the total CCRP (Fig. 3C, inp) was precipitated by the anti-hsp90 antibody, indicating that only a small fraction of CCRP formed the complex with hsp90 under the experimental conditions. Therefore, in the presence of CCRP, mCAR formed a complex with hsp90 that is composed of one molecule each of mCAR and CCRP.

Detection of the CAR-CCRP-hsp90 complex. Cytosolic fractions were prepared from HepG2 cells after being transiently transfected with pcDNA3.1/V5-His-mCAR and pcDNA3.1/-CCRP. These cytosolic proteins (1 mg) were immunoprecipitated using anti-mCAR (A), anti-CCRP (B), or anti-hsp90 antibodies (C), separated on 4 to 12% gradient gels, blotted, and stained with anti-V5 antibody. inp, total cytosolic fraction that was run for comparison.
Domain-Based Binding. Coexpression analysis was used to determine the domain of mCAR to which CCRP bound (Fig. 4A). The putative ligand binding-domain (LBD) of mCAR was expressed in the cytosolic fraction of HepG2 cells, as was full-length mCAR. Coexpression of CCRP increased the level of LBD in the cytosolic fraction to a level similar to that observed with full-length mCAR. Removing the AF2 domain of mCAR did not affect the CCRP-dependent increase of the receptor. Thus, CCRP seemed to bind to the LBD to accumulate mCAR in the cytosolic fraction, independent of the AF2 domain. The putative DNA binding-domain (DBD) of mCAR was neither effectively expressed nor accumulated in the HepG2 cells, suggesting that the DBD was not involved in the interaction of mCAR with CCRP. These results suggest that the LBD of mCAR is responsible for forming the mCARCCRP-hsp90 complex. To further confirm the results, the LBD was coexpressed with CCRP in HepG2 cells. Cytosolic fraction was prepared from the cells and was incubated with anti-CCRP antibody. Subsequent Western blot analysis of the resulting immunoprecipitates clearly showed that anti-CCRP precipitates the LBD protein, thus confirming the presence of a LBD-CCRP complex (Fig. 4B).

Domain analysis of CAR for its binding to CCRP. A, expression vectors for the full-length and truncated CARs, pcDNA3.1/V5-His-CAR, pcDNA3.1/V5-His-LBD, pcDNA3.1/V5-His-ΔAF2, and pcDNA3.1/V5-His-DBD, were cotransfected with pcDNA3.1/V5-His-CCRP into HepG2 cells. Cytosolic fractions prepared from these cells were analyzed by Western blot using anti-V5 antibody. The approximate molecular masses of the recombinant CAR proteins are: full-length, 50 kDa; ΔAF2, 44 kDa; LBD, 32 kDa; and DBD, 15 kDa. B, HepG2 cells were cotransfected with pcDNA3.1/V5-His-LBD and pcDNA3.1/V5-CCRP. The cytosolic fraction from the cells was incubated with anti-CCRP antibody and the resulting immunoprecipitate was subjected to Western blot analysis using anti-V5 antibody. Total cytosolic fraction (inp) was run for comparison.
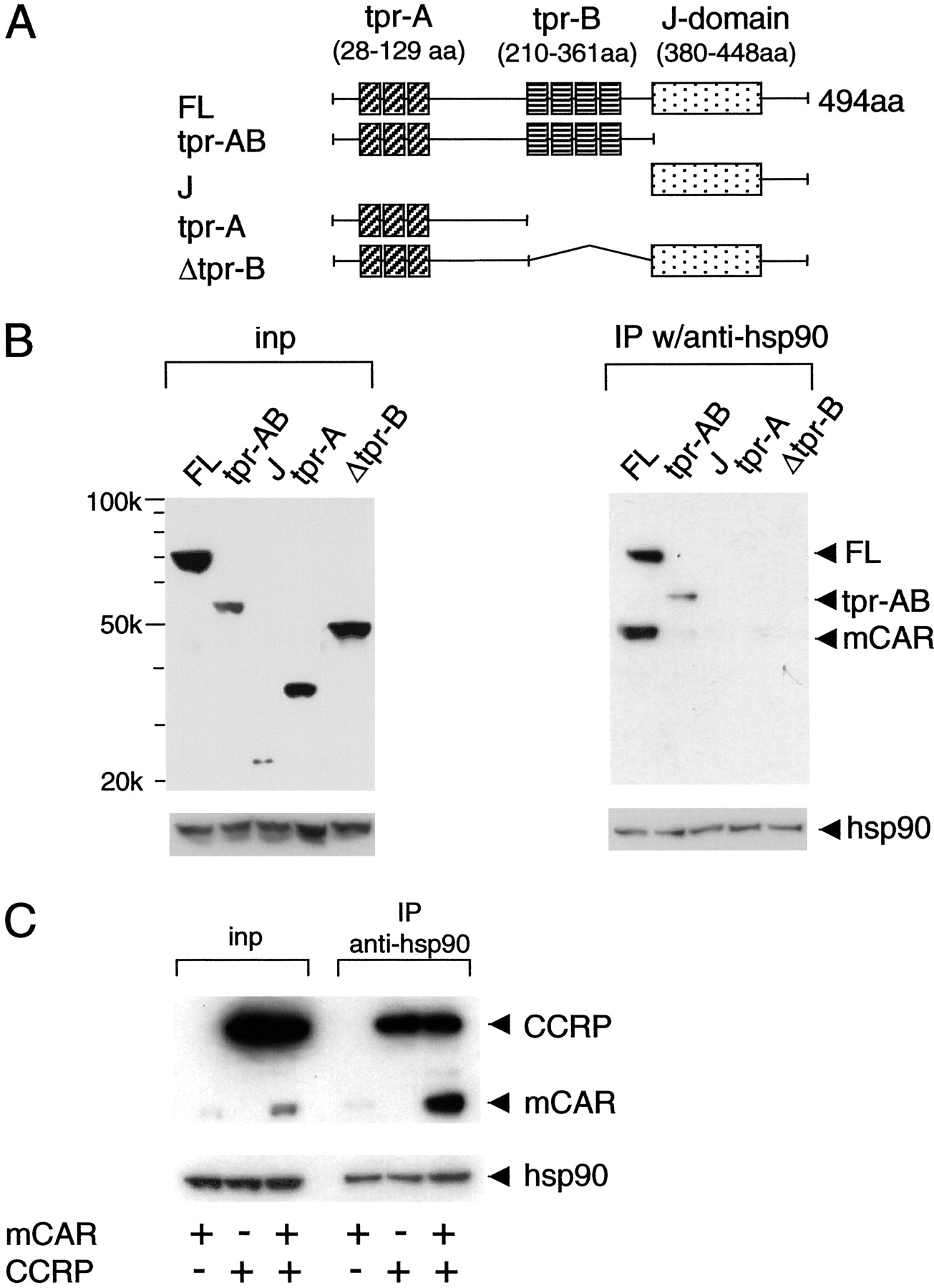
CCRP-Mediated Interaction of mCAR to Hsp90. CCRP consists of three separate domains: two TPR domains in the N-terminal and middle regions of the molecule (termed tpr-A and tpr-B, respectively) and the C-terminal J domain (Fig. 5A). The N-terminal TPR domain contains three TPR motifs and the middle domain contains four such motifs. Four CCRP deletion constructs were used to investigate which domain of CCRP was responsible for forming the complex with CAR and hsp90. These deletion mutants were well expressed with respect to the full-length CCRP, except that the expression level of the J domain was low (the left of Fig. 5B). As expected, anti-hsp90 antibody coprecipitated mCAR with full-length CCRP. Coprecipitation was insignificant when any of the deletion mutants was used (the right of Fig. 5B), suggesting that all three domains may be required for CCRP recruitment of mCAR into the hsp90 complex. The hsp90 antibody precipitated tpr-AB that contained both TPR domains. Thus, in the presence of both TPR domains, CCRP was capable of binding to hsp90.

Domain analysis of CCRP for its ability to form the CAR/hsp90 complex. A, schematic representation of structures of CCRP and its truncated mutants. The estimated molecular masses of recombinant CCRPs are: full-length,65 kDa; trp-AB, 55 kDa; J, (25 kDa; trp-A, 37 kDa; and Δtpr-B, 50 kDa. B, pcDNA3.1/V5-His-CCRP, pcDNA3.1/V5-His-tpr-AB, pcDNA3.1/V5-His-J, pcDNA3.1/V5-His-tpr-A, and pcDNA3.1/V5-His-(Δtpr-B) each was cotransfected with pcDNA3.1/V5-His-mCAR into HepG2 cells. The cytosolic fractions were prepared and subjected to Western blot analysis using anti-V5 antibody (inp). The cytosolic proteins were precipitated from the cytosolic fractions with anti-hsp90 antibody and subjected to Western blot analysis using anti-V5 antibody as described under Materials and Methods. C, cytosolic fractions were prepared from HepG2 cells that were transfected with pcDNA3.1/V5-His-CCRP or pcDNA3.1/V5-His-CAR alone or were cotransfected with both plasmids. Total cytosolic fractions (inp, 10 μg of protein) and immunoprecipitates (75% of immunoprecipitated sample from 1 mg of cytosolic protein), by the hsp90 antibody from the fractions were subjected to Western blot analysis using anti-V5 antibody.
Given the results indicating that tpr-AB could bind to hsp90 but could not recruit mCAR to the hsp90 complex, we further examined the hypothesis that mCAR interacted with hsp90 through its binding to CCRP. When CCRP alone was expressed, it precipitated with hsp90, indicating the direct interaction between CCRP and hsp90 (Fig. 5C). When CCRP was coexpressed with mCAR, anti-hsp90 antibody coprecipitated CCRP and mCAR, resulting in marked enrichment of mCAR. On the other hand, when mCAR was expressed alone, anti-hsp90 antibody did not enrich mCAR, although a subtle amount of mCAR was present in the precipitate (Fig. 5C). These results indicate that mCAR bound directly to CCRP but not to hsp90 and that CCRP was capable of binding to both mCAR and hsp90.
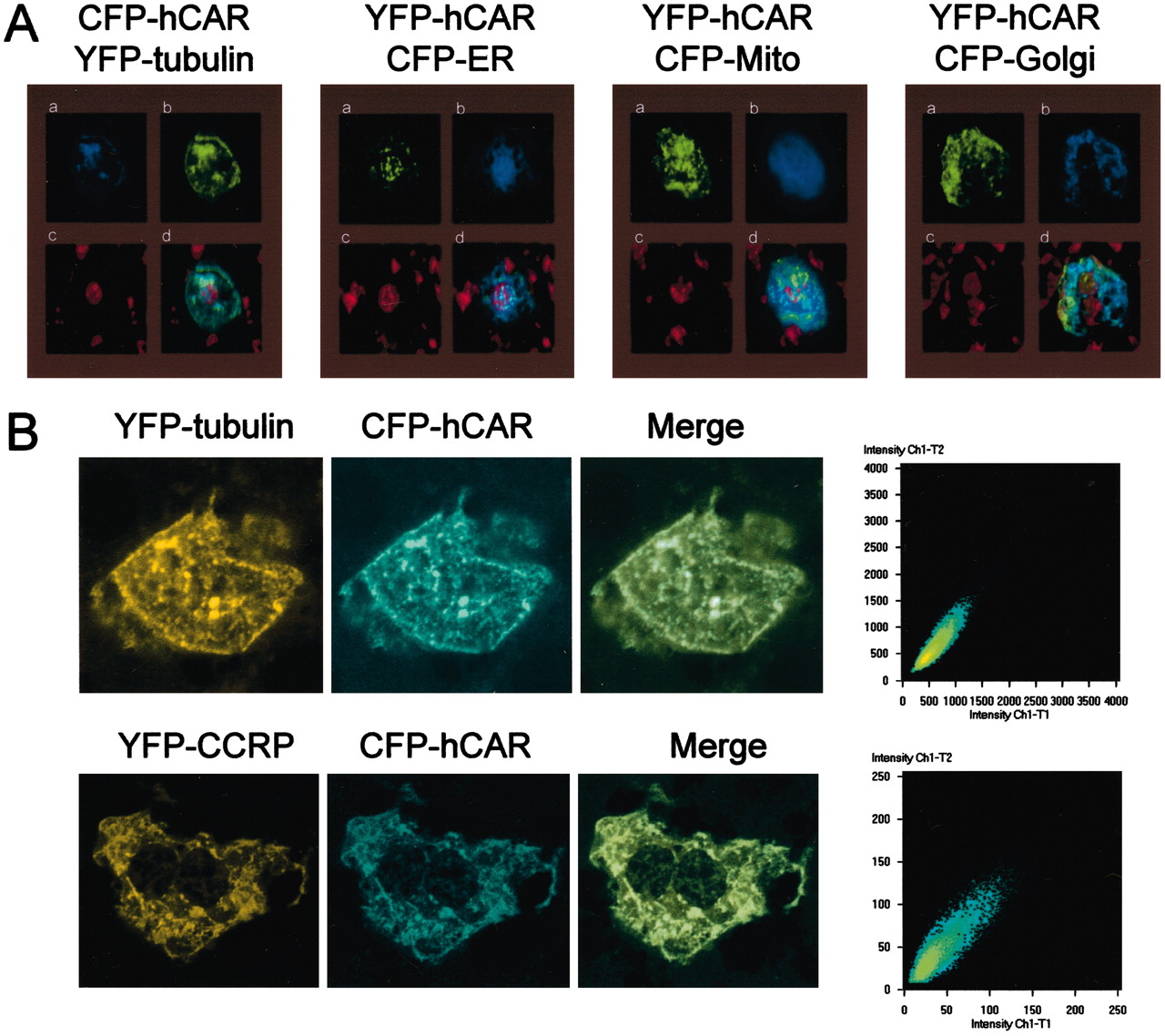
Colocalization of CAR with CCRP in Liver in Vivo. Fluorescent protein-tagged hCAR can be visualized in the cytoplasm of mouse liver after tail-vein injection of its expression plasmid. Tagging with two different fluorescent proteins CFP and YFP, we investigated intracellular localization of CAR and CCRP in mouse liver cells in vivo. First, colocalization of CAR with intracellular markers, such as tubulin, endoplasmic reticulum, mitochondria, and Golgi apparatus, was examined using conventional fluorescent microscope (Fig. 6A). CAR was colocalized with only the tubulin maker. Colocalization of CAR with the tubulin marker was further confirmed using confocal microscopy (Fig. 6B). Next, CFP-hCAR and YFP-CCRP were coexpressed to examine their localization in liver cells in vivo. Confocal analysis of their expression revealed that the receptor and CCRP were colocalized (Fig. 6B). Immuno-precipitation was attempted to show the presence of an endogenous CAR:CCRP complex in the mouse liver cytosols. However, the extremely low cytosolic content of the endogenous CAR (less than one hundredth of GR) proved challenging, affecting the success of the experiment. This and additional experimental difficulties prevented the detection of the endogenous complex. Nevertheless, the present results obtained from the coexpression experiments suggest that CAR may form a complex with CCRP in liver in vivo and that the CAR-CCRP complex is associated with microtubules.

In vivo coexpression of fluorescent protein-tagged CAR and CCRP with intracellular makers. A, fluorescent protein-tagged hCAR was coexpressed with tubulin, endoplasmic reticulum (ER), mitochondria (Mito), or Golgi apparatus (Golgi) marker and was visualized under conventional fluorescent microscope. a, fluorescent protein-tagged hCAR (CFP-hCAR or YFP-hCAR). b, YFP-tubulin, CFP-ER, CFP-Mito, or CFP-Golgi, from left to right. c, stained nuclei. The three images (a, b, and c) are merged in d. B, the CFP-tagged hCAR was coexpressed with YFP-tagged tubulin or CCRP. Left, YFP-tubulin (top) and YFP-CCRP (bottom). Center left, CFP-hCAR. Center right, merge of left and center left images. Their expressions and colocalizations were analyzed by confocal microscope and shown at far right according to manual provided by Zeiss. The brightness level of the YFP images pixels was taken as the x coordinate (Ch1-T1) and that of the CFP image pixel as the y coordinate (Ch1-T2) of the scatter diagram.
Discussion
The TPR protein named CCRP is now identified as a factor that associates the nuclear receptor CAR in the cytoplasm of HepG2 cells and possibly of liver cells. Unlike FKBP and XAP-2, CCRP is not an immunophilin or an immunophilinlike protein. Instead, it was first characterized as the mouse homolog of hsp40/DnaJ (Ohtsuka and Hata, 2000) and is characterized by two structural signatures, a TPR motif and a J-domain. The TPR motif, a 34-residue peptide forming a pair of anti-parallel α helices, mediates an array of protein-protein bindings via TPR-TPR as well as TPR-non-TPR interactions (Lamb et al., 1995). Therefore, two separate regions, tpr-A and tpr-B, which contain seven TPR motifs can provide CCRP with seven different protein-protein binding sites. CCRP formed a complex with hsp90 in the absence of CAR, for which both TPR regions were required. Of the seven motifs, however, only a few may be direct binding sites to hsp90. Given the fact that CAR contains no TPR motif, the receptor does not seem to bind to CCRP through a TPR-TPR interaction. CAR formed a complex with hsp90 only in the presence of CCRP. Apparently, CCRP acts as a bifunctional linker protein that recruits CAR to the CCRP-hsp90 complex. This mode of CAR-CCRP-hsp90 complex formation differs from those of the GR-FKBP-hsp90 or AhR-XAP-2-hsp90 complexes. GR and AhR directly bind to hsp90 (Perdew, 1992; Dittmar et al., 1997); moreover, binding of GR to hsp90 recruits FKBP into the hsp90 complex (Dittmar et al., 1997). Nevertheless, CCRP, acting as a mediator, is capable of generating a CAR-hsp90 complex, thus resulting in the stable accumulation of CAR in the cytoplasm. Our preliminary study revealed that mCAR was accumulated in HepG2 cells treated with proteasome inhibitor MG132 (data not shown). Whether the role of the CAR-CCRP-hsp90 complex is to protect CAR from proteolysis and/or is in the process of the nuclear translocation remains investigated.
CCRP can also be considered as a member of the J domain family. In general, J domain directly binds the hsp70 or hsp40 class of chaperones and binding recruits regulatory factors to a given J domain family protein (Kelley, 1998). For example, the inhibitory protein P58IPK regulates the interferon-induced protein kinase. P58IPK consists of TPR motifs and a J domain. In response to signals, the J domain binds to P52rIPK, resulting in the recruitment of interferon-induced protein kinase to the TPR motifs of P58IPK (Gale et al., 1998). P52rIPK is also identified as hsp40 (Melville et al., 1997). Our present study shows that when both tpr-A and tpr-B are present, CCRP binds to hsp90 but fails to recruit CAR to its hsp90 complex. Only when all the TPR regions and the Jdomain are present is CCRP able to recruit CAR to the hsp90 complex. Because the J domain is primarily a recognition site for heat-shock proteins, it is unlikely that the domain binds directly to CAR, although this possibility can not be totally eliminated at the present time. Requirement of the J domain in the CCRP-dependent recruitment of CAR implies that CAR recruitment is a regulated sequential process involving multiple proteins and their interaction with CCRP. It is known that hsp70 and hsp40 are also components of GR- or progesterone receptor-hsp90 complexes (Pratt, 1993; Hernandez et al., 2002). In another example, simian virus 40 small T antigen binds to hsp70 via its J domain, leading to subsequent binding of protein phosphatase 2A (PP2A) to a zinc-finger-like domain residing adjunct to the J domain. This potential ability of TPR motif to recruit PP2A is intriguing, because the protein phosphatase inhibitor okadaic acid suppresses PB-elicited nuclear translocation of CAR in mouse primary hepatocytes (Kawamoto et al., 1999); our recent work suggests that PP2A is recruited to the cytoplasmic CAR-hsp90 complex in liver in vivo after PB induction (Yoshinari et al., 2003). We do not know whether hsp70 and/or hsp40 are included in the CAR-CCRP-hsp90 complex that is formed in HepG2 cells. Future investigations may resolve these aspects of the CAR complex, helping us to understand the cytoplasmic retention and nuclear translocation mechanisms.
Recently, XAP-2 was implicated as a protein that mediates association of the AhR-hsp90 complex with cytoskeletons (Berg and Pongratz, 2002). The GR-hsp90 complex is colocalized with microtubules; apparently, the replacement of FKBP52 with FKBP51 recruits dynein to the GR-hsp90 complex, initiating nuclear translocation (Davies et al., 2002). Our in vivo expression study now shows colocalization of CAR and CCRP with tubulin in liver, thus indicating association of the CAR-hsp90 complex with microtubules. Whether CCRP plays a role in the association of CAR with microtubules remains an unanswered question. The lack of an in vitro system that properly retains CAR in the cytoplasm has hindered us from rapid progress in understanding the molecular mechanisms of CAR regulation. With CCRP, for the first time it has been possible to create the experimental conditions in which CAR can be stably retained in the cytoplasm. However, the present study also reveals that overexpression of CCRP seems to repress the TCPOBOP-elicited receptor translocation in HepG2 cells. An additional factor may be required to confer the translocation capability to the cells. Once the role of CCRP in the formation and cytoplasmic retention of the CAR-hsp90 complex become evident, we may become closer to elucidating the regulatory mechanisms of CAR activation. Moreover, an in vitro cell system mimicking the in vivo active nuclear translocation of CAR may eventually be constructed.
Footnotes
-
ABBREVIATIONS: CAR, constitutive active (or androstane) receptor; PB, phenobarbital; GR, glucocorticoid receptor; CCRP, cytoplasmic CAR retention protein; AhR, aryl hydrocarbon receptor; hsp90, 90-kDa heat shock protein; FKBP, FK-binding protein; XAP2, X-associated protein 2; TPR, tetratricopeptide repeat; YFP, yellow fluorescent protein; CFP, cyan fluorescent protein; GST, glutathione S-transferase; TCPOBOP, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene; LBD, ligand binding domain; DBD, DNA binding domain; PAGE, polyacrylamide gel electrophoresis.
- Received May 6, 2003.
- Accepted August 4, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}