


Abstract
Exciting advances have been made in the discovery of selective positive allosteric modulators of the metabotropic glutamate receptor (mGluR) mGluR5. These compounds may provide a novel approach that could be useful in the treatment of certain central nervous system disorders. However, because of their low potencies, previously described mGluR5 potentiators are not useful for functional studies in native preparations. In addition, binding sites at which these compounds act have not been identified. It has been suggested that two allosteric potentiators, 3,3′-difluorobenzaldazine and 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB), act by binding to the same allosteric site as the negative allosteric modulators of mGluR5 such as 2-methyl-6-(phenylethynyl)pyridine (MPEP). However, another mGluR5 potentiator, N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)m-ethyl]phenyl}-2-hydroxybenzamide, does not bind to this site, bringing this hypothesis into question. We have synthesized a series of CDPPB analogs and report that these compounds bind to the MPEP site with affinities that are closely related to their potencies as mGluR5 potentiators. Furthermore, allosteric potentiation is antagonized by a neutral ligand at the MPEP site and reduced by a mutation of mGluR5 that eliminates MPEP binding. Together, these data suggest that interaction with the MPEP site is important for allosteric potentiation of mGluR5 by CDPPB and related compounds. In addition, whole-cell patch-clamp studies in midbrain slices reveal that a highly potent analog of CDPPB, 4-nitro-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (VU-29), selectively potentiates mGluR5 but not mGluR1-mediated responses in midbrain neurons, whereas a previously identified allosteric potentiator of mGluR1 has the opposite effect.
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system. In addition to eliciting fast excitatory synaptic responses, glutamate has important neuromodulatory effects by the activation of G protein-coupled receptors (GPCRs) termed metabotropic glutamate receptors (mGluRs). The mGluRs play important roles in a broad range of central nervous system functions and have potential as novel targets for the development of new therapeutic agents for a number of neurological and psychiatric disorders, including Parkinson's disease (Marino and Conn, 2002b), epilepsy (Doherty and Dingledine, 2002), Alzheimer's disease (Wisniewski and Carr, 2002), pain (Varney and Gereau, 2002), schizophrenia (Marino and Conn, 2002a), depression (Palucha and Pilc, 2002), anxiety disorders (Chojnacka-Wojcik et al., 2001; Pilc, 2003), and others.
The mGluRs are family C GPCRs and include eight subtypes termed mGluR1 to mGluR8. These receptors have been classified into three groups based on sequence homology, primary G protein coupling, and pharmacological properties. Group I mGluRs (mGluR1 and mGluR5) couple to Gαq/11 and activate phospholipase C. Group II (mGluR2 and mGluR3) and group III (mGluR4, mGluR6, mGluR7, and mGluR8) mGluRs couple to effectors through Gαi/o (Conn and Pin, 1997; Coutinho and Knopfel, 2002). Since the initial discovery of the mGluRs, there has been an increasing focus on developing subtype-selective modulators of these receptors for use as potential clinical agents and as pharmacological tools that could aid in developing a better understanding of mGluR function.
Although mGluRs have a seven transmembrane (7TM)-spanning domain similar to other GPCRs (Conn and Pin, 1997; Bhave et al., 2003), glutamate binds these receptors on a large N-terminal extracellular glutamate binding domain that is composed of two globular domains and a hinge region (O'Hara et al., 1993; Jingami et al., 2003). As expected for a region involved in binding a common endogenous agonist, the glutamate binding sites share high homology across the mGluR subtypes relative to other regions of the receptor (Conn and Pin, 1997). Based on this, we and others have begun to take a novel approach and develop compounds that interact with potentially less evolutionary conserved allosteric sites of mGluRs (Knoflach et al., 2001; Gasparini et al., 2002; Marino et al., 2003; May and Christopoulos, 2003; O'Brien et al., 2003, 2004; Schaffhauser et al., 2003). For instance, we have developed DFB, CPPHA, and CDPPB as three distinct structural classes of allosteric potentiators of mGluR5 (O'Brien et al., 2003, 2004; Kinney et al., 2005). These compounds do not activate mGluR5 directly but potentiate the response of mGluR5 to glutamate, inducing a leftward shift of the glutamate concentration-response curve. It is noteworthy that these allosteric modulators do not affect binding of ligands to the orthosteric glutamate binding site. Thus, in contrast to known allosteric modulators of family A GPCRs, they do not act by altering agonist affinity. However, competition binding with [3H]methoxyPEPy, an analog of the allosteric mGluR5 antagonist MPEP, reveals that two potentiators, DFB and CDPPB, displace binding to this site. This led to the suggestion that allosteric potentiators and allosteric antagonists act at overlapping sites in the transmembrane domain. However, whereas CDPPB fully displaces [3H]methoxyPEPy binding, it is not clear whether this compound interacts competitively with [3H]methoxyPEPy at this site. Furthermore, the potency of CDPPB as an allosteric potentiator of mGluR5 is more than one magnitude higher than the apparent affinity of this compound at the [3H]methoxyPEPy site. Finally, at least one mGluR5 allosteric potentiator, CPPHA, has been identified that does not displace [3H]methoxyPEPy binding (O'Brien et al., 2003, 2004; Kinney et al., 2005). Based on this, it is unclear whether the allosteric potentiator activity of CDPPB requires interaction with the site occupied by [3H]methoxyPEPy. In addition, the majority of studies that have been focused on characterizing mGluR5 potentiators have relied on cultured cell lines rather than native neuronal populations. Thus, it is unclear whether mGluR5 potentiators will selectively potentiate the regulation of mGluR5 neuronal excitability by native neurons.
We report studies in which we use synthetic chemistry, along with molecular pharmacology approaches, to systematically examine the relationship between interaction of CDPPB and related compounds to the allosteric MPEP site and allosteric potentiator activity. Our studies suggest that activities of CDPPB and its analogs as allosteric potentiators are closely related to their affinities for the MPEP site. Furthermore, the discovery of an analog of CDPPB (VU-29) with low nanomolar potency provides an excellent tool for determining the effects of allosteric potentiators on excitation of neurons by mGluR5 and its closest relative, mGluR1. These compounds selectively potentiate mGluR5-mediated responses in midbrain slices without altering responses that are mediated by mGluR1.
Materials and Methods
Mutagenesis and Transient Transfection. HEK293A cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 1 mM l-glutamine, and 1× antibiotic-antimycotic (all from Invitrogen, Carlsbad, CA). Cells were collected and plated in clear-bottomed 96-well plates (Costar; Corning Life Sciences, Acton, MA) pretreated with poly(d-lysine) (Sigma, St. Louis, MO) in normal growth medium with a density of 40,000 cells/well overnight before transfection. Cells were transiently transfected with wild-type and mutant forms of rat mGluR5a cDNA using the pRK5 vector (Clontech, Mountain View, CA). Point mutations were generated using the QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). All mutations were verified by sequencing. The transfection plasmid was prepared using Sigma Maxi Prep kit (Sigma-Aldrich, St. Louis, MO). Cells were transfected with Lipofectamine (Invitrogen) for 6 h according to the manufacturer's instructions (80 ng of DNA and 0.2 μl of Lipofectamine per well) before switching to normal growth medium. Rat GLAST pCDNA3.1 (20 ng/well) was coexpressed with mGluR5 pRK5 to reduce extracellular glutamate concentration. Glutamate/glutamine-free medium (glutamine-free DMEM plus 10% dialyzed fetal bovine serum; Invitrogen) was applied to substitute growth medium at least 4 h before performing functional assays. Cell culture, transfection, and starving were performed at 37°C in an atmosphere of 95% air plus 5% carbon dioxide. Transfected cells were tested approximately 48 h after transfection. Rat mGluR2 and human mGluR4 were coexpressed with Gqi5, which enables coupling to the calcium mobilization as reported by Galici et al. (2006).
Secondary Rat Astrocytes Culture. Secondary rat cortical astrocytes were prepared as described previously (Peavy et al., 2001; Zhang et al., 2005). Astrocytes were plated into poly(d-lysine)-coated 96-well plates with a density of 30,000 cells/well on day 0 in DMEM containing 10% FBS, 1 mM l-glutamine (Invitrogen), and 1× antibiotic-antimycotic (Invitrogen) overnight. Then G-5 supplement (Invitrogen), which contains epidermal growth factor (10 ng/ml), basic fibroblast growth factor (5 ng/ml), insulin (5 μg/ml), and other factors, was added to the growth medium on day 1 and switched to glutamine-free DMEM with 10% dialyzed FBS on day 3. Calcium mobilization assay was performed on day 4. Cell culture and starvation were performed at 37°C with 5% carbon dioxide.
Calcium Fluorescence Measurement. Cells were loaded with calcium-sensitive dye according to the manufacturer's instructions (Calcium 3 kit; Molecular Devices, Sunnyvale, CA) after incubation in glutamate/glutamine-free medium (DMEM and 10% dialyzed fetal bovine serum) for 5 h. Compound A (1 ml) from Calcium 3 kit was dissolved in 20 ml of 1× Hanks' balanced salt solution (HBSS; Invitrogen) containing 2.5 mM probenecid (Sigma), adjusted to pH 7.4. Cells were loaded for 50 min at 37°C with 5% carbon dioxide. Dye was then carefully removed, and cells were washed with HBSS containing probenecid. Cells were maintained in the same buffer at room temperature for the following assay. For calcium fluorescence measurement of rat cortical astrocytes, allosteric modulators were added 5 min before the addition of agonist manually. For transient transfected cells, allosteric modulators were added 1 min before the addition of agonist using Flexstation II (Molecular Devices). Agonist was added at a speed of 52 μl/s, and calcium flux was measured using Flexstation II at 25°C. All of the peaks of the calcium response were normalized to the maximum response to a saturated dose of glutamate (10 μM). The submaximal concentration (EC20 value) of glutamate was determined for every separate experiment, allowing for a response varying from 10 to 30% of the maximum peak.
Radioligand Binding Assays. The MPEP analog [3H]methoxyPEPy was used to test the binding of MPEP site on mGluR5 (Cosford et al., 2003). Membranes were prepared from stable rat mGluR5-HEK293A cells (Rodriguez et al., 2005). [3H]methoxyPEPy was incubated with membrane (10 μg/well) in the binding buffer (50 mM Tris/0.9% NaCl, pH 7.4) with the presence or absence of CDPPB analogs at room temperature for 1 h with shaking. Then the membrane-bound ligand was separated from free ligand by filtration through 96-well glass-fiber filter plates (Unifilter-96 GF/B; PerkinElmer Life and Analytical Sciences, Boston, MA) and washed three times with binding buffer (Brandel Cell Harvester; Brandel Inc., Gaithersburg, MD). Scintillation fluid (30 μl) was added to each well, and the membrane-bound radioactivity was determined by scintillation counting (TopCount; PerkinElmer Life and Analytical Sciences). Nonspecific binding was estimated using 5 μM MPEP. For Scatchard analysis, [3H]methoxyPEPy concentrations of 2.5, 5, 10, 20, and 40 nM were used, whereas 2 nM concentration of [3H]methoxyPEPy was used for competition binding assay. The KD value of [3H]methoxyPEPy by saturation binding was 3.4 nM.
Compound Preparation and Application. 5MPEP, CDPPB, VU-20 to VU-24, VU-28, VU-29, VU-35, and VU-36 were synthesized as described previously (Lindsley et al., 2004; Rodriguez et al., 2005; de Paulis et al., 2006). Compounds were dissolved in dimethyl sulfoxide (Sigma) and stored at –80°C. Stock solutions were dissolved in 1× HBSS containing 0.1% bovine serum albumin (Sigma) on the day of experiment. Final dimethyl sulfoxide concentration was 0.12 to 0.15% for all of the assays.
N-Terminal Truncated mGluR5 and Inositol Phosphate Determination. Construction of the N-terminal truncated mutant of mGluR5 and inositol phosphate (IP) accumulation measurement were performed as reported by Goudet et al. (2004). In brief, the mGluR5 mutant possesses the signal peptide of the wild-type mGluR5 followed by the hemagglutinin epitope and the coding sequence of the 7TM region starting at Pro568 and terminating at Leu864. IP measurements were performed after transient transfection by electroporation of HEK293A cells with the plasmid expressing the truncated mGluR5. The cells were incubated overnight with [myo-3H]inositol (23.4 Ci/nmol; GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). After washing, cells were stimulated with the indicated compounds for 30 min in the presence of 10 mM LiCl. Inositol phosphate accumulated was recovered by ion-exchange chromatography using a Dowex resin (Bio-Rad Laboratories, Hercules, CA) in 96-well microfilter plates. Results are expressed as the ratio between IP and the total radioactivity (IP fraction plus the radioactivity in the membranes).
Electrophysiology in Subthalamic Nucleus and Substantia Nigra Neurons. Whole-cell recordings were performed using midbrain brain slices prepared from 12- to 18-day-old male Sprague-Dawley rats, as described previously (Awad et al., 2000; Marino et al., 2001). After decapitation, brains were rapidly removed and submerged in an ice-cold choline replacement solution containing 126 mM choline chloride, 2.5 mM KCl, 1.2 mM NaH2PO4, 1.3 mM MgCl2, 8 mM MgSO4, 10 mM glucose, and 26 mM NaHCO3, equilibrated with 95% O2/5% CO2. Sagittal brain slices (350 μm) containing subthalamic nucleus and substantia nigra were cut using a microtome (Leica Microsystems, Nussloch, Germany) and transferred to a holding chamber containing artificial cerebrospinal fluid (ACSF) composed of 124 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4, 1.0 mM NaH2PO4, 2 mM CaCl2, 20 mM glucose, and 26 mM NaHCO3, equilibrated with 95% O2/5% CO2, and maintained at room temperature. For all experiments, both choline replacement buffer and holding chamber ACSF buffer were supplemented with 5 μM glutathione, 500 μM pyruvate, and 250 μM kynurenic acid to increase slice viability.
After 1 h of recovery in the holding chamber, brain slices were then transferred to the slice recording chamber and maintained fully submerged with continuous perfusion of ACSF (2–3 ml/min). Neurons in the subthalamic nucleus (STN) or substantia nigra pars reticulata (SNr) were visualized with a 40× water immersion lens with Hoffman modulation contrast optics. Patch electrodes were pulled from borosilicate glass on the Narishige (Greenvale, NY) vertical patch pipette puller and filled with internal solution (125 mM potassium gluconate, 4 mM NaCl, 6 mM NaH2PO4, 1 mM CaCl2, 2 mM MgSO4, 10 mM BAPTA-tetrapotassium salt, 10 mM HEPES, 2 mM magnesium-ATP, and 0.3 mM Na2-GTP; pH adjusted to 7.3 with 1 N KOH). Electrode resistance was 3 to 7 MΩ. All whole-cell patch-clamp recordings were performed using a MultiClamp 700B amplifier (Molecular Devices). Data were digitized with DigiData 1322A, filtered (2 kHz), and acquired by the pClamp 9.2 program (both from Molecular Devices). After formation of a whole-cell configuration, the recorded neurons were current-clamped to –60 mV. Membrane potentials of STN or SNr neurons were recorded. All compounds were applied by adding into perfusion solution. Data were analyzed using Clampfit 9.2 (Molecular Devices). All results are expressed as mean ± S.E.M., and statistical significance was determined using Student's t test.
Results
CDPPB Displaces [3H]MethoxyPEPy Binding on mGluR5 Competitively. We reported previously that CDPPB completely displaces binding of the allosteric site ligand [3H]methoxyPEPy to membranes from cells stably expressing mGluR5 (Kinney et al., 2005). We now performed saturation binding experiments with increasing concentrations of [3H]methoxyPEPy in the presence or absence of two concentrations of CDPPB, and data were transformed using a Scatchard analysis to determine whether this is consistent with a competitive interaction of CDPPB with the [3H]methoxyPEPy binding site (Fig. 1). Nonspecific binding was defined as binding in the presence of 5 μM MPEP and subtracted from total binding. In the absence of CDPPB, Scatchard analysis of [3H]methoxyPEPy binding reveals a straight line (r2 = 0.77), demonstrating interaction at a single binding site. The X-intercept indicates a binding density (Bmax) of approximately 2300 fmol/mg of protein in this membrane preparation, and the slope reveals an apparent KD value of 6.2 nM, consistent with previous results. Addition of CDPPB induced a shift in the slope of the regression line but no effect on the X-intercept, suggesting that CDPPB has no effect on the receptor density. However, the apparent affinity of [3H]methoxyPEPy was reduced by CDPPB, with KD values of 8.3 and 12 nM for 1 and 2.5 μM CDPPB, respectively. The maintenance of a linear Scatchard regression (r2 = 0.74 and 0.69 for 1 and 2.5 μM CDPPB, respectively) with change in apparent KD and no change in Bmax is consistent with the hypothesis that CDPPB competitively displaces [3H]methoxyPEPy binding at the MPEP binding site.
Potencies of CDPPB Analogs at Potentiating mGluR5 Responses Correlate Significantly with Their Affinities at the [3H]MethoxyPEPy Binding Site. The finding that CDPPB displaces [3H]methoxyPEPy binding in a manner that is consistent with competitive interaction with this allosteric MPEP binding site raises the possibility that binding to this site is necessary for allosteric potentiator activity. However, another allosteric potentiator of mGluR5, CPPHA, does not bind to this site (O'Brien et al., 2004). In addition, the potency of CDPPB as the mGluR5 allosteric potentiator is more than 1 order of magnitude higher than its apparent affinity at the MPEP binding site (Kinney et al., 2005; de Paulis et al., 2006). Thus, it is possible that CDPPB-induced potentiation of mGluR5 responses is unrelated to its interaction with the allosteric MPEP site. To address this, we synthesized a series of structural analogs of CDPPB to determine whether affinities of these compounds at the MPEP site are closely related to their potencies at potentiating mGluR5 (de Paulis et al., 2006). We selected 10 of these compounds based on their close structural similarity to CDPPB and with no changes to the diphenylpyrazole portion of the molecule (de Paulis et al., 2006). Concentration-response analysis revealed that these compounds potentiate calcium mobilization responses to the mGluR5 agonist glutamate with potencies that range from 9 to 228 nM as mGluR5 allosteric potentiators (Fig. 2 and Table 1). One compound, VU-137, was inactive as an mGluR5 potentiator. VU-29 was the most potent allosteric potentiator in this group with potency of 9 nM. None of these 10 compounds showed significant activities as allosteric potentiators of mGluR1 concentrations up to 10 μM (Hemstapat et al., 2006). Radioligand binding studies revealed that 9 of the 10 CDPPB analogs displace [3H]methoxyPEPy in a concentration-dependent manner (data not shown). It is interesting that the one compound that was inactive at displacing [3H]methoxyPEPy binding, VU-137, was also inactive at potentiating mGluR5-mediated calcium mobilization responses (Fig. 2). Similar to CDPPB, the potencies of multiple compounds in the CDPPB series at potentiating glutamate-mediated functional responses were higher than their affinities at displacing [3H]methoxyPEPy from its binding site (Table 1). However, regression analysis of the affinities at the MPEP site versus allosteric potentiator potencies revealed that there is a close correlation between binding affinities to this site and potentiator activity (Fig. 3) (r = 0.89; p ≤ 0.001). This, together with the finding that VU-137 is inactive in either binding to the MPEP site or as an allosteric potentiator, is consistent with the hypothesis that binding to this site is required for allosteric potentiator activity. Having identified VU-29 as a highly potent allosteric potentiator of mGluR5, we used this compound in further studies aimed at characterizing this response.
Affinities of CDPPB analogs at the allosteric mGluR5 antagonist MPEP binding site obtained from [3H]methoxyPEPy competition binding using membrane prep from the rmGluR5 stable cell line and their potencies as allosteric mGluR5 potentiators from glutamate-induced calcium release in cultured rat cortical astrocytes
Data are average ± S.E.M. from three or four independent experiments in duplicate.

CDPPB reduces [3H]methoxyPEPy binding to mGluR5 in a competitive manner. Scatchard analysis showed that CDPPB dose-dependently decreases [3H]methoxyPEPy binding affinity but does not alter maximum binding. Saturation binding on membranes from mGluR5 stable HEK cell line was performed in the absence or presence of CDPPB. X-intercepts showed maximum binding under different binding conditions. In the absence of CDPPB, Bmax was 2312 ± 355 fmol/mg of protein, with 1 or 2.5 μM CDPPB, Bmax was 2350 ± 366 or 2135 ± 251 fmol/mg of protein, respectively, showing no significant differences in Bmax [3H]methoxyPEPy (Student's t test). Linear regression lines were generated from four independent experiments in duplicate. Error bars represent S.E.M.
5MPEP Antagonizes VU-29-Mediated Potentiation of mGluR5 Response. We reported recently the discovery and characterization of a novel compound that is a positional isomer of MPEP, 5MPEP, that acts as a neutral ligand at the allosteric MPEP site and blocks responses of both allosteric antagonists and potentiators (Rodriguez et al., 2005). Consistent with previous results, 10 μM 5MPEP completely blocked the potentiation of the calcium mobilization response to glutamate by 60 nM VU-29 (p < 0.001; Fig. 4, A and B). Concentration-response analysis revealed that blockade of the response to VU-29 by 5MPEP is concentration-dependent with an IC50 value of 710 ± 170 nM for 5MPEP (Fig. 4C), which is consistent with the IC50 of 5MPEP at blocking the antagonist effect of MPEP (Rodriguez et al., 2005).

CDPPB analogs have a range of potencies on secondary cultured rat astrocytes as mGluR5 allosteric potentiators. A, chemical structures of selected CDPPB analogs. B, intracellular calcium mobilization responses were measured in response to an EC20 concentration of glutamate in the absence or presence of different concentrations of CDPPB analogs. The EC20 concentration for glutamate was determined each day and ranged from approximately 270 to 320 nM. CDPPB and its analogs induced a concentration-dependent increase in the response to this EC20 glutamate concentration. Data were normalized to the maximum response of each reaction determined by 10 μM glutamate. Concentration-response curves were generated from three independent experiments each performed in duplicate. Error bars represent S.E.M.
VU-29 Is an Agonist of N-Terminal Truncated mGluR5. If the allosteric potentiator of VU-29 and related compounds is due to actions in the 7TM spanning domain, in which the allosteric MPEP binding site resides, it is possible that this compound could retain activity at a truncated mutant of mGluR5 in which the N-terminal extracellular domain, including the glutamate binding domain, is missing. We have reported recently that MPEP and other allosteric modulators of mGluR5 can retain their activity in cells expressing this truncated form of the receptor (Goudet et al., 2004). However, with the glutamate binding site absent, this activity does not depend on the presence of glutamate, and these ligands behave in a manner similar to orthosteric ligands at family A GPCRs. Consistent with our previous studies, in HEK cells transiently expressing an N-terminal truncated mutant of mGluR5, MPEP behaved as an inverse agonist and inhibited baseline accumulation of inositol phosphates, a measure of coupling of mGluR5 to phosphoinositide hydrolysis (Goudet et al., 2004) (Fig. 5A). Consistent with its activity as a neutral ligand at the MPEP site, 5MPEP did not behave as an inverse agonist of the truncated receptor and also did not activate the truncated mutant (Fig. 5A). It is interesting that VU-29 behaved as an agonist in this system and directly activated phosphoinositide hydrolysis in a concentration-dependent manner in cells expressing the N-terminal truncated form of mGluR5 (Fig. 5A). The agonist effect of VU-29 was inhibited by 5MPEP (Fig. 5B), which is consistent with the hypothesis that activation of phosphoinositide hydrolysis in the cells is mediated by actions of VU-29 on the 7TM binding domain of mGluR5.

The potencies of CDPPB analogs as mGluR5 allosteric potentiators significantly correlate with their affinities at the [3H]methoxyPEPy binding site (r = 0.89, p < 0.001). Membranes were prepared from rmGluR5 stable Chinese hamster ovary cell line; 2 nM [3H]methoxyPEPy was used for the competition binding assay, and 5 μM MPEP was used to determine nonspecific binding. Data were obtained from three or four separate experiments each performed in duplicate.
A Mutation that Eliminates Binding of Allosteric Antagonists to the MPEP Binding also Reduces the Potentiation of mGluR5 by VU-29. Mutation of alanine at the 809 position of mGluR5 to valine mGluR5 (A809V) reduces binding of MPEP to mGluR5 and severely reduces the potency of MPEP as an mGluR5 allosteric antagonist (Pagano et al., 2000; Malherbe et al., 2003). In contrast, mutation of a neighboring amino acid, M801T, which is also in the 7TM domain, has little effect on the affinity and function of MPEP (Pagano et al., 2000; Malherbe et al., 2003). If the allosteric potentiator activity of CDPPB and VU-29 requires interaction with the allosteric MPEP site, a mutation that reduces binding of ligands to the MPEP site should also reduce activity of these compounds as allosteric potentiators, whereas mutation of this neighboring amino acid should not. Consistent with the previous report, the potency of MPEP on mGluR5 (A809V) was approximately 10-fold lower than its potency on the wild-type receptor, whereas the potency of MPEP on mGluR5 (M801T) remained intact (Pagano et al., 2000; data not shown). Consistent with the hypothesis that allosteric potentiator activity of these compounds requires binding to the MPEP site, both CDPPB and VU-29 were inactive as allosteric potentiators at mGluR5 (A809V), whereas both compounds retained activity at mGluR5 (M801T). Thus, as demonstrated in stable mGluR5-expressing cell lines and astrocytes, CDPPB (1 μM) and VU-29 (200 nM) induced parallel leftward shifts of the glutamate concentration-response curves in cells transiently transfected with wild-type mGluR5 (Fig. 6A) or with mGluR5 (M801T) (Fig. 6B). In contrast, neither compound induced a significant shift in the glutamate concentration-response curve of mGluR5 (A809V) (Fig. 6C).

5MPEP is a neutral antagonist of VU-29. A, calcium fluorescence responses of cortical astrocytes to an EC20 concentration of glutamate added in the absence and presence of VU-29 and, in some cases, the neutral allosteric site ligand 5-MPEP. Glutamate was added (arrow) 5 min after the addition of 5MPEP and/or VU-29. B, mean (± S.E.M.) calcium fluorescence responses for each condition. 5MPEP (10 μM) did not alter the EC20 glutamate response. VU-29 enhanced the EC20 glutamate response approximately 3-fold (Student's t test, one tail, p < 0.001). 10 μM 5MPEP completely blocked the enhancement by 60 nM VU-29 to the baseline (Student's t test with VU-29 potentiated response, one tail, p < 0.001). C, concentration-response relationship for inhibition of the response to VU-29 by 5MPEP. The response to 60 nM VU-29 plus and EC20 concentration of glutamate in the absence of 5MPEP is shown on the left. 5MPEP dose-dependently antagonized VU-29-induced potentiation to a level similar to that of an EC20 concentration of glutamate in the absence of VU-29 (shown at right). 5MPEP, 10 μM; VU-29, 60 nM; EC20 glutamate, approximately 300 nM. Data were obtained from three or four separate experiments each performed in duplicate.

VU-29 is an agonist of N-terminal truncated mGluR5. A, VU-29 directly activated N-terminal truncated mGluR5. MPEP was an inverse agonist and 5MPEP did not activate or antagonize the truncated mutant. B, 5MPEP antagonized VU-29-induced IP accumulation through truncated mGluR5. Error bars represent S.E.M.
VU-29 Is Selective for mGluR5 Relative to Other mGluR Subtypes. Discovery of VU-29 as an allosteric potentiator of mGluR5 with low nanomolar potency provides one of the most potent allosteric potentiators of mGluR5 to date. The potency and solubility properties of this compound make VU-29 well-suited to use in functional studies aimed at determining the physiological effects of allosteric potentiators of mGluR5. However, before using this compound to probe mGluR5 function, it is important to determine the selectivity of VU-29 for mGluR5 relative to other mGluR subtypes. We reported previously that this compound is without allosteric potentiator activity on mGluR1 (Hemstapat et al., 2006). We now determined the effects of this compound on mGluR2 and mGluR4 as representative members of the other major subgroups of mGluRs (groups II and III, respectively). For measurement of responses to activation of mGluR2 and mGluR4, these receptors were cotransfected with chimeric G protein-Gqi5, which allows coupling of these receptors to activation of phospholipase C and calcium mobilization. A concentration of VU-29 (1 μM) that is capable of inducing maximal potentiation of mGluR5-mediated responses did not potentiate responses to activation of mGluR2 (Fig. 7A) or mGluR4 (Fig. 7B).
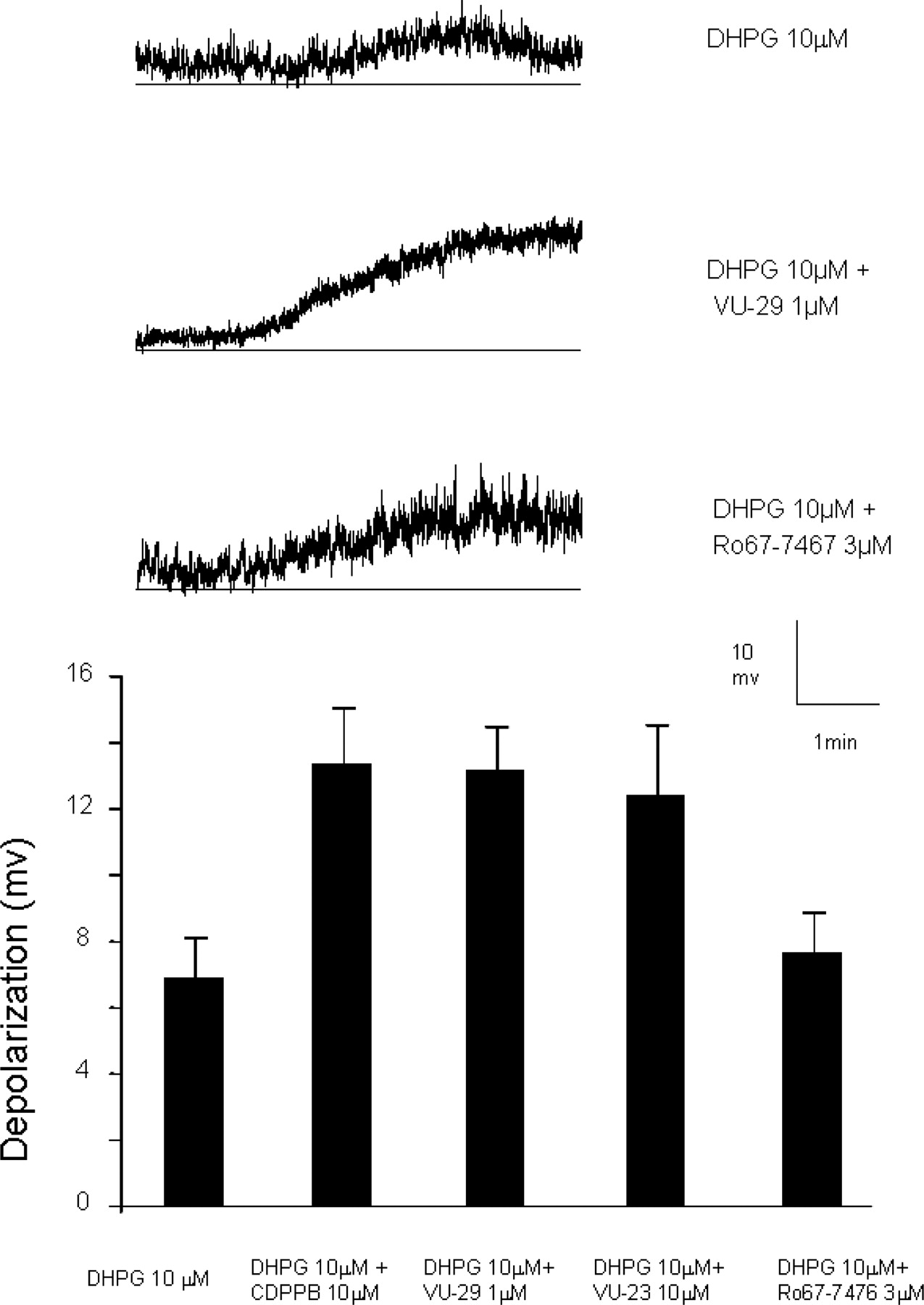
CDPPB and Its Analogs Potentiate Excitatory Effects of DHPG on Neurons in the STN Neurons but Not the SNr. Previous studies reveal that the metabotropic receptor subtypes mGluR5 and mGluR1 are postsynaptically localized on neurons in the STN and SNr. Activation of mGluR5 by DHPG elicits depolarization in STN neurons, which can be blocked by the mGluR5-selective antagonist MPEP (Awad et al., 2000), whereas mGluR1 plays a predominant role in regulating activity of neurons in the SNr (Marino et al., 2001). However, both of these neuronal populations express both mGluR1 and mGluR5, and previous studies suggest that each of these receptor subtypes can contribute to depolarization of either neuronal population under certain pathological conditions such as dopamine depletion (Marino et al., 2002). Thus, although it is possible that selective mGluR5 potentiators will selectively potentiate responses that are normally mediated by the respective mGluR subtype, it is also possible that these compounds could recruit mGluR5 activity in neurons in which this receptor is present but does not normally contribute to neuronal depolarization. Whole-cell recordings were performed in STN and SNr neurons in rat midbrain slices to determine whether CDPPB and its analogs selectively potentiate mGluR5-mediated responses in STN neurons but exert no effect on DHPG-induced depolarization of neurons in the SNr. Consistent with previous reports, bath application of 10 μM DHPG induced a depolarization in STN neurons (6.9 ± 1.2 mV, n = 6 cells; Fig. 8), and CDPPB (10 μM) enhanced the DHPG-induced responses (13.4 ± 1.5 mV, n = 8 cells; Fig. 8). The higher-potency CDPPB analog VU-29 (1 μM) had no effect on the membrane potential of STN neurons during the 10- to 15-min application period. However, in the presence of 1 μM VU-29, DHPG (10 μM) elicited stronger depolarization in STN neurons (13.2 ± 1.3 mV, n = 8 cells; Fig. 8) compared with the effect of DHPG (10 μM) alone. In addition, VU-23, the 3-nitro analog of CDPPB, potentiated DHPG-induced responses in STN neurons at a concentration of 10 μM (12.4 ± 2.1 mV, n = 7 cells). In contrast to CDPPB and its analogs, the mGluR1-selective allosteric potentiator Ro 67-7476 (Knoflach et al., 2001) (3 μM) did not potentiate DHPG-induced (10 μM) depolarization in STN neurons (7.64 ± 1.23 mV, n = 9 cells; Fig. 8), suggesting that the mGluR1 potentiator does not recruit mGluR1-mediated depolarization of these cells and that the mGluR5-mediated response in STN neurons is specifically enhanced by the mGluR5 allosteric modulators.

Single point mutation that eliminates radiolabeled MPEP binding also eliminates CDPPB- or VU-29-induced potentiation of mGluR5-mediated calcium mobilization in transiently transfected HEK293A cells. A, CDPPB analogs shifted glutamate concentration-response curves of wild-type rmGluR5 2.8 ± 0.6-fold (CDPPB, 1 μM) and 2.5 ± 0.5-fold (VU-29, 200 nM) to the left, respectively. B, CDPPB analogs shifted glutamate concentration-response curves of rmGluR5(M801T) 3.16 ± 0.91-fold (CDPPB, 1 μM) and 3.55 ± 0.54-fold (VU-29, 200 nM) to the left. C, CDPPB (1 μM) and VU-29 (200 nM) did not shift the glutamate concentration-response curves of rmGluR5(A809V). Concentration-response curves were generated from three or four independent experiments performed in duplicate. Error bars represent S.E.M.

VU-29 does not potentiate mGluR1-, -2-, or -4-mediated responses. A concentration of VU-29 that maximally potentiates mGluR5 responses (0.5 μM) neither potentiates nor antagonizes mGluR1- (A), mGluR2- (B), and mGluR4-mediated (C) responses in transiently transfected HEK 293A cells as measured using the calcium mobilization assay. Concentration-response curves were generated from three to four independent experiments performed in duplicate. Error bars represent S.E.M.
We next determined the effect of CDPPB analogs on DHPG-induced depolarization of SNr neurons, a response that is normally mediated exclusively by mGluR1 (Marino et al., 2001). Consistent with previous reports (Marino et al., 2001), DHPG (3 μM) induced a depolarization in SNr neurons (5.45 ± 0.93 mV, n = 6; Fig. 9), and the DHPG-induced response was blocked by the mGluR1-selective antagonist 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester (data not shown). In contrast to its effects in STN neurons, VU-29 (1 μM) did not potentiate DHPG-induced (3 μM) depolarization in SNr neurons (7.7 ± 1.9 mV, n = 11 cells; Fig. 9). However, the mGluR1 potentiator Ro 67-7476 (3 μM) enhanced DHPG-induced (3 μM) depolarization in SNr neurons (13.3 ± 2.7 mV, n = 7 cells; Fig. 9). Taken together, these data indicate that mGluR5 allosteric modulator CDPPB and its analogs act as potent and selective positive allosteric modulators of native mGluR5 in STN neurons, whereas mGluR1 allosteric modulator Ro 67-7476 selectively potentiates mGluR1-mediated responses in SNr neurons.

CDPPB and its analogs potentiate DHPG-induced depolarization in STN neurons. A, representative traces show depolarization elicited by application of DHPG (10 μM) alone, DHPG plus VU-29 (1 μM), and DHPG plus Ro 67-7476 (3 μM) in STN neurons. B, bar graph (mean ± S.E.M.) illustrates depolarization values elicited by DHPG (n = 6 cells), DHPG plus CDPPB (n = 8 cells), DHPG plus VU-29 (n = 8 cells), DHPG plus VU-23 (n = 7 cells), and DHPG in the presence of plus Ro 67-7476 (n = 9 cells) in STN neurons. *, p < 0.01, Student's t test.
Discussion
Three families of mGluR5 selective allosteric potentiators have now been reported from distinct structural groups. These are represented by DFB, CPPHA, and CDPPB (O'Brien et al., 2003, 2004; Kinney et al., 2005). DFB and CDPPB inhibit binding of ligands to the site labeled by allosteric antagonists, such as [3H]methoxyPEPy and [3H]MPEP. However, CPPHA does not displace binding to this site, suggesting that allosteric potentiator activity does not always require interaction with the MPEP site. Furthermore, we reported recently that three structurally distinct families of allosteric potentiators of mGluR1 do not share a common binding site with allosteric antagonists at this receptor (Hemstapat et al., 2006). Finally, the potency of CD-PPB at displacing antagonist binding to the MPEP site is considerably lower than the potency of this compound as an mGluR5 allosteric potentiator. Together, these data raise the question of whether displacement of [3H]methoxyPEPy binding is important for the allosteric potentiator activity of this compound. The present results provide strong evidence that CDPPB inhibits allosteric antagonist binding to the MPEP site in a manner consistent with a competitive interaction and that interaction with this site is important for allosteric potentiator activity. Scatchard analysis reveals the competitive nature of CDPPB interactions with this site. This is consistent with the hypothesis that CDPPB is not binding to a separate site to allosterically reduce [3H]methoxyPEPy binding (Limbird, 1996). Furthermore, our results show that there is a close correlation between the apparent affinities of several structural CDPPB analogs at the MPEP site and their activities as allosteric potentiators of mGluR5, suggesting that these two activities are interrelated. In addition, point mutations that reduce binding of MPEP and related compounds to the allosteric mGluR5 antagonist site also reduce the ability of CDPPB and its analogs to potentiate mGluR5 responses. Taken together, these data suggest that CDPPB and its analogs share a common or partly overlapping binding site with mGluR5 allosteric antagonists and that binding to this site is important for both the activity of CDPPB and related allosteric potentiators and previously known allosteric mGluR5 antagonists.

CDPPB analogs do not potentiate DHPG-induced depolarization in SNr neurons. A, representative traces showing depolarization of SNr neurons by application of DHPG (3 μM), DHPG (3 μM) plus Ro 67-7476 (3 μM), and DHPG plus VU-29 (1 μM) in SNr neurons. B, bar graph (mean ± S.E.M.) illustrates depolarization of SNr neurons by DHPG (n = 6 cells), DHPG plus Ro 67-7476 (n = 7 cells), and DHPG in the presence of VU-29 (n = 11 cells). *, p < 0.01, Student's t test.
Given the finding that interaction with the MPEP site is critical for allosteric potentiator activity, it is interesting that allosteric potentiators of mGluR5 consistently have higher potencies at potentiating responses to glutamate than their apparent affinities at the MPEP binding site. This is the case in multiple systems, including astrocytes studied here, cell lines stably expressing mGluR5 (Kinney et al., 2005), and cells transiently transfected with mGluR5 (Y. Chen and P. J. Conn, unpublished data). Thus, full allosteric potentiator activity can be achieved with concentrations of allosteric potentiators that displace a relatively small fraction of ligand binding to the MPEP sites. It is noteworthy that CDPPB can also directly activate mGluR5 in some systems. For instance, unlike the situation in astrocytes, we reported previously that higher concentrations of this compound can directly activate mGluR5, as measured by calcium fluorescence, in the absence of glutamate in cell lines stably expressing mGluR5 (Kinney et al., 2005). It is interesting that the EC50 value for direct activation of mGluR5 by CDPPB in the absence of glutamate is in the low micromolar range, which is closer to the CDPPB apparent Ki value at mGluR5. We have observed a similar activation of mGluR5-mediated calcium responses by CDPPB and its analogs in another stable cell line with consistently lower potencies relative to potentiation of responses to glutamate in the same cell line (Y. Chen, A. Rodriguez, and P. J. Conn, unpublished data). Likewise, we report here that VU-29 directly activates a truncated mutant form of mGluR5 in which the extracellular glutamate binding domain has been deleted. The EC50 value of VU-29 at activating the truncated receptor is similar to its apparent Ki value at the allosteric MPEP site. The simplest explanation for the difference between modulator potency when tested against glutamate and modulator affinity for the free receptor is that the former parameter is influenced by the positive cooperative interaction between the modulator and glutamate when both occupy the receptor simultaneously. The greater the positive cooperativity, the greater the degree of leftward shift in modulator potency. In contrast, when the modulator is tested on its own as an agonist, the potency derived from this latter type of experiment will simply reflect the affinity of the modulator for the free receptor and the strength of stimulus-response coupling. Because CDPPB is a very weak agonist in its own right, coupling efficiency is very low, and hence, the potency of CDPPB as an agonist should be close to its binding affinity.
Current studies indicate that there are multiple allosteric potentiation sites on mGluR5. Among the current three families of mGluR5 allosteric potentiators, DFB has been shown to interact at the MPEP binding site, as evidenced by competitive [3H]methoxyPEPy binding and by point mutations that eliminate [3H]MPEP binding and reduce DFB potentiation (O'Brien et al., 2003; Chen et al., 2004). Here, we have confirmed that the CDPPB series of allosteric potentiators also acts through interaction with the same site as MPEP. Giving the fact that CPPHA does not inhibit [3H]methoxyPEPy binding to its site up to 100 μM (O'Brien et al., 2004), it is clear that CPPHA does not interact with the MPEP binding site. Thus, we propose that there are multiple allosteric potentiator sites on mGluR5. Allosteric potentiators that bind to different sites may regulate mGluR5 activity differentially. A recent study has shown that CPPHA and DFB have different modulatory profiles on mGluR5-mediated extracellular signal-regulated kinases 1 and 2 phosphorylation in secondary cultured rat cortical astrocytes (Zhang et al., 2005). Based on this, it is possible that allosteric modulators that potentiate activity of a single receptor by different mechanisms may have distinct physiological effects. The finding that the CDPPB series of allosteric potentiators act through a shared binding site with MPEP does not necessarily imply that CDPPB analogs interact with identical amino acid residues as MPEP. It is more likely that they are interacting through binding to different amino acids in a largely overlapping pocket. Several residues in transmembranes 3, 6, and 7 have been mapped out as crucial interaction sites for MPEP (Pagano et al., 2000; Malherbe et al., 2003). However, the exact amino acid residues that constitute the binding pockets for MPEP or CDPPB are not currently known.
It is interesting that we have now identified ligands that interact with the MPEP site that have a range of activities from allosteric antagonists to allosteric potentiators and include neutral ligands that interact with this allosteric site but have no intrinsic activity. This is in some ways analogous to the range of activities of ligands at orthosteric sites that includes agonists, inverse agonists, and neutral antagonists. However, it is interesting to note that compounds within a single chemical series that span this entire range of activity seem to be rare. For instance, an analysis of the effects of almost 50 analogs of CDPPB has not revealed any compounds in this series that act as allosteric antagonists or neutral ligands (de Paulis et al., 2006; Hemstapat et al., 2006). Likewise, many members of the CDPPB series can also act as allosteric potentiators of mGluR1, but none of these compounds has neutral or allosteric antagonist activity at mGluR1 (Hemstapat et al., 2006). Furthermore, analysis of analogs of MPEP has yielded many allosteric antagonists and a small number of neutral ligands but no allosteric potentiators in the MPEP series (Gasparini et al., 1999; Alagille et al., 2005a,b; Rodriguez et al., 2005; Iso et al., 2006). This suggests clear differences in the structural requirements of different activities at allosteric sites on mGluRs. However, it is also important to note that the benzaldazine series of compounds, exemplified by 3,3′-difluorobenzaldazine (DFB), also interacts with the MPEP site and includes closely related members that act as allosteric antagonists, allosteric potentiators, and neutral ligands (O'Brien et al., 2003).
Discovery of VU-29 as an allosteric potentiator of mGluR5 with nM potency provides a useful tool for studying the physiological impact of selective potentiation of this receptor subtype. It is noteworthy that VU-29 was found to be selective for mGluR5 relative to mGluRs 1, 2, and 4. In recent years, it has become clear that multiple neuronal populations express both mGluR1 and mGluR5 but that these receptors have distinct physiological effects. For instance, neurons in the STN express both of these mGluR subtypes, but under normal conditions, only mGluR5 participates in depolarization of STN neurons by the mGluR1/5 agonist DHPG. However, under some conditions, activation of mGluR1 can substitute for mGluR5 and depolarize STN neurons in response to DHPG (Awad et al., 2000; Marino et al., 2001, 2002a). The opposite is true in SNr projections neurons. Only mGluR1 is involved in DHPG-induced depolarization under normal conditions, but mGluR5 is also present and can induce calcium transients and can depolarize these cells under some conditions (Marino et al., 2001, 2002a). In theory, it is possible that VU-29 and other allosteric potentiators could selectively potentiate responses to DHPG that are normally mediated by mGluR5. However, it is also possible that by potentiating mGluR5 activity, these compounds could also lead to coupling of mGluR5 to responses in which it does not normally participate in cells that express this receptor. The finding that VU-29 and related mGluR5 potentiators enhanced DHPG-induced depolarization of STN neurons, but not of SNr neurons, and that the mGluR1 potentiator Ro 67-7476 had the opposite effects suggests that, in these cells, these compounds potentiate the normal response to mGluR5 or mGluR1 activation but maintain the normal physiological roles of these receptor subtypes. This is of critical importance in considering the physiological impact of these compounds in intact systems. In future studies, these compounds will provide excellent tools for understanding the impact of selective potentiation of mGluR1- and mGluR5-mediated responses in a range of neuronal populations and circuits.
Acknowledgments
We thank Dr. Vsevolod V. Gurevich and Dr. Yongqin Zhang for assistance with the experimental design and interpretation of the Scatchard analysis studies. We thank Dr. Jinling Xie for the help with the mutagenesis and Dr. Samir Saleh for the synthesis of VU-137. Finally, we thank Dr. Arthur Christopoulos (Monash University, Melbourne, Australia) for helpful discussions of the data and comments on the manuscript.
Footnotes
-
This work is supported by grants from the National Institute of Mental Health, National Institute of Neurological Disorders and Stroke, the Stanley Foundation, and the National Alliance for Research on Schizophrenia and Affective Disorders. Vanderbilt is a center within the National Institutes of Health-supported Molecular Libraries Screening Centers Network.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.106.032425.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; CDPPB, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide; CPPHA, N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide; DFB, 3,3′-difluorobenzaldazine; DMEM, Dulbecco's modified Eagle's medium; DHPG, (S)-3,5-dihydroxyphenylglycine; FBS, fetal bovine serum; HBSS, Hanks' balanced salt solution; 7TM, seven-transmembrane; MPEP, 2-methyl-6-(phenylethynyl)pyridine; methoxyPEPy, 3-methoxy-5-(2-pyridinylethynyl)pyridine; mGluR, metabotropic glutamate receptor; SNr, substantia nigra pars reticulata; STN, subthalamic nucleus; 5MPEP, 5-methyl-2-(phenylethynyl)pyridine, VU-29, 4-nitro-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide; HEK, human embryonic kidney; IP, inositol phosphate; ACSF, artificial cerebrospinal fluid; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; Ro 67-7476, (S)-2-(4-fluorophenyl)-1-(toluene-4-sulfonyl)pyrrolidine.
- Received November 7, 2006.
- Accepted February 13, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}