


Abstract
Drug efficacy is typically considered an intrinsic property of a ligand/receptor couple. However, recent observations suggest that efficacy may also be influenced by the signaling effectors engaged by a unique receptor. To directly and systematically test this possibility, we assessed the ability of a panel of β-adrenergic ligands to modulate the activity of two effector systems, the adenylyl cyclase (AC) and the mitogen-activated protein kinase (MAPK), via β1 and β2 adrenergic receptors. Although some compounds displayed similar efficacies toward the two pathways, others showed complex efficacy profiles. For example, compounds that are inverse agonists for the AC activity were found to be either agonists, neutral antagonists, or inverse agonists for the MAPK pathway. Likewise, agonists for the AC were either agonists or neutral antagonists for MAPK. Given this complexity, we propose a Cartesian representation of the efficacies that takes into account the activities of the different effectors that can be engaged by a given receptor. In addition, compounds considered as nonselective for β1 and β2 adrenergic receptors, based on their binding affinities, showed distinct relative efficacy profiles toward AC and MAPK, adding a new dimension to the concept of ligand selectivity. Taken together, the results suggest that binding of different ligands promote distinct conformational changes leading to specific signaling outcomes. Our data therefore clearly illustrate that efficacy is a pluridimensional parameter that is not an intrinsic characteristic of a ligand/receptor couple. This should have important implications for the future design of screening assays used in drug discovery campaigns.
G protein-coupled receptors (GPCRs) represent the largest class of proteins involved in signal transduction across biological membranes. As such, they are among the main molecular targets considered for the development of therapeutic agents. Drugs acting on GPCR have traditionally been classified into two main categories: agonists and antagonists, which promote or block receptor activation, respectively. In the last 10 years, however, the recognition that GPCR can exhibit constitutive activity led to the discovery of a third class of compounds that can decrease such spontaneous activity and are known as inverse agonists. In the framework of an allosteric model whereby receptors are in equilibrium between inactive (R) and active (R*) conformations, agonists and inverse agonists are believed to stabilize R* and R, respectively. Neutral competitive antagonists, for their part, presumably compete for the binding of agonists or inverse agonists but do not affect the equilibrium and thus have no intrinsic activity (for review, see Bond, 1997; Strange, 2002; Milligan, 2003).
It is noteworthy that the extended and cubic ternary complex models, which were developed to formalize ligand behaviors, included terms that qualified the affinity of the activated receptor for the G protein, opening the possibility that various ligands may stabilize different active conformations resulting in distinct signaling properties (for review, see Kenakin 2004). Consistent with this theoretical possibility, some studies reported that the order of potency/efficacy of compounds acting through a unique receptor can be different depending of the effector system considered (Spengler et al., 1993; Kenakin, 1995; Berg et al., 1998; Hall et al., 1999, Kurrasch-Orbaugh et al., 2003; Gay et al., 2004; Moniri et al., 2004; Krueger et al., 2005; McLaughlin et al., 2005). This phenomenon, often referred to as “ligand-directed stimulus trafficking”, “functional selectivity”, or “biased agonism”, has been taken as evidence that more than one active receptor conformation exists (Kenakin, 2002; Urban et al., 2006). Particularly striking in this respect are recent studies reporting that ligands can have opposite efficacies toward two different signaling pathways. For example, ICI118,551 and propranolol, which act as inverse agonists on the β2-adrenergic receptor (β2AR) toward the adenylyl cyclase (AC) signaling pathway, were shown to be partial agonists when tested on the extracellular signal-regulated kinase (ERK) activity (Azzi et al., 2003; Baker et al., 2003). Similar dual efficacies for distinct signaling pathways were also reported for ligands acting on the H3-histamine receptor (Gbahou et al., 2003), the δ-opioid receptor (Audet et al., 2005), and the serotonin 5-HT2C receptor (Werry et al., 2005).
Taken together, these observations suggest that efficacy is a more complex parameter than was originally anticipated and that the effector systems may need to be included in its description. When considering two distinct signaling pathways modulated by a single receptor, multiple efficacy combinations are theoretically possible. Compounds could be agonist for the two pathways, inverse agonist for the two pathways, or have opposite efficacies on each of the pathways. The present study, therefore, was initiated to test whether these different theoretical efficacy profiles can exist. For this purpose, the ability of various β-adrenergic ligands to modulate the activity of the adenylyl cyclase and ERK1/2 was assessed in cells expressing either the human β1 adrenergic receptor (β1AR) or β2AR. In the case of each receptor subtype, ligands that activate both, inhibit both, or have opposite effects on each of the two pathways were indeed identified. The wide diversity of efficacy and potency profiles revealed by our study clearly illustrates the notion of signaling pluridimensionality that complicates the classification of ligands according to unique efficacy terms. The systematic comparison of the efficacy profiles for the two closely related receptor subtypes also demonstrated that drug efficacy can only be considered in the context of the diverse signaling pathways that can be engaged by a specific receptor subtype.
In addition to shedding light on the concept of drug efficacy, these results could have important clinical implications because the distinct efficacy profiles of β-blockers, which are widely used in the treatment of hypertension and heart failure, could underlie differences in therapeutic and side effect patterns. Additional studies linking defined physiological actions of various compounds to their efficacies toward specific effector systems should allow to explore the potential therapeutic impact of pluridimensional signaling efficacy.
Materials and Methods
Reagents. (-)-Isoproterenol, labetalol, dl-propranolol, S(-)-atenolol, and (±)-metoprolol were purchased from Sigma-Aldrich (St Louis, MO), and (±)-bisoprolol was from Tocris Cookson Inc. (Ellisville, MO). Carvedilol and bucindolol were a generous gift from GlaxoSmithKline (Research Triangle Park, NC) and Dr. Michael Bristow (University of Colorado Health Sciences Center, Denver, CO), respectively. Mouse anti-phosphorylated ERK1/2 and rabbit anti-ERK1/2 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase (HRP)-anti-mouse and HRP-anti-rabbit polyclonal antibodies were from GE Healthcare (Baie d'Urfé, QC, Canada). All other reagents were of analytical grade and obtained from various suppliers.
Stable Cell Lines and Cell Culture. Stable cell lines expressing human-β1AR and human-β2AR were generated by transfection of pcDNA3.1-HA-β1AR or pcDNA3.1-HA-β2AR plasmids (Lavoie et al., 2002) into human embryonic kidney (HEK) 293S cells (Reeves et al., 1996) using the calcium-phosphate precipitation method. Isolates that stably incorporated the plasmids were selected on the basis of their resistance to G418 by treating cells with G418 (400 μg/ml). Receptor level expression was deduced from binding experiments carried out on whole cells using 125I-cyanopindolol as radioligand. The two cell lines used expressed 7 to 8 pmol and 4 to 5 pmol receptor/mg of protein for the β1AR and β2AR, respectively.
Cells were routinely grown in Dulbecco's modified Eagle's medium supplemented with 5% fetal bovine serum, 100 U/ml penicillin and streptomycin, 2 mM l-glutamine, and 400 μg/ml G418 in a 37°C humidified 5% CO2 atmosphere.
Quantification of cAMP Accumulation. Cells at 80% confluence were serum-starved for 16 h. The day of the experiment, cells were resuspended in phosphate-buffered saline (PBS)/0.1% glucose/1 mM 3-isobutyl-1-methylxanthine and then treated for 30 min at 37°C with the indicated drugs. Compounds behaving as inverse agonists were tested in the presence of 0.3 μM forskolin to increase the window of inhibition. After drug treatment, cells were immediately lysed, and cAMP levels were measured using the CatchPoint cAMP Kit (Molecular Devices, Sunnyvale, CA) according to manufacturer's recommendations. In brief, cell lysates were incubated in 384-well plates coated with anti-cAMP antibodies in the presence of known amounts of HRP-cAMP. cAMP from cell lysates was allowed to compete with the HRP-cAMP for 2 h, and the remaining peroxidase activity was measured after 3 washes. The cAMP generated under the different conditions was interpolated from a cAMP standard curve generated in parallel for each experiment. Triplicates were used for each condition, and all experiments were repeated at least three times. For the determination of the EC50, data are expressed as a percentage of the maximal response reached for each compound. For the determination of the relative activities, maximal agonist responses (Emax) are expressed as the percentage of maximal (10 μM) isoproterenol-promoted stimulation. For the compounds behaving as inverse agonists, data were expressed in percentage of forskolin (0.3 μM) inhibition.
Detection of Phosphorylated ERK1/2. Cells expressing β1AR or β2AR were seeded in poly-d-lysine-coated six-well plates. The day after, cells were washed once with PBS and incubated with serumfree media for 16 hours. Cells at 80% confluence were then stimulated for the indicated times at 37°C. The media were then rapidly removed and cells were washed with ice-cold PBS before being lysed using 100 μl per well of Laemmli sample buffer (62.5 mM Tris-HCl, 2% SDS, 10% glycerol, 50 mM dithiothreitol, and 0.1% bromphenol blue, pH 6.8). Whole-cell lysates were sonicated, resolved by SDS-PAGE, and transferred onto nitrocellulose. The blots were blocked at room temperature for 1 h with TBS-T buffer [50 mM Tris, pH 7.4, 150 mM NaCl, and 0.1% (v/v) Tween 20], 5% fat-free milk. Phospho-ERK1/2 was detected using mouse polyclonal anti-phospho p42/p44 ERK-specific antibody (1:3000, overnight at 4°C in TBS-T, 5% fatfree milk). The immunoreactivity was revealed using a secondary HRP-conjugated anti-mouse antibody (1:10,000, 1 h at room temperature in TBS-T, 5% fat-free milk) and the peroxidase activity detected by chemiluminescence (PerkinElmer Life and Analytical Sciences, Boston, MA). Blots were stripped and re-probed for total ERK1/2 with rabbit polyclonal anti-ERK1/2 antibody (1:25,000, 1 h at room temperature in TBS-T, 5% fat-free milk) followed by HRP-anti-rabbit antibody (1:20,000, 1 h at room temperature in TBS-T, 5% fat-free milk). Films were scanned, and band intensities were quantified using Quantity One (Bio-Rad Laboratories, Hercules, CA) software. ERK1/2 phosphorylation was normalized according to the loading of proteins by expressing the data as a ratio of P-ERK1/2 over total ERK1/2.
Statistical Analysis. Statistical analysis and curve fitting were done using Prism 2.01 (GraphPad Software, San Diego, CA). Statistical significance of the differences was assessed using one-way analysis of variance (ANOVA) and post hoc Bonferonni or Dunnett's test.
Results
The efficacy and potency profiles of eight β-adrenergic ligands were tested on both AC and ERK signaling pathways in cells expressing either the human β1- or β2-adrenergic receptors (βARs). The compounds were selected based on their prevalent use in pharmacological studies and clinical settings. Their reported affinities for both β1 and β2AR are listed in Table 1. Five of the compounds bind with similar affinities to both receptors, whereas three display a strong selectivity toward β1AR.
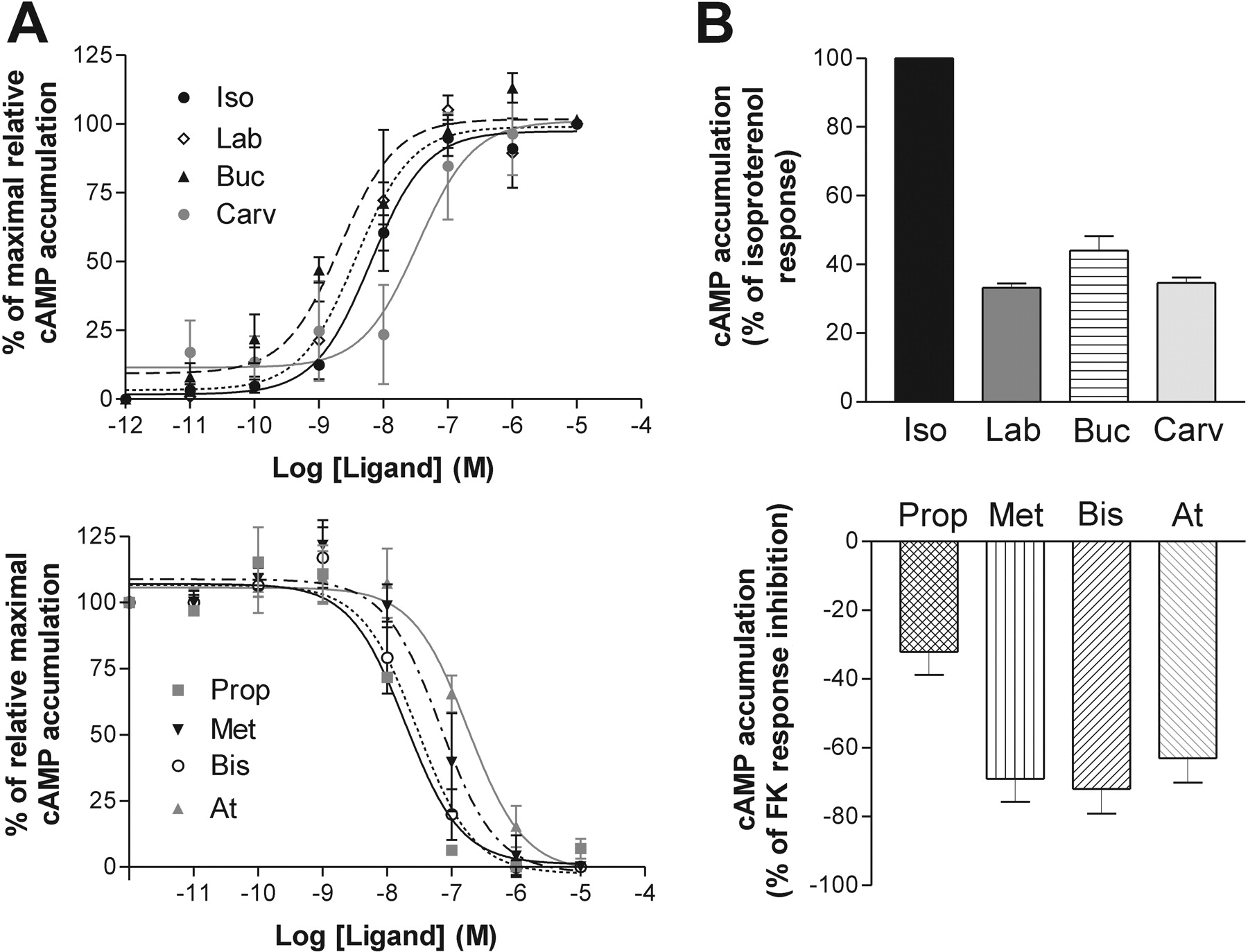
Ligand Profiles on the β1 Adrenergic Receptor. The eight selected compounds were first tested for their ability to modulate cAMP production in HEK293S cells stably expressing the human-β1AR. In a first round, their activity was assayed to establish whether they behave as agonists, neutral antagonists, or inverse agonists (data not shown). In the case of antagonists and inverse agonists, their potency and efficacy were tested in the presence of 0.3 μM forskolin to enhance the response window. As shown in Fig. 1, isoproterenol, labetalol, bucindolol, and carvedilol behaved as agonists, whereas propranolol, metoprolol, bisoprolol, and atenolol were inverse agonists for the AC pathway. Using maximal concentration (at least 10× the EC50 for each compound determined from dose response experiments; Fig. 1A and Table 2, left column), the efficacy of each compound was determined. When considering the agonists, isoproterenol was the most efficacious compound, whereas labetalol, bucindolol, and carvedilol had equivalent partial efficacy, corresponding to 33 to 44% of the maximal isoproterenol-stimulated response (Fig. 1B, top, and Table 3, left column). For inverse agonists, metoprolol, bisoprolol, and atenolol had similar high inverse efficacies. Propranolol behaved as a partial inverse agonist, leading to an inhibition of only 32% of the forskolin-stimulated AC compared with the 63 to 72% inhibition observed for the other three compounds (Fig. 1B, bottom).
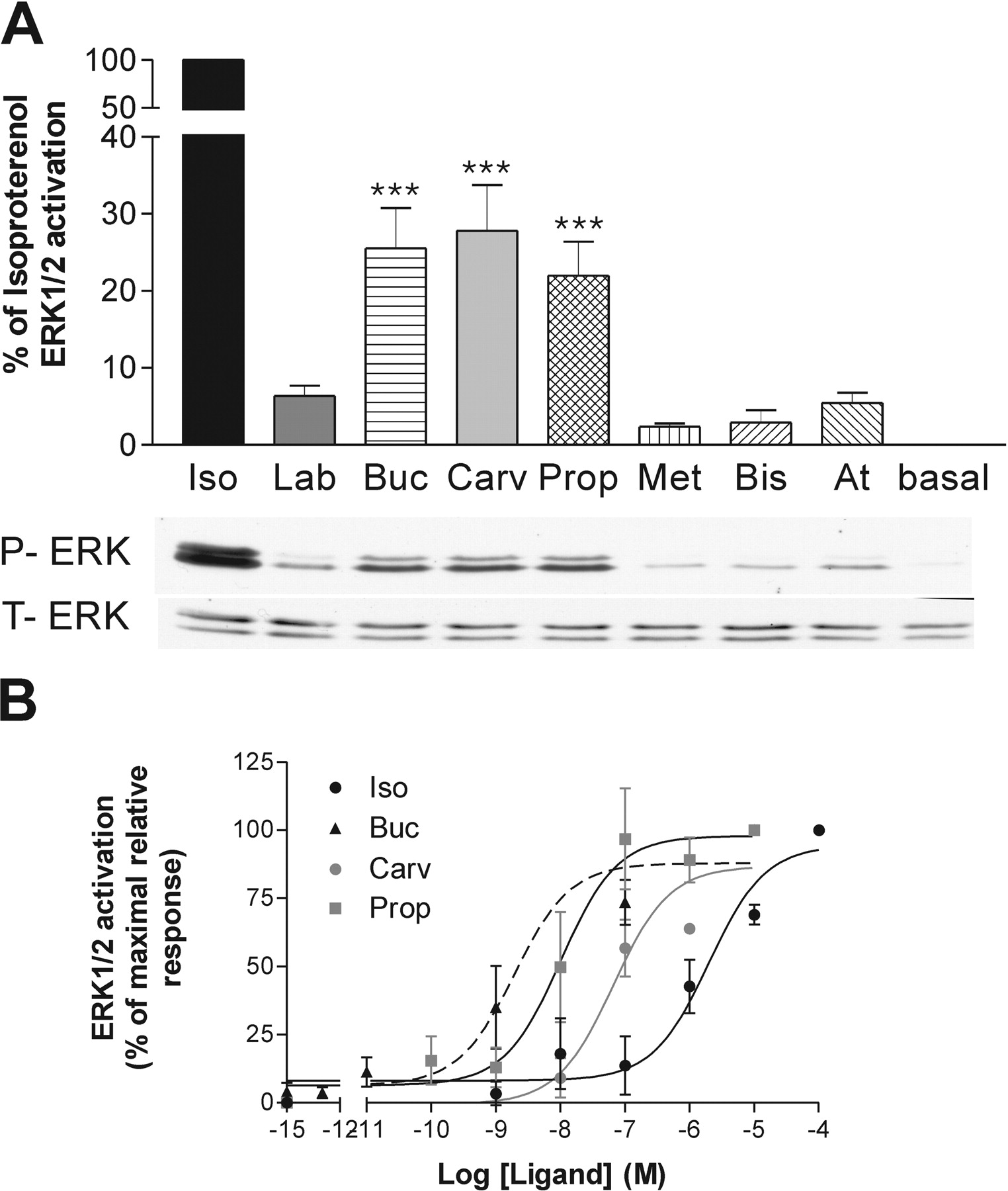
The same compounds were then assessed for their ability to modulate the ERK1/2 activity in the β1AR-expressing cells. Because ERK1/2 activation is known to be fast and transient, we first performed time course experiments to determine the time of maximal activation for each compound. These times of activation (between 2 and 4 min depending on the compound) were then selected to assess relative efficacies. As shown in Fig. 2, the efficacy pattern found in the ERK1/2 assay was significantly different from that observed for AC activation. When considering the compounds that were agonists for the AC pathway, isoproterenol, bucindolol, and carvedilol also acted as agonists and partial agonists for ERK1/2; bucindolol and carvedilol showed 25 to 28% of isoproterenol's efficacy (Fig. 2A and Table 3, right column). Labetalol, however, which was as efficacious as bucindolol and carvedilol for the AC, did not significantly stimulate ERK1/2 phosphorylation (the small increase seen in Fig. 2A did not reach statistical significance). For the inverse agonists in the AC assay, three compounds (metoprolol, bisoprolol, and atenolol) acted as neutral antagonists (the small increases did not reach statistical significance) on the ERK1/2 pathway. Propranolol showed an opposite efficacy, stimulating ERK1/2 phosphorylation as efficiently as bucindolol and carvedilol and reaching 22% of the isoproterenol efficacy (Fig. 2A and Table 3, right column).

Profile of the different ligands on the β1AR adenylate cyclase pathway. cAMP accumulation experiments were performed in HEK293S cells stably expressing β1AR. A, EC50 of compounds were obtained using increasing concentrations of ligands in the presence (bottom) or absence (top) of 0.3 μM forskolin. B, Emax of compounds were tested using maximal concentrations of ligands, determined from EC50 curves, with (bottom) or without (top) 0.3 μM forskolin. Data represent the mean ± S.E.M. of at least three experiments performed in triplicates. Iso, isoproterenol; Lab, labetalol; Buc, bucindolol; Carv, carvedilol; Prop, propranolol; Met, metoprolol; Bis, bisoprolol; At, atenolol.
When considering the potency of the compounds in the two signaling pathways (AC and ERK1/2), similar EC50 values were found for all compounds except isoproterenol (Table 2). Even propranolol, which acted as an inverse agonist on AC but as antagonist on ERK1/2, did so with similar potency. In contrast, isoproterenol activated ERK1/2 with a potency markedly lower (450×) than that observed for its stimulation of AC (Fig. 2B, Table 2). The possible meanings of this difference are considered under Discussion.
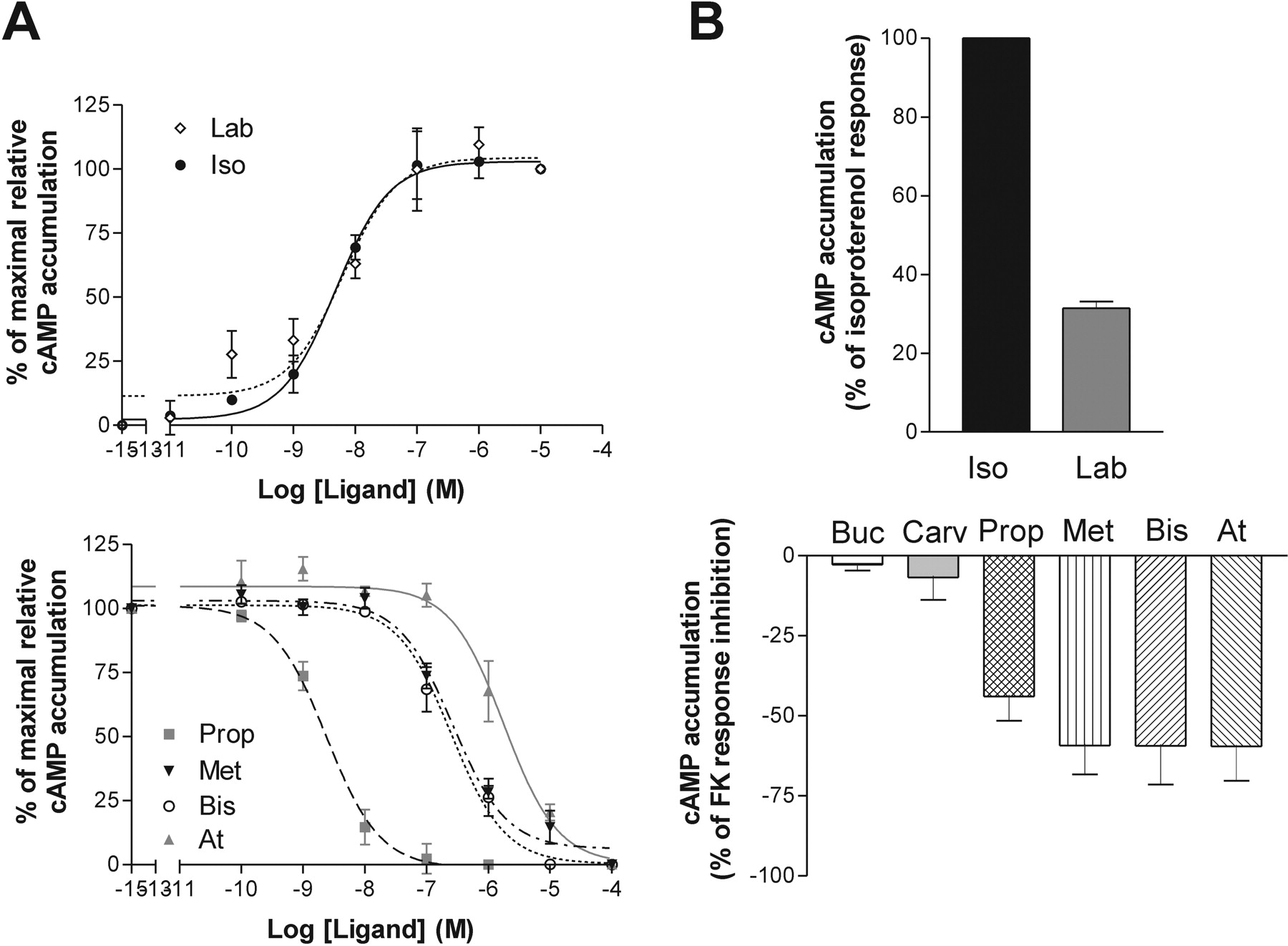
Ligand Profiles on the β2 Adrenergic Receptor. The β-adrenergic ligands were then assessed for their ability to modulate AC and ERK1/2 activity in HEK293S cells expressing the human β2AR. As shown in Fig. 3, only isoproterenol and labetalol were able to increase cAMP levels. Isoproterenol behaved as an agonist, whereas labetalol acted as partial agonist for this pathway, possessing an efficacy of ∼25% of isoproterenol. Propranolol, metoprolol, bisoprolol, and atenolol all acted as inverse agonists on the cyclase response (Fig. 3 A and B, bottom). Their efficacy to inhibit forskolin-stimulated AC was similar, ranging from 44 to 59% (Table 5, left column). Bucindolol and carvedilol showed no significant efficacy toward the AC pathway up to a concentration of 10-5 M.
Significant differences in the potency of the compounds to modulate cAMP production were observed in cells expressing the β2AR (Table 4, left column). Indeed, whereas the nonselective β-adrenergic ligands showed similar high potencies to either stimulate (isoproterenol and labetalol) or inhibit (propranolol) AC activity, the β1AR selective ligands (metoprolol, bisoprolol, and atenolol) showed poor potency to inhibit AC (Fig. 3A and Table 4, left column).
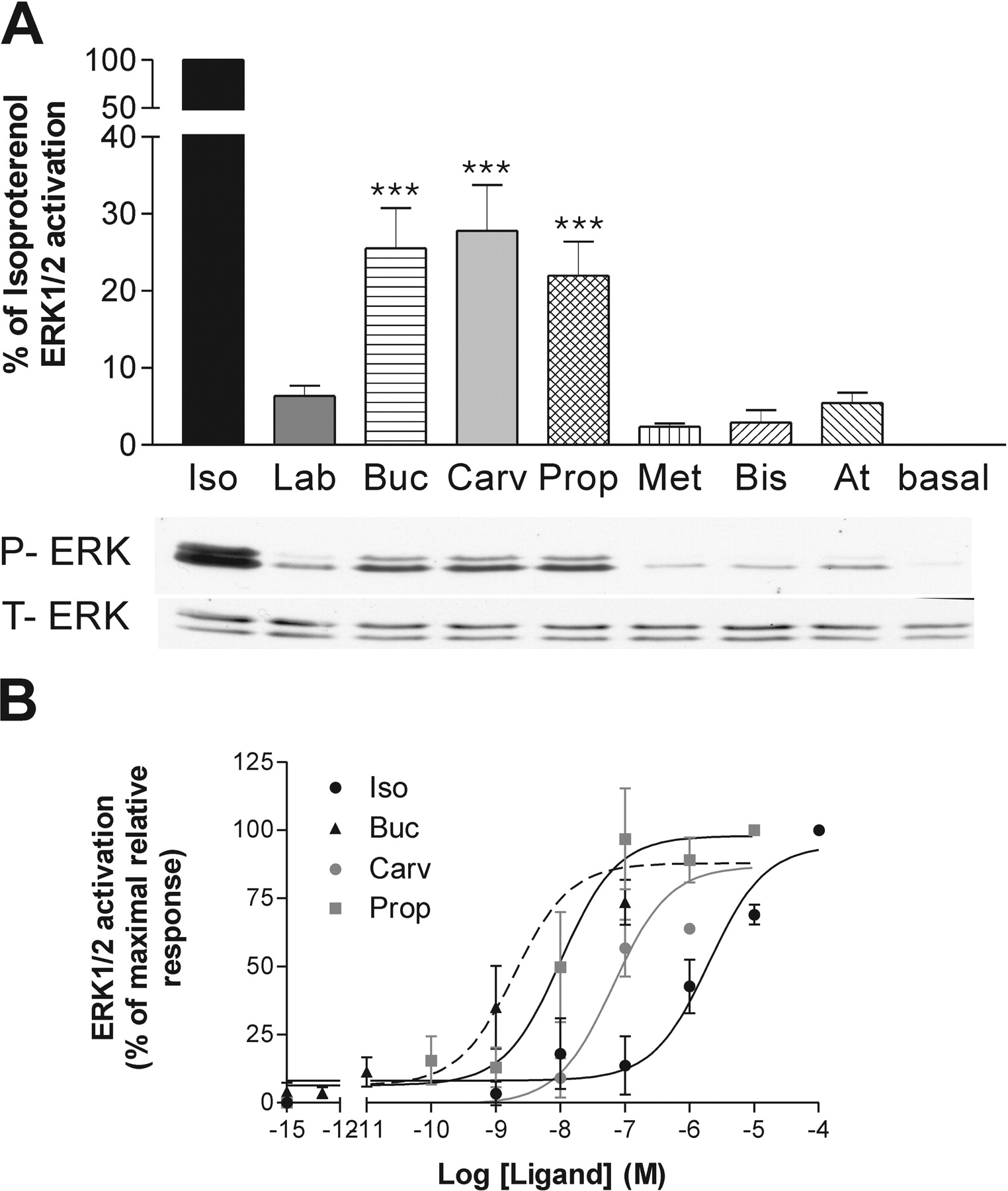
Profile of the different ligands on the β1AR MAPK ERK1/2 pathway. ERK1/2 phosphorylation was assessed in HEK293S cells stably expressing β1AR. Compounds were tested at their appropriate maximal stimulation time (4 min for isoproterenol and 2 min for other compounds). A, Emax of compounds were obtained using maximal ligand concentration (10-4 M for isoproterenol and 10-5 M for other ligands). B, EC50 of compounds were obtained using increasing concentrations of ligands. Data represent the mean ± S.E.M. of at least four experiments. Statistical significance was determined by two-way ANOVA, followed by a Dunnett test with basal as control column. ***, p < 0.001.
After determination of the time of maximal activation for each compound (1 or 2 min, depending of the compounds), the relative efficacies and potencies of the compounds to modulate ERK1/2 activity were assessed in the β2AR-expressing cells. As shown in Fig. 4, and similar to what was observed in β1AR-expressing cells, the efficacy pattern found in the ERK1/2 assay was significantly different from that observed for AC activation. In addition to isoproterenol and labetalol (which acted as agonists for the AC pathway), bucindolol and carvedilol (which did not have efficacy for the AC pathway) were also able to significantly increase the ERK1/2 phosphorylation (Fig. 4A). Moreover, propranolol, which was an inverse agonist for the AC, was able to stimulate the ERK1/2 signaling pathway (Fig. 4A). Labetalol, bucindolol, carvedilol, and propranolol were thus classified as partial agonists on the ERK1/2 pathway, their maximal stimulatory activity reached between 38% and 64% of isoproterenol efficacy (Fig. 4A; Table 5, MAPK). Metoprolol, bisoprolol, and atenolol, which were efficacious inverse agonists on the AC, did not seem to modulate ERK1/2 activity. However, because the basal level of ERK1/2 phosphorylation was close to the detection limit, inverse efficacy could have gone unnoticed. Hence, the efficacies of these compounds were reassessed in the presence of phorbol 12-myristate 13-acetate (PMA), a compound known to elevate ERK1/2 activity as a result of protein kinase C stimulation. Under these conditions, metoprolol, bisoprolol, and atenolol significantly decreased the PMA-stimulated ERK1/2 phosphorylation, thus acting as inverse agonists for this pathway (Fig. 4A, inset). This inverse agonist effect was also observed when these compounds were tested after elevation of the ERK1/2 activity by a different activator, the epidermal growth factor (data not shown). The revealed inverse efficacy of metoprolol, bisoprolol, and atenolol did not result from the artificially elevated ERK1/2 activity, because propranolol still acted as an agonist on the PMA-stimulated ERK1/2 activity (data not shown). Moreover, PMA treatment did not reveal inverse efficacy toward the β1AR-regulated ERK1/2 activity for any of the compounds (data not shown), clearly indicating the subtype selectivity of the effect observed.
As was the case for the β1AR-expressing cells, isoproterenol had a much lower (43×) potency to activate ERK1/2 than AC in cells expressing β2AR (Table 4). In contrast, labetalol and propranolol had similar potencies for the two signaling pathways. It is noteworthy that bucindolol and carvedilol, which had no detectable efficacy toward the AC pathway, activated the ERK1/2 with high potency.
Discussion
Combining the use of multiple ligands, two closely related receptor subtypes, and two effector systems, the present study, in line with other recent observations, illustrates a novel level of complexity in the definition of signaling efficacy and selectivity. When considering a single receptor, several ligands showed a complex efficacy profile, in some cases resulting in the opposite regulation of the two pathways considered by the same ligand. Our results also clearly indicate the existence of subtype-dependent efficacy profiles toward distinct signaling pathways that are not a simple reflection of the binding affinities of the ligands for each of the receptor subtype. Taken together, our data forcefully support the emerging notion that signaling efficacy can no longer be defined as a function of a ligand/receptor couple but needs to include the specific effector(s) considered, thus revealing the pluridimensionality of efficacy.
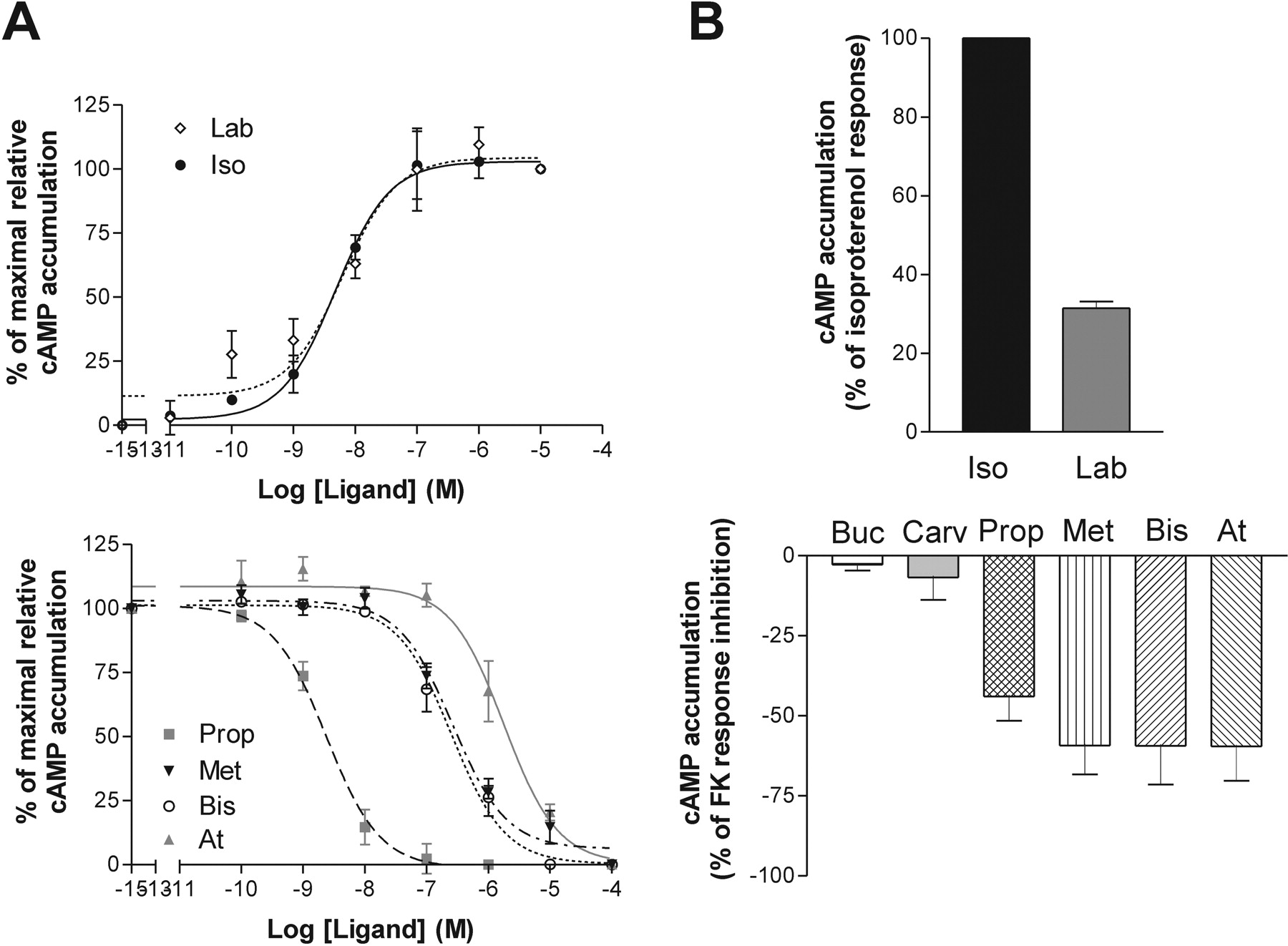
Profile of the different ligands on the β2AR adenylate cyclase pathway. cAMP accumulation experiments were performed in HEK293S cells stably expressing β2AR. A, EC50 of compounds were obtained using increasing concentrations of ligands in the presence (bottom) or absence (top) of 0.3 μM forskolin. B, Emax of compounds were tested using maximal concentrations of ligands, determined from EC50 curves, with (bottom) or without (top) 0.3 μM forskolin. Data represent the mean ± S.E.M of at least three experiments performed in triplicate.
One of the most striking observations of this study is that, although some ligands have similar efficacies toward ERK and AC, others displayed effectors-specific efficacies. In some cases, effector-specific efficacy profiles were revealed by different rank orders of efficacy for the two pathways. For example, when considering the β1AR, labetalol is an agonist as efficacious as bucindolol and carvedilol for AC, whereas it is much less efficacious than these two compounds toward the ERK1/2 stimulation. In other cases, the efficacy of a given compound is completely different, depending on the pathway considered. This is particularly striking for propranolol, which is an inverse agonist for AC but an agonist of the ERK1/2 pathway for both β1AR and β2AR. The effector-specific efficacy profiles for the two receptors can easily be appreciated by a Cartesian representation of the data that displays the relative efficacy of each compound toward the two signaling pathways (Fig. 5). As can be seen, three of the four possible scenarios have been observed: ACago-ERKago, ACinv-ERKago, and ACinv-ERKinv. Although the case scenario ACago-ERKinv was not observed in the present study, such possibility cannot be excluded on theoretical grounds and is likely to be observed for other ligands and/or other receptors. We and others have previously shown that β2-adrenergic, V2-vasopressin, serotonin 5-HT2C, and δ-opioid receptor ligands can act as inverse agonists on the adenylyl cyclase pathway but as agonists for the mitogen-activated protein kinase (MAPK) (Azzi et al., 2003; Baker et al., 2003; Audet et al., 2005; Werry et al., 2005). Likewise, Gbahou et al. (2003) reported that proxyfan, a high-affinity histamine H3 receptor ligand, is a partial agonist on AC and ERK1/2 but acts as an inverse agonist for the arachidonic acid release pathway. Although these studies introduced the concept of dual-efficacy ligands, the study of several ligands presented here clearly illustrates the pluridimensionality of ligand efficacy. In the present study, the efficacy description relied on two signaling pathways leading to a bidimensional representation, resulting in four possible efficacy quadrants (Fig. 5). However, more complete descriptions could take into account multiple possible signaling pathways and could thus offer a theoretical representation of efficacy in “n” dimension, resulting in “2”n efficacy quadrants. In addition to defining the qualitative nature of the efficacy toward various signaling pathways, such graphical representation allows us to attribute quantitative terms to these efficacies by using the spatial coordinates of the point corresponding to a given ligand, thus conferring a new meaning to the term efficacy. An example of how efficacies could be reported using such a coordinate system is presented at the bottom of Fig. 5.

Profile of the different ligands on the β2AR MAPK ERK1/2 pathway. ERK1/2 phosphorylation was assessed in HEK293S cells stably expressing β2AR. Compounds were tested at their appropriate maximal stimulation time (1 min for isoproterenol and 2 min for other compounds). A, Emax of compounds were obtained by the use of a maximal ligand concentration (10-5 M for all compounds). Inset, inverse efficacy of compounds was tested in the presence of 100 ng/ml PMA, 90 s. B, EC50 of compounds were determined using increasing concentrations of ligands. Data represent the mean ± S.E.M. of at least four experiments. Statistical significance was determined by two-way ANOVA, followed by a Dunnett test with basal (A) or with PMA (A, inset) as control column. ***, p < 0.001.
The distribution of the efficacy points presented in Fig. 5 do not follow the theoretical diagonal line predicted by a classic two-state model in which the receptor is either active or inactive for all the effectors considered. The dispersed distribution rather supports the emerging concept that multiple discrete conformations can be active for one pathway but inactive for another and that these conformations can be differentially stabilized/induced by distinct ligands (for review, see Kenakin, 1995, 2001, 2003). This is in line with the concept of ligand-directed stimulus trafficking that was first introduced based on studies reporting that a given agonist can activate two different signaling pathways with distinct orders of potency while acting on a single receptor. Similar differences in the orders of potency were also observed in the present study. For example, the rank order of potency for the β2AR-modulated AC (established independently of their efficacy; ie, combining agonists and inverse agonists) was propranolol ≥ isoproterenol = labetalol  carvedilol = bucindolol, whereas it was bucindolol ≥ carvedilol = propranolol = labetalol > isoproterenol for ERK1/2. The case of isoproterenol is particularly interesting when considering that its potencies to stimulate the two pathways are greatly different. Whereas its potency toward ERK1/2 (EC50 = 214 nM) is in line with its affinity for the receptor (Ki ∼450 nM; see Table 1), its potency to promote AC activation is much higher (EC50 = 5.0 nM, see Table 4). This suggests that the conformation stabilized/induced by isoproterenol favors the coupling to the AC signaling pathway so that a smaller fraction of the receptor population needs to be in this active conformation to fully activate the AC than to activate the ERK pathway. When considered in the context of the “spare receptors” concept, the isoproterenol-promoted conformation leads to a greater number of spare receptors for the AC than the ERK pathway.
carvedilol = bucindolol, whereas it was bucindolol ≥ carvedilol = propranolol = labetalol > isoproterenol for ERK1/2. The case of isoproterenol is particularly interesting when considering that its potencies to stimulate the two pathways are greatly different. Whereas its potency toward ERK1/2 (EC50 = 214 nM) is in line with its affinity for the receptor (Ki ∼450 nM; see Table 1), its potency to promote AC activation is much higher (EC50 = 5.0 nM, see Table 4). This suggests that the conformation stabilized/induced by isoproterenol favors the coupling to the AC signaling pathway so that a smaller fraction of the receptor population needs to be in this active conformation to fully activate the AC than to activate the ERK pathway. When considered in the context of the “spare receptors” concept, the isoproterenol-promoted conformation leads to a greater number of spare receptors for the AC than the ERK pathway.
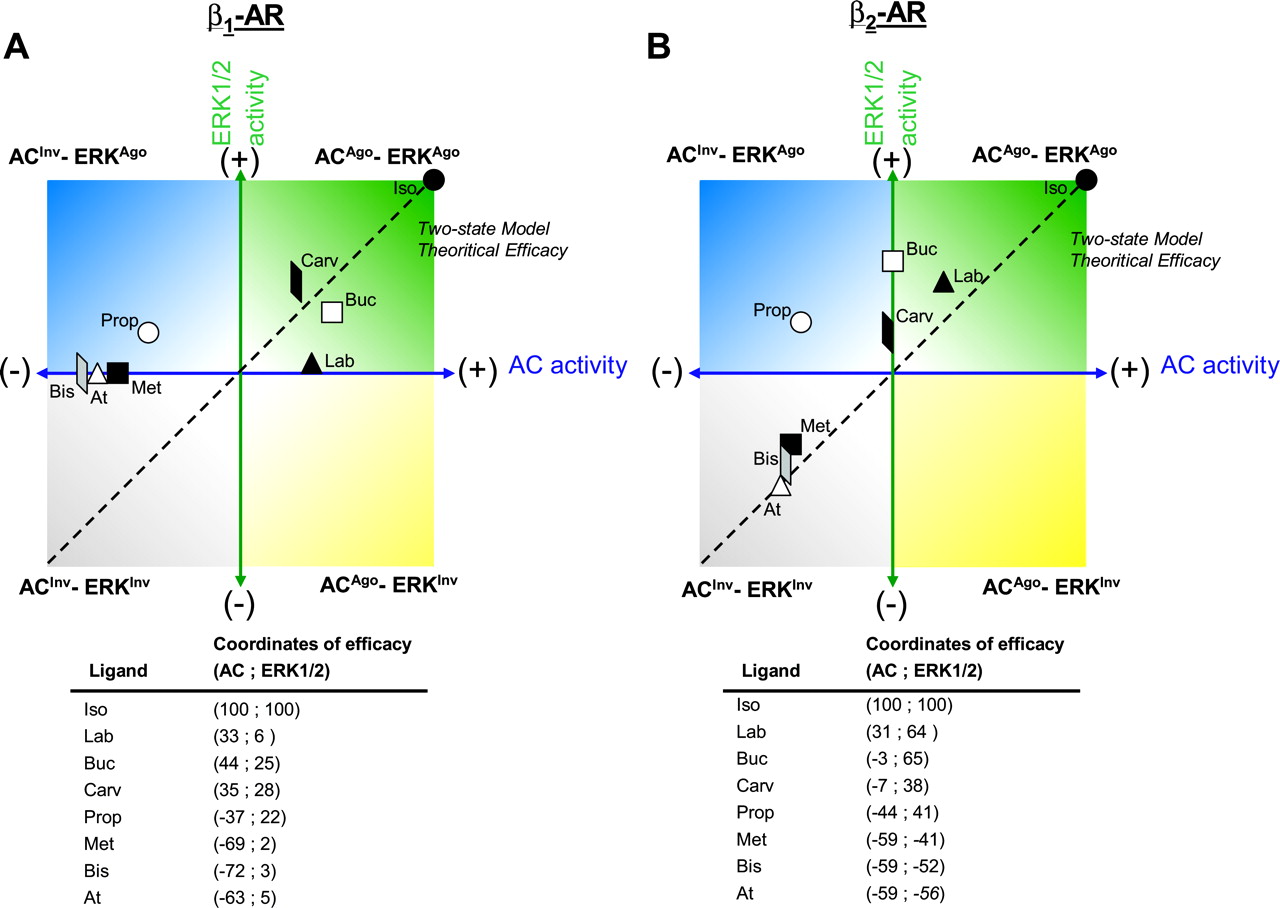
Cartesian representation (AC pathway in abscise and MAPK ERK1/2 pathway in ordinate) of compounds efficacy profiles and their appropriate efficacy coordinates. A, efficacy profile and coordinates of the compounds tested on the β1AR. B, efficacy profile and coordinates of the compounds tested on the β2AR. ACago-ERKago, compounds that are agonists on the AC and on the ERK1/2; ACinv-ERKago, compounds that are inverse agonists on the AC and agonists on the ERK1/2; ACinv-ERKinv, compounds that are inverse agonists on the AC and on the ERK1/2; ACago-ERKinv, compounds that are agonist on the AC and inverse agonists on the ERK1/2. Coordinates are obtained from Tables 3 and 5.
Overall, our data are consistent with the emerging concept that different ligands can stabilize distinct receptor conformations (Vauquelin and Van, 2005) that may differ in their signaling partner preference (Kenakin, 2002). Such behavior can easily be formalized using either the extended or the cubic ternary complex models of G protein activation. Indeed, by including factors controlling the affinity of the receptor for its cognate G protein, these models allow different ligands to induce/stabilize distinct receptor/G protein affinity states (Kenakin, 2004). Given that many receptors can couple to more than one G protein, it can easily be foreseen that the different ligand-promoted receptor conformations could yield differential signaling efficacies through distinct effector systems. Such behavior has indeed been observed in native tissues supporting its potential physiological relevance. For instance, whereas several β-adrenergic agonists (e.g., salbutamol, terbutaline, procaterol, zinterol) have been found to promote both Gi and Gs activation, fenoterol, which is an efficacious agonist for the Gs pathway, was found to be inactive toward Gi in rat cardiomyocytes (Xiao et al., 2003; Ponicke et al., 2006). There is no theoretical reason to limit this conformationally based selection to G proteins. It could therefore be proposed that the individual conformations stabilized by each ligand could display a specific set of affinities for the various partners involved in the different behaviors of the receptor, including signaling via multiple effectors, receptor phosphorylation, endocytosis, and desensitization. In this context, it is not surprising that signaling efficacies are often found to be context-dependent. Indeed, the different levels of expression or subcellular distribution of specific partners among cells or tissues would be predicted to affect the signaling profiles observed in these individual systems (Watson et al., 2000).
The capacity of different ligands to promote distinct conformational rearrangements has been clearly confirmed by biophysical studies monitoring the fluorescent properties of intramolecular probes within purified β2AR (Ghanouni et al., 2001; Swaminath et al., 2005). However, the link between the conformations stabilized by specific ligands and their efficacy pattern toward different signaling pathways remains to be established. Speaking intuitively, compounds with the greatest chemical similarity would be expected to stabilize similar conformation, thus resulting in comparable efficacy patterns. As can be seen in Table 6, this prediction seems to be borne out, because compounds with identical efficacy patterns present similar chemical structures.
Structures of the ligands tested and their efficacy pattern towards AC and ERK1/2 pathways on the two β-adrenergic subtypes.
The comparison between β1AR and β2AR led to the observation that in addition to their traditionally defined subtypespecific affinities, β-adrenergic ligands also display subtypespecific efficacy patterns (Fig. 5). This is particularly striking when considering labetalol. This compound binds β1AR and β2AR with similar affinities (Table 1) and has similar potencies and efficacies toward the AC pathway for the two receptor subtypes (Figs. 1 and 3, and Tables 3 and 5). However, labetalol is β2AR-selective ERK1/2 agonist is unable to activate this pathway in β1AR-expressing cells. This clearly indicates that subtype specificity cannot be established only on the basis of ligand binding affinities or the relative potency determined for a single signaling pathway. To be pharmacologically exhaustive, the receptor subtype selectivity would therefore need to be determined for each effector system that can be engaged by the receptors considered.
Although the complexity of signaling efficacy and selectivity can be more easily dissected in heterologous expression systems in which specific receptor expression can be controlled, efficacy profiles that differ depending of the effector systems considered have also been observed in native environments. For instance, propranolol was found to act as an inverse agonist toward the AC pathway while being an agonist on the ERK1/2 activity in canine cardiomyocytes, S49 lymphoma cells, and Cos cells endogenously expressing the βAR (Azzi et al., 2003). Likewise, proxyfan was found to be a partial agonist when assessed in the context of H3 histaminereceptor-promoted histamine release but an inverse agonist in a GTPγS binding assay (Gbahou et al., 2003). These observations suggest that the pluridimensionality of signaling efficacies observed herein is not simply an artifact of overexpression in artificial systems but could be physiologically relevant. Additional studies, however, are needed to assess the extent to which the effector-dependent signaling efficacies will be detectable in normal and pathophysiological conditions.
It should also be noted that primary drug screenings are often carried out using unique cell-based assays, taking advantage of robust signals detected in heterologous expression systems. The observation that different efficacies can be observed depending on the signaling outcome considered has obvious implication on the conclusions that can be drawn from such screening campaigns. On the one hand, it stresses the importance of selecting an effector pathway that is pertinent for the pathological condition considered. On the other hand, taking the pluridimensionality of efficacy into account, testing the signaling efficacies of compounds into multiple assay systems could allow us to link specific signaling properties to given therapeutic activities or side effects, thus increasing the chances of identifying compounds with efficacy profiles that may have better therapeutic outcomes. A better description of the efficacy profiles for each member of a drug family could also help us understand why individual members of a drug class sometimes have different therapeutic indications. For example, clinical trials assessing the efficacy of β-blockers in the treatment of congestive heart failure revealed that carvedilol, metoprolol, and bisoprolol, but not bucindolol, decreased mortality, and the reason for the lack of beneficial action of bucindolol remains elusive. In fact, although carvedilol and metoprolol are approved in many countries for the treatment of heart failure, the molecular basis of their beneficial effects is still poorly understood. Based on the efficacy profiles determined for two receptor subtypes and two signaling pathways, the present study did not provide a signature that could predict efficacy in heart failure. Whether increasing the number of signaling modalities measured for each compounds will allow such predictive signatures to emerge remains to be investigated.
One of the major conclusions of the present study is that efficacy and selectivity toward GPCRs cannot be defined based simply on a ligand/receptor couple but should also include the effector system in its operational definition. Given the increasing number of signaling possibilities that have recently surfaced for GPCRs, the pluridimensionality of efficacy will certainly become more and more evident and will be integrated in our way of thinking about drug action and classification.
Acknowledgments
We thank GlaxoSmithKline (Research Triangle Park, NC) and Dr. Michael Bristow (University of Colorado Health Sciences Center, CO) for kindly providing carvedilol and bucindolol, respectively. We are grateful to Dr. Monique Lagacé for her critical reading of the manuscript.
Footnotes
-
This work was supported by grants from the Canadian Institute for Health Research and from the Heart and Stroke Foundation of Canada. M.B. holds a Canada Research Chair in Signal Transduction and Molecular Pharmacology.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; ICI118,551, (±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol; AR, adrenergic receptor; AC, adenylyl cyclase; ERK, extracellular signal-regulated kinase; HRP, horseradish peroxidase; HEK, human embryonic kidney; PBS, phosphate-buffered saline; TBS-T, Tris-buffered saline-Tween 20; PMA, phorbol 12-myristate 13-acetate; MAPK, mitogen-activated protein kinase; ANOVA, analysis of variance; CGP-12177, 4-[3-[(1,1-dimethylethyl)amino]-2-hydroxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one; Iso, isoproterenol; Lab, labetalol; Buc, bucindolol; Carv, carvedilol; Prop, dl-propanolol; Bis, bisoprolol; Met, (±)-metoprolol; At, S(-)-atenolol.
- Received May 15, 2006.
- Accepted August 10, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}