


Abstract
Parkinson's disease (PD) is caused by the death of dopamine neurons in the basal ganglia and results in motor symptoms such as tremor and bradykinesia. Activation of metabotropic glutamate receptor 4 (mGluR4) has been shown to modulate neurotransmission in the basal ganglia and results in antiparkinsonian effects in rodent PD models. N-Phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide (PHCCC) is a positive allosteric modulator (PAM) of mGluR4 that has been used to further validate the role of mGluR4 in PD, but the compound suffers from a lack of selectivity, relatively low potency, and poor solubility. Via high-throughput screening, we discovered more than 400 novel PAMs of mGluR4. Compounds derived from a novel chemical scaffold were characterized in vitro at both rat and human mGluR4 using two distinct assays of mGluR4 function. The lead compound was approximately 8-fold more potent than PHCCC, enhanced the potency of glutamate at mGluR4 by 8-fold, and did not show any significant potentiator or antagonist activity at other mGluR subtypes. Resolution of the regioisomers of the lead revealed that the cis regioisomer, (±)-cis-2-(3,5-dichlorphenylcarbamoyl)cyclohexanecarboxylic acid (VU0155041), contained the majority of the mGluR4 PAM activity and also exhibited partial agonist activity at mGluR4 at a site that was distinct from the glutamate binding site, suggesting that this compound is a mixed allosteric agonist/PAM of mGluR4. VU0155041 was soluble in an aqueous vehicle, and intracerebroventricular administration of 31 to 316 nmol of VU0155041 dose-dependently decreased haloperidol-induced catalepsy and reserpine-induced akinesia in rats. These exciting results provide continued support for mGluR4 as a therapeutic target in PD.
Metabotropic glutamate receptors (mGluRs) play important roles in a broad range of central nervous system functions and have therapeutic potential in a variety of neurological and psychiatric disorders (Niswender et al., 2005). mGluRs are G protein-coupled receptors (GPCRs) classified into three major groups, groups I, II, and III, based on their sequence homology, signal transduction profile, and ligand binding specificity. The group III mGluRs (mGluRs 4, 6, 7, and 8) are coupled to Gi/o G proteins and are predominantly expressed presynaptically where they regulate the release of both glutamate and GABA (Conn and Pin, 1997).
Three group III mGluR subtypes, mGluRs 4, 7, and 8, are expressed in the basal ganglia, a group of brain nuclei that are involved in the control of motor function and are critical to the motor deficits observed in Parkinson's disease (PD). It is interesting that activation of mGluR4 reduces transmission at a key basal ganglia synapse (the striatopallidal synapse) that is believed to be overactive in patients with PD, and this effect is lost in mGluR4 knockout animals (Valenti et al., 2003). Furthermore, intracerebroventricular (icv) injection of the nonselective group III agonist l-(+)-2-amino-4-phosphonobutryic acid (l-AP4) can reverse motor defects in preclinical models of PD (Valenti et al., 2003), suggesting that activation of a group III mGluR may reduce motor symptoms of PD. In addition, previous studies have led to the hypothesis that activation of mGluR4 could be useful as a disease-modifying strategy for PD by reducing the release of glutamate and excitotoxicity in degenerating substantia nigra neurons (Valenti et al., 2005).
Unfortunately, the high conservation of the glutamate binding site makes it difficult to develop highly selective orthosteric ligands for individual mGluR subtypes. To improve specificity for the individual receptors, we and others have developed ligands that interact at sites other than the orthosteric (glutamate) binding site. For mGluR4, these efforts have resulted in the discovery of N-phenyl-7-(hydroxyimino)cyclopropa[b]-chromen-1a-carboxamide (PHCCC), a positive allosteric modulator (PAM) of mGluR4 (Maj et al., 2003; Marino et al., 2003). Alone, PHCCC has no observed effect on mGluR4 but potentiates mGluR4 responses to glutamate. It is encouraging that icv infusion of PHCCC in rats has been shown to reverse reserpine-induced akinesia (Marino et al., 2003), a rodent model of PD. Furthermore, PHCCC has been shown to reduce dopamine neuron degeneration in the substantia nigra in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine preclinical model of PD (Battaglia et al., 2006). These promising results suggest that mGluR4 is a candidate for both symptomatic and disease-modifying treatment in PD.
Although PHCCC represents a major breakthrough, this compound suffers from low potency (approximately 4 μM in vitro; Marino et al., 2003), poor aqueous solubility, and is also an mGluR1 antagonist with similar potencies at mGluR4 and mGluR1 (Annoura et al., 1996). Thus, there is a critical need for more potent and selective mGluR4 ligands that can be used to further explore the physiological roles of this receptor and the potential of mGluR4 activation for treatment of PD.
In an effort to improve the properties of PHCCC, we created a small library of analogs and tested them for their ability to potentiate the glutamate response at mGluR4. Unfortunately, none of these synthesized compounds were more potent or effective than PHCCC. This prompted us to undertake a high-throughput screening (HTS) approach to search for novel mGluR4 PAMs. We identified a number of new ligands with the ability to potentiate both rat and human mGluR4 responses, and one such scaffold is highlighted here. The lead from this cluster of structurally related compounds, VU0155041, is highly selective for mGluR4, exhibits an improved potency at mGluR4 versus PHCCC, and shows a significant improvement in aqueous solubility. Furthermore, this lead compound possesses intrinsic allosteric agonist activity at mGluR4 in vitro, a property which further differentiates it from PHCCC. It is noteworthy that this novel mGluR4 allosteric activator has robust activity in two rodent models of PD. Discovery of these compounds provides fundamental new insights into the pharmacological properties of allosteric modulators of mGluR4 and provides strong support for the hypothesis that selective activation of this receptor has potential as a novel therapeutic strategy for the treatment of PD.
Materials and Methods
Cell Line Creation and Culture of the Human mGluR4/Gqi5/CHO Line
Human mGluR4 (hmGluR4)/CHO cells were stably transfected with the chimeric G protein Gqi5 (Conklin et al., 1993) in pIRESneo3 (Invitrogen, Carlsbad, CA), and single neomycin-resistant clones were isolated and screened for mGluR4-mediated calcium mobilization using the method described below. hmGluR4/CHO cells were cultured in 90% Dulbecco's modified Eagle's medium (DMEM), 10% dialyzed fetal bovine serum (FBS), 100 U/ml penicillin/streptomycin, 20 mM HEPES, pH 7.3, 1 mM sodium pyruvate, 2 mM glutamine, 400 μg/ml G418 sulfate, 20 μM proline (Mediatech, Inc., Herndon, VA), and 5 nM methotrexate (Calbiochem, EMD Chemicals, Gibbstown, NJ). Culture of human embryonic kidney (HEK) 293 cell lines coexpressing rat mGluR4 and the G protein-regulated inwardly rectifying K+ channel (GIRK) have been described in detail elsewhere (Niswender et al., 2008). Culturing conditions for other mGluR cell lines are described below. All cell culture reagents were purchased from Invitrogen unless otherwise noted.
Primary High-Throughput Screening
Assays were performed within Vanderbilt University's High-Throughput Screening Center. Human mGluR4/Gqi5/CHO cells (30 × 103 cells/20 μl/well) were plated in black-walled, clear-bottomed, TC-treated, 384-well plates (Greiner Bio-One, Monroe, NC) in DMEM containing 10% dialyzed FBS, 20 mM HEPES, 100 U/ml penicillin/streptomycin, and 1 mM sodium pyruvate (plating medium). The cells were grown overnight at 37°C in the presence of 5% CO2. The next day, the medium was removed using a VSpin (Velocity 11, Menlo Park, CA) fitted with a modified bucket allowing the 384-well plate to be mounted inverted over a catch basin and spun at 80g for 10 s with 40% acceleration and deceleration.
The medium was replaced, using a Thermo Fisher Combi (Thermo Fisher Scientific, Waltham, MA), with 20 μl of 1 μM Fluo-4/acetoxymethyl ester (Invitrogen, Carlsbad, CA) prepared as a 2.3 mM stock in DMSO and mixed in a 1:1 ratio with 10% (w/v) Pluronic acid F-127 and diluted in assay buffer (Hanks' balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid; Sigma-Aldrich, St. Louis, MO) for 45 min at 37°C. Dye was removed using the VSpin and replaced, using a Combi, with 20 μl of assay buffer. Test compounds were transferred to daughter plates using an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA) and then diluted into assay buffer, using a Combi, to generate a 20 μM stock solution. Ca2+ flux was measured using the Functional Drug Screening System 6000 (FDSS; Hamamatsu, Tokyo, Japan). Baseline readings were taken (10 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm) and then 20 μl/well test compounds were added using the FDSS's integrated pipettor. For the primary screen, cells were incubated with test compounds (final concentration, 10 μM) for 2.5 min, and then an EC20 concentration of glutamate was applied; 2 min later, an EC80 concentration of glutamate was added. The overall assay protocol was automated using the instruments noted above integrated with a Thermo Fisher F3 robotic arm (Thermo Fisher Scientific) under the control of a Polara scheduler (Thermo Fisher Scientific). All data were recorded to instruments' local drives and later were migrated to a network drive. FDSS data were analyzed using a custom analysis application and were associated with unique compound identifiers based on liquid handler transfer logs and plate barcode readings captured by the Echo and by Polara. Potentiator “hits” were selected by comparing the amplitude of the responses at the time of EC20 addition plus and minus test compounds. Wells with responses that differed from vehicle wells by 3 S.D. were selected as hits for further study.
For initial concentration-response curve experiments, compounds were serially diluted 1:3 into 10-point concentration-response curves and were transferred to daughter plates using the Echo. Test compounds were again applied and followed by EC20 concentrations of glutamate. Curves were fitted using a four-point logistical equation using Microsoft XLfit (IDBS, Bridgewater, NJ). Subsequent confirmations of concentration-response parameters were performed using independent serial dilutions of source compounds, and data from multiple days experiments were integrated and fit using a four-point logistical equation in Prism (GraphPad Software, Inc., San Diego, CA).
Confirmation/Selectivity Studies
Rat M1 Muscarinic Receptor. CHO cells expressing the rat M1 muscarinic receptor were purchased from the American Type Culture Collection (Manassas, VA) and cultured in Ham's F-12 medium with 10% FBS, 20 mM HEPES, and 50 μg/ml G418. For calcium assays, cells were plated at 104 cells/well in plating medium, and dye loading was as above for mGluR4. Compounds were added 2.5 min before an EC20 concentration of the muscarinic agonist carbachol followed 2 min later by an EC80 concentration of carbachol. Raw data from the FDSS were imported into Microsoft Excel (Microsoft, Redmond, WA). Maximum change in fluorescence, compared with vehicle control wells, was calculated in the presence of the EC20 agonist concentration.
Rat mGluRs 1 and 5. Rat mGluR1 and 5 cells were cultured as described by Hemstapat et al. (2007). Calcium fluorescence assays were used for counterscreening rat mGluR1/baby hamster kidney (mGluR1/BHK) and rat mGluR5/HEK cells using a similar triple-addition protocol using appropriate EC20 and EC80 glutamate concentrations for each receptor, the exceptions being that cells were plated at 15 and 20 × 103 cells/well in black-walled, poly(d-lysine)-coated 384-well plates (Greiner Bio-One) in plating medium, respectively, and calcium assays proceeded as above. Maximum calcium fluorescence, compared with control, was calculated for the EC20 and EC80 peaks, respectively, after exporting raw FDSS data to Microsoft Excel.
Human mGluR2. Membrane preparation and GTPγS binding assays for mGluR2 were performed as described by Hemstapat et al. (2007) and stored as frozen aliquots. Membranes were thawed and homogenized using a glass homogenizer in ice-cold binding buffer containing 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 150 mM NaCl, 1 mM EDTA, 10 μg/ml saponin, and 1 μM GDP. Assay mixtures contained 10 μg of membrane protein, test compound, glutamate, 0.1 nM [35S]GTPγS, and assay buffer to yield a total volume of 100 μl. Nonspecific binding was determined in the presence of 10 μM unlabeled GTPγS. Assay mixtures were incubated at room temperature with shaking for 60 min, and the reaction was terminated by rapid filtration through Unifilter-96 GF/B filter plates (presoaked with ice-cold binding buffer), and the filter plates were washed three times with ice-cold binding buffer using a 96-well Brandel harvester (Brandel Inc., Gaithersburg, MD). Filter plates were dried and filled with 40 μl MicroScint-20, and radioactivity was counted using a TopCount NXT microplate scintillation counter (PerkinElmer Life and Analytical Sciences, Waltham, MA).
Rat mGluRs 4, 7, and 8. Compound activity at the rat group III mGluRs was assessed using thallium flux through GIRK channels, a method that has been described in detail by Niswender et al. (2008). These cell lines were grown in growth media containing 45% DMEM, 45% Ham's F-12, 10% FBS, 20 mM HEPES, 2 mM l-glutamine, antibiotic/antimycotic, nonessential amino acids, 700 μg/ml G418, and 0.6 μg/ml puromycin at 37°C in the presence of 5% CO2. In brief, mGluR4, -7, or -8 GIRK cells were plated into 384-well, black-walled, clear-bottomed, poly(d-lysine)-coated plates at a density of 15 × 103 cells/20 μl/well in plating medium and incubated overnight at 37°C in the presence of 5% CO2. The following day, the medium from the cells and 20 μl/well of 1.7 μM concentration of the indicator dye BTC-AM (Invitrogen) in assay buffer was added. Cells were incubated for 1 h at room temperature and the dye was replaced with 20 μl/well of assay buffer. For these assays, compounds were added at two times the final concentration, and then 2.5 min later, either an EC20 or EC80 concentration of glutamate (mGluR4 or 8) or l-AP4 (mGluR7) was added using the FDSS 6000. Agonists were diluted in thallium buffer (125 mM sodium bicarbonate, 1 mM magnesium sulfate, 1.8 mM calcium sulfate, 5 mM glucose, 12 mM thallium sulfate, and 10 mM HEPES) at five times the final concentration to be assayed. Five frames of data were collected (excitation, 470 ± 20 nm; emission, 540 ± 30 nm) at 0.5 Hz before compound addition. Data collection continued at 0.5 Hz until 10 s before agonist addition, when the rate was increased to 1 Hz for 2 min after agonist addition. Data were analyzed as described by Niswender et al. (2008).
Striatal Slice Electrophysiology
Coronal striatal slices were prepared from Sprague-Dawley rats (postnatal day 14-16). Rats were anesthetized with isoflurane and decapitated. The brain was rapidly removed from the skull and submerged in ice-cold modified artificial cerebrospinal fluid (ACSF), which was oxygenated with 95% O2/5% CO2 and composed of 230 mM sucrose, 2.5 mM KCl, 0.5 mM CaCl2, 8 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM d-glucose. The brain was then blocked in the coronal plane, glued to the stage of a vibratome (Vibratome, St. Louis, MO) that was filled with ice-cold modified ACSF, and cut at a thickness of 290 μm. Slices were then incubated in oxygenated normal ACSF (126 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1.5 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM d-glucose) at 31 to 32°C for 0.5 h and maintained at room temperature afterward until transferred individually to a fully submerged recording chamber, which was continuously perfused with oxygenated ACSF at ∼30°C.
Whole-cell recordings were made from visually identified medium spiny neurons in the dorsolateral striatum under an Olympus BX50WI upright microscope equipped with a 40× water immersion objective, Hoffman optics, and video system (Olympus, Lake Success, NY). A MultiClamp amplifier (Molecular Devices, Sunnyvale, CA) was used for voltage-clamp recordings. Patch pipettes were prepared from borosilicate glass (World Precision Instrument, Sarasota, FL) using a Narashige vertical patch-pipette puller (Narashige, Tokyo, Japan) and filled with the pipette solution containing 125 mM cesium methanesulfonate, 5 mM NaCl, 10 mM tetraethylammonium chloride, 10 mM HEPES, 0.1 mM EGTA, 5 mM QX-314, 4 mM Mg-ATP, 0.3 mM Na-GTP, and 10 mM phosphocreatine. The pH of the pipette solution was adjusted to 7.3 with 1 M CsOH, and osmolality was adjusted to 290 to 295 mmol/kg. The patch pipette had resistance of 4 to 6 MΩ when filled with the above solution. N-Methyl-d-aspartate (NMDA) receptor-mediated currents were induced by pressure ejection of 1 mM NMDA to the soma of the recorded neurons through a patch pipette using a Picospritzer II (General Valve, Fairfield, NJ). The neuron was typically voltage-clamped at -60 mV. Tetrodotoxin (1 μM) was routinely included in the perfusate to block voltage-gated sodium channels. Data were acquired using a Digidata 1322A interfaced to a PC computer equipped with pClamp 9.2 software (Molecular Devices), analyzed using Clampfit and Microsoft Excel, and presented as percentage of control value.
Reversal of Haloperidol-Induced Catalepsy and Reserpine-Induced Akinesia
Animals. Third ventricle cannulated (TVC) male Sprague-Dawley rats weighing between 225 and 255 g (Taconic Farms, Inc., Hudson, NY) were used for the behavioral studies and were maintained in accordance with American Association for the Accreditation of Laboratory Animal Care guidelines under a 12-h light/dark cycle (lights on at 6 AM, lights off at 6 PM) with free access to food and water. The experimental protocols, which were performed during the light cycle, were approved by the Institutional Animal Care and Use Committee of Vanderbilt University and conformed to the guidelines established by the National Research Council Guide for the Care and Use of Laboratory Animals.
Induction and Measurement of Catalepsy. Catalepsy was assessed using a horizontal bar placed 6 cm from the testing surface. The forepaws of each rat were placed gently on the bar with the body positioned at an angle of ∼45° to the testing surface. The latency in seconds required for the rat to remove one or both forepaws from the bar was manually measured. Any rat that remained on the bar between 45 and 60 s was considered to be cataleptic. TVC rats, randomly assigned to treatment groups, were injected with haloperidol (1.5 mg/kg i.p., dissolved in 0.2% lactic acid) and monitored for catalepsy 2 h later. Cataleptic rats were subsequently reexamined 15, 30, and 60 min after intracerebroventricular administration of either l-AP4 (100-1000 nmol/10 μl), VU0155041 (31 or 93 nmol/10 μl), or vehicle. l-AP4 was prepared in artificial cerebrospinal fluid (Harvard Apparatus, Holliston, MA). VU0155041 was dissolved in 1 N sodium hydroxide, brought to 8 ml with double-distilled water, pH adjusted to 7.4 with HCl, and then brought to final volume with double-distilled water.
Induction and Measurement of Akinesia. TVC rats were injected with reserpine (5 mg/kg, subcutaneously, dissolved in 1% acetic acid) and kept in their home cages for 2 h after injection. Activity was measured by placing rats in photocell activity cages (Hamilton-Kinder, Poway, CA) equipped with 16 × 16 infrared beams. After a 30-min baseline period, rats were given a single intracerebroventricular injection of either l-AP4 (100, 300, or 1000 nmol), VU0155041 (93 or 316 nmol), or corresponding vehicles, and motor activity was recorded for an additional 30 min.
Compounds
l-Glutamate, PHCCC, and Ro25-6981 were purchased from Tocris Bioscience (Ellisville, MO). l-AP4 was purchased from Ascent Scientific (Weston-SuperMare, UK). Haloperidol lactate was purchased from Abraxis (Schaumburg, IL). NMDA and reserpine were purchased from Sigma. The Vanderbilt High-Throughput Screening Center compound collection was obtained from ChemBridge Corporation (San Diego, CA) and ChemDiv, Inc. (San Diego, CA) and was stored in barcoded, 384-well, U-bottomed, standard volume polypropylene plates (Corning, Corning, NY). The plates were thermally sealed with peelable seals using a PlateLoc (Velocity 11). Groups of 10 plates were vacuum-packed in thermally sealed freezer bags (FoodSaver; Jarden Corporation, Cleveland, OH) and stored frozen at -80°C. Primary hits identified in the screen were reordered from ChemBridge or ChemDiv as 10 mM DMSO stocks; these orders were accompanied by NMR spectra to confirm compound identity. Compounds in Table 1 were then also confirmed by liquid chromatography/mass spectrometry at Vanderbilt University. Synthesis of PHCCC analogs, VU0155040, and VU0155041 and chiral resolution of VU0155041 are described in Supplemental Methods.
Potencies, maximal responses, and -fold shifts of glutamate concentration-response curves induced by structurally related compounds identified in the mGluR4 PAM HTS campaign
Potencies of compounds were determined in the presence of an EC20 concentration of glutamate at human or rat mGluR4 using either Gq15-mediated calcium mobilization or GIRK-mediated thallium flux. % Max represents the maximal potentiated EC20 response compared with the response generated in the presence of a maximal glutamate concentration alone. -Fold shift experiments were performed using a 10-point glutamate concentration-response curve with and without a 2.5-min preincubation with a 30 μM concentration of PAM. Values represent mean ± S.E.M., n = three to four independent replicates performed in triplicate. Compounds correspond to commercial compounds from either ChemDiv (CD) or ChemBridge (CB) and correspond as follows: 2a, CD K826-0011; 2b, CD 2267-0368; 2c, CB 5541532; 2d, CB 6542351; 2e, CB 5265419; 2f, CB 6149989; 2g, CB 5264082; 2h, CB 7307507.
Results
Lack of Structure-Activity Relationship for mGluR4 Potentiation among Synthesized PHCCC Analogs. To determine whether PHCCC represented a starting point to generate PAMs with higher potency and efficacy for potentiation of mGluR4 activity, we synthesized a series of compounds based on the PHCCC scaffold (Supplemental Table 1). These compounds were tested at 30 μM for the potentiation of an EC20 glutamate concentration response using a human mGluR4 CHO cell line in which we had stably transfected the chimeric G protein Gqi5 (Conklin et al., 1993). This chimera permits the coupling of Gi/o-coupled receptors to the phospholipase C pathway, resulting in calcium mobilization. As shown in Fig. 1, none of the synthesized compounds potentiated mGluR4 responses to the same extent as PHCCC. In addition to poor efficacy, full concentration-response curves revealed that none of the synthesized compounds exhibited improved potency compared with PHCCC (data not shown).
Novel Modulators of mGluR4 Were Identified via High-Throughput Screening. Although the current studies do not represent an exhaustive analysis of potential changes to the PHCCC scaffold, we turned to HTS to identify new chemical scaffolds with mGluR4 PAM activity. For these studies, we used the hmGluR4/Gqi5/CHO cell line described above and measured receptor-induced intracellular calcium mobilization using a kinetic-imaging plate reader that simultaneously monitors changes in fluorescence in each well of a 384-well microplate. Upon initiation of the screen, a baseline measurement was taken, and then either vehicle or a test compound (10 μM final nominal concentration) was added to hmGluR4/Gqi5 cells. After a 2.5-min incubation period, a submaximally effective (EC20) concentration of glutamate was added, and this was followed 2 min later by an EC80 concentration agonist addition.
Raw kinetic data from the screen were normalized by dividing all of the fluorescence readings of the trace by the minimum data point occurring 2 to 5 s before the EC20 glutamate concentration addition. This step corrected for well-to-well differences in cell number, dye loading, nonuniform illumination/imaging, and to permit retention of information for compounds that were slightly fluorescent or that induced subtle changes in the baseline trace. Vehicle, EC20, and EC80 controls were included on each plate, and data were analyzed on a plate-by-plate basis. As an example of the uniformity of the data for the EC20 window, control values (mean ± CV) for eight random plates, taken on different days throughout the screen, were 1.6 ± 0.3%, indicating that the signal was very uniform across plates and among wells. At least 20% of the plates were visually spot-checked to ensure the quality of the data and to validate these set points for hit-picking, prevent the loss of compounds with weak activity, and eliminate compounds giving apparent nonspecific or spurious signals such as compounds with obvious saturating fluorescence. Figure 2 shows example traces for either screening controls (Fig. 2A), a trace obtained in the presence of the control PAM PHCCC (Fig. 2B), or a trace obtained in the presence of a compound identified during the primary HTS (Fig. 2C). Approximately 16 × 104 compounds were screened in the primary screen, and 1490 “potentiator” hits were identified (0.9% hit rate). Of the primary hits, 1355 PAM hits (the remainder being unavailable for commercial reorder) were formatted into 10-point concentration-response curves and tested for concentration-dependent activity on mGluR4. Compounds were also screened against a CHO cell line expressing the M1 muscarinic receptor to determine whether their action was via a nonspecific mechanism. A total of 434 compounds were confirmed as having concentration-dependent PAM activity, producing a retest rate of approximately 32%. Sixty-five (15%) of the compounds potentiated (9 compounds) or antagonized (56 compounds) the response of M1-expressing cells to ACh, indicating that approximately 85% of the compounds were selective for mGluR4 over M1. Of these 434 PAMs, initial concentration-response curves indicated that 179 compounds exhibited a potency of less than approximately 5 μM, and 23 compounds were less than 1 μM in potency. An assessment of confirmed PAM hits from the screen quickly revealed that many of the compounds shared common chemical scaffolds; one of these scaffolds is highlighted here.
A Newly Identified mGluR4 PAM Cluster from the HTS Revealed a Robust Structure-Activity Relationship. The cluster chosen for further exploration, composed of eight HTS hits, was represented by a cyclohexyl amide moiety joined to a substituted phenyl ring (Table 1); the majority of the compounds also contained a carboxylic acid at position 1 of the cyclohexane. The potencies of compounds in this cluster were assessed at both human mGluR4 and rat mGluR4. For human mGluR4, the assay used was the chimeric G protein approach used for HTS. For the rat receptor, a cell line was used in which rat mGluR4 was coexpressed with GIRK. This assay relies on the activation of GIRK1/2 channels via the Gβγ subunits of Gi/o G proteins and exploits the ability of the GIRK channel to conduct ions of thallium through the channel pore in response to agonist activation of a GPCR (Niswender et al., 2008). One advantage of the assay is that it does not require cotransfection of a chimeric or promiscuous G protein to induce coupling to a non-native signaling pathway. Because the group III mGluRs have been shown to regulate GIRK in electrophysiological studies (Saugstad et al., 1996, 1997) and ion channels in neurons (Guo and Ikeda, 2005; Bertaso et al., 2006; Pelkey et al., 2006), presumably via Gβγ subunits, this technique represents a mechanism to screen or confirm activity of compounds using an assay that monitors activity through G proteins representative of those normally used by the receptor. This technique is also an easy and efficient method to examine an alternate signaling pathway downstream of mGluR4 and confirm activity of compounds as general mGluR4 PAMs.

The chemical modifications of the PHCCC scaffold depicted here do not result in improved potency or efficacy for potentiation of mGluR4 activity. Structures of synthesized compounds are shown in Supplemental Fig. 1. Chemical synthesis is described in Supplemental Methods. Compounds a to e and k to p were synthesized according to the method of Annoura et al. (1998). Compounds f to j, l, and q were synthesized from compound g by the same methods. Compound g was prepared from 2-hydroxyacetophenone as described in Silva et al. (1998). A 30 μM final concentration of each compound was added to human mGluR4/Gqi5 cells; 2.5 min later, a submaximal (EC20) concentration of glutamate was added, and changes in calcium-mediated fluorescence were measured. Results represent mean ± S.E.M. of three independent experiments performed in triplicate.
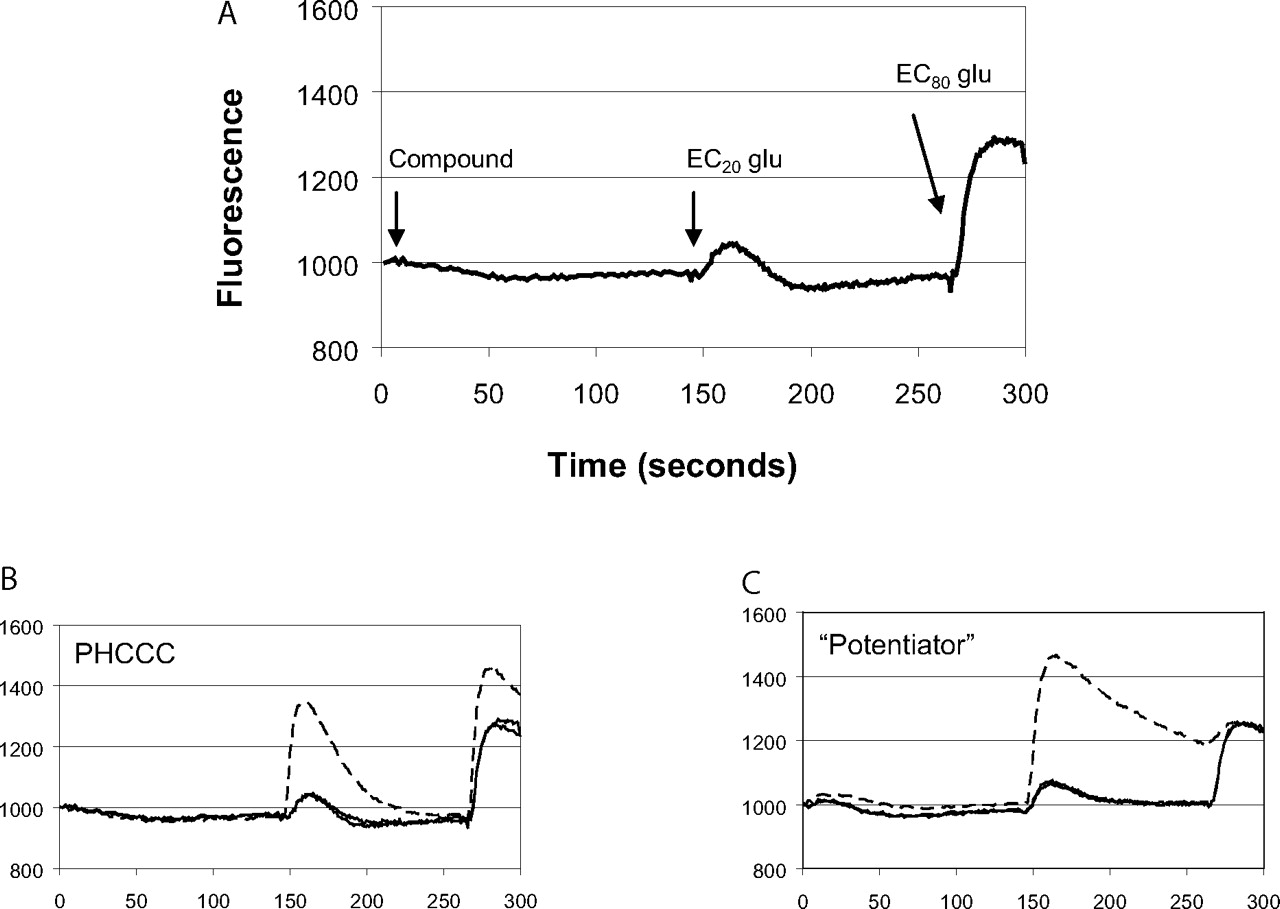
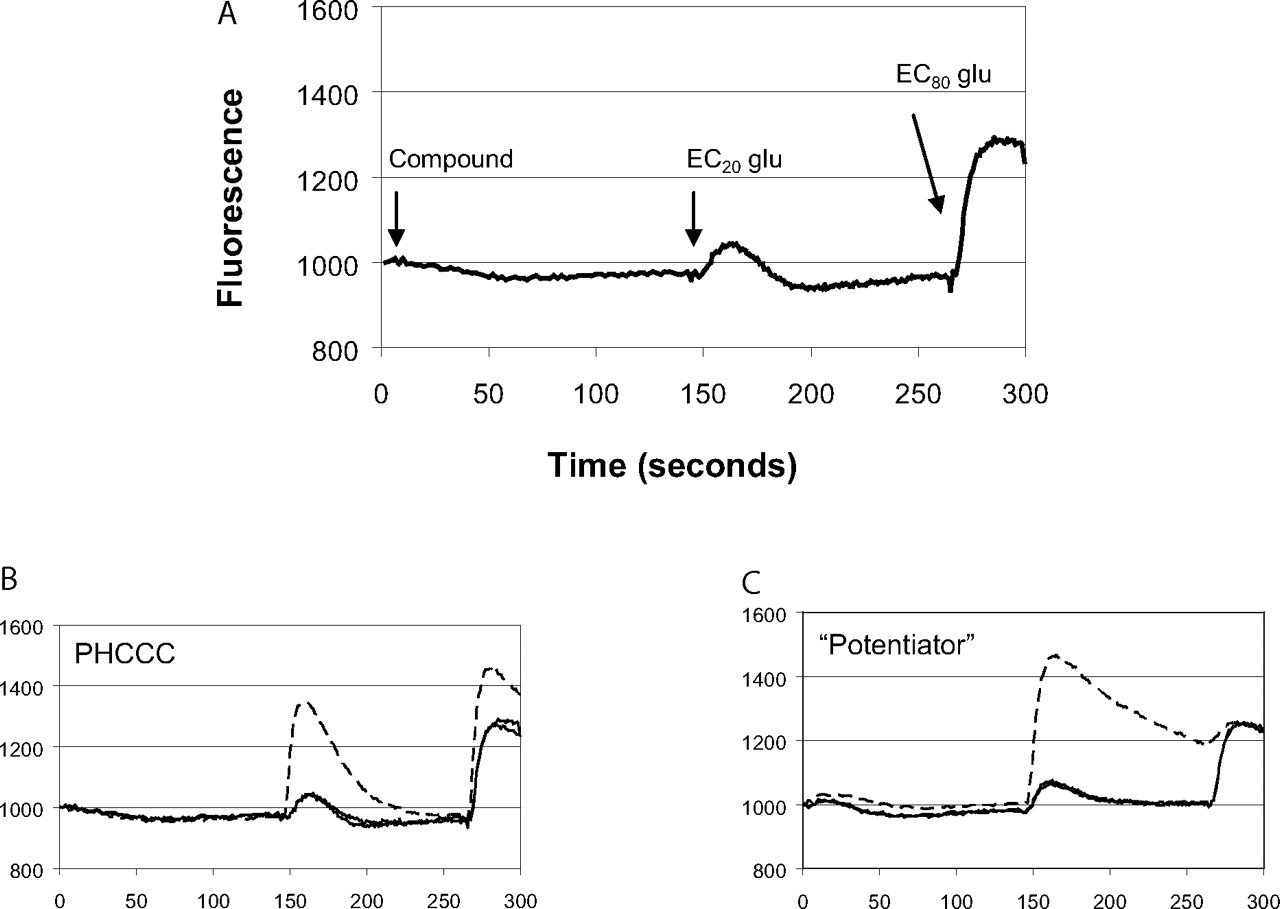
HTS assay design and fluorescence traces of potential mGluR4 PAMs measured during the HTS campaign. A, HTS assay design. Human mGluR4/Gqi5 cells were loaded with Fluo-4 calcium-indicator dye as described under Materials and Methods. A baseline fluorescence measurement was taken for 3 s and then either vehicle or a 10 μM concentration of compound was added. Approximately 2.5 min later (time, 146 s), an EC20 concentration of glutamate (2 μM final) was added followed at 266 s by an EC80 concentration of glutamate (20 μM final). B and C, representative traces of control compounds or compounds identified during HTS. B, control trace measured in the presence (broken line) or absence (solid line) of 10 μM PHCCC. C, trace observed in the presence of a novel compound (broken line) that potentiates the response of glutamate at mGluR4.

Novel PAM identified via HTS was significantly more potent than PHCCC. A and B, increasing concentrations of compound 2h or PHCCC were preapplied to either human mGluR4/Gqi5 cells (A) or rat mGluR4/GIRK cells (C); an EC20 concentration of glutamate was added approximately 2.5 min later, and responses were measured as described under Materials and Methods. Potencies in the calcium assay (mean ± S.E.M.) were the following: PHCCC, >10 μM; 2h, 750 ± 200 nM; and for the GIRK assay: PHCCC, 4.9 ± 1.3 μM; 2h, 560 ± 100 nM.
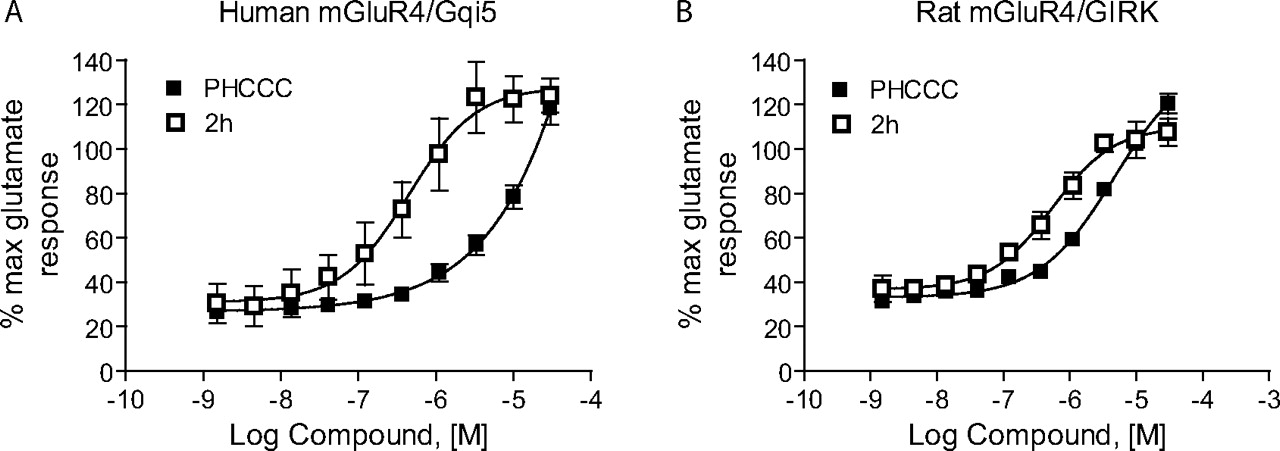
Table 1 shows the structures, potencies, and efficacies of compounds identified via HTS. Viewed from a structural standpoint, compounds 2e, 2g, and 2h were highly similar, with the positions of the dichloro-substitutions of 2h being preferred for both potency and efficacy. Compound 2f, which lacks the carboxylic acid present in every other member of this series, was very similar in activity to 2e, suggesting that the carboxylic acid group is not absolutely required for activity as an mGluR4 PAM. In Fig. 3, concentration-response curves for the most potent compound in this cluster (2h, corresponding to ChemBridge compound 7307507) are shown in comparison to PHCCC; Fig. 3 shows that this compound is more potent than PHCCC in both the calcium and thallium flux/GIRK assays. This compound was subsequently reordered and confirmed for mGluR4 PAM activity; at this point, the compound was given the identifier VU0003423 (2h/7307507). The above studies indicate that we have identified a new structural class of mGluR4 PAMs with the lead compound exhibiting improved potency (approximately 8-fold) compared with PHCCC.
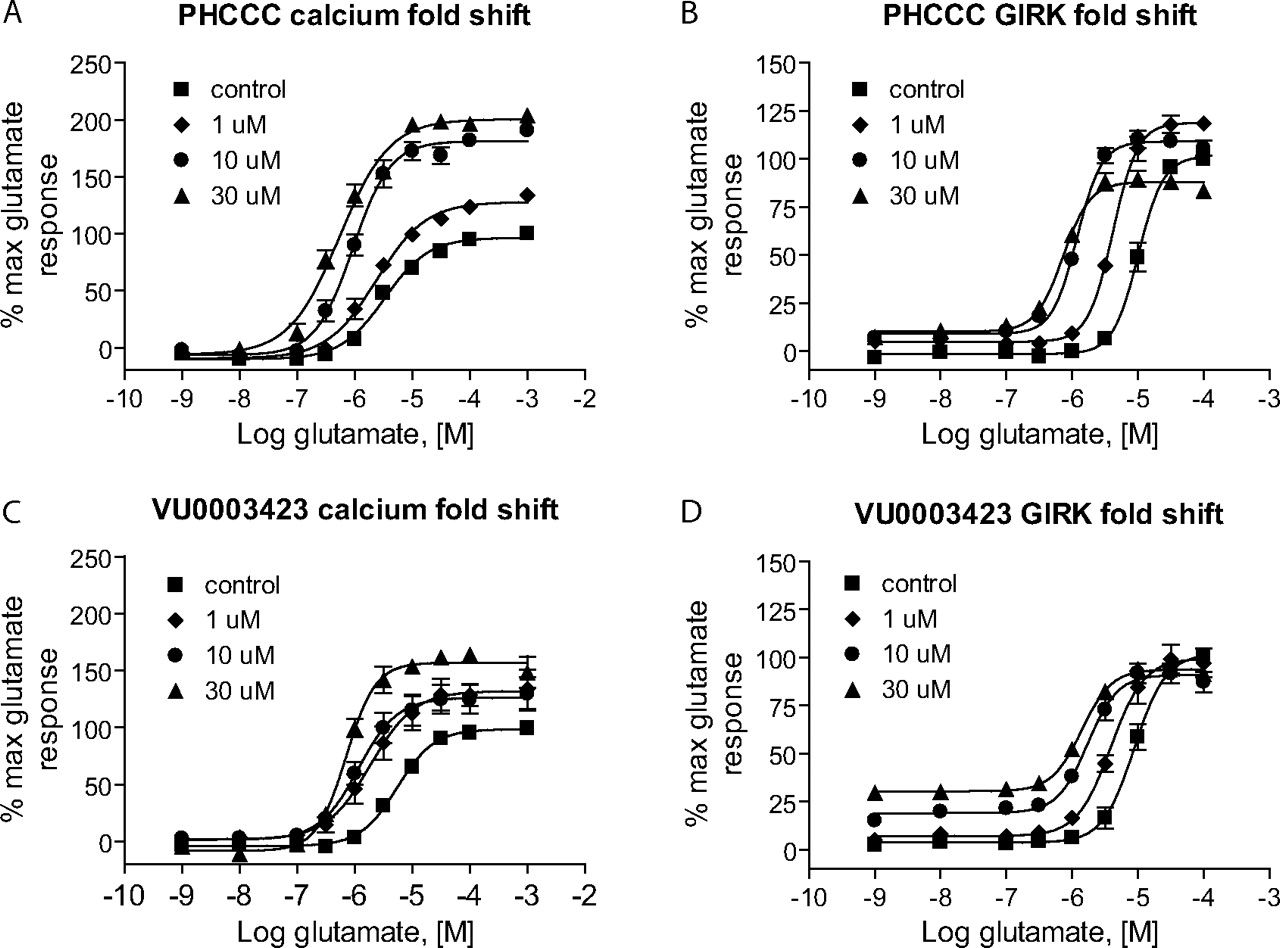
Further Structure-Activity Relationships Were Revealed Using Efficacy Determinations. To most effectively examine the efficacy of PAMs, it is important to perform concentration-response curves of agonist in the presence of a fixed concentration of PAM. We assessed the activity of PAMs from this series by performing 10-point glutamate concentration-response curves in the presence of 30 μM concentrations of each compound (Table 1). PHCCC induced shifts of the glutamate concentration-response curve of 6.7 ± 0.8- and 14.2 ± 1.7-fold (mean ± S.E.M., n = 3-4 independent experiments) in the hmGluR4/Gqi5 and rmGluR4/GIRK assays, respectively (30 μM concentration; Fig. 4). As might be expected by its potency compared with other compounds in this cluster, VU0003423 was also the most efficacious compound at shifting the concentration-response curve for glutamate when tested at a 30 μM concentration.
VU0003423 was further compared with PHCCC using progressive concentrations of each PAM for both the calcium (Fig. 4, A and C) and thallium flux/GIRK (Fig. 4, B and D) assays. For the calcium studies, each compound not only shifted the glutamate concentration-response to the left but also increased the maximal response. For the thallium flux assay, we found that the compounds induced a leftward shift with less effect on the maximum response. These studies verify activity of VU0003423 at both the rat and human receptor and confirm PAM activity in two independent assays of mGluR4 function.
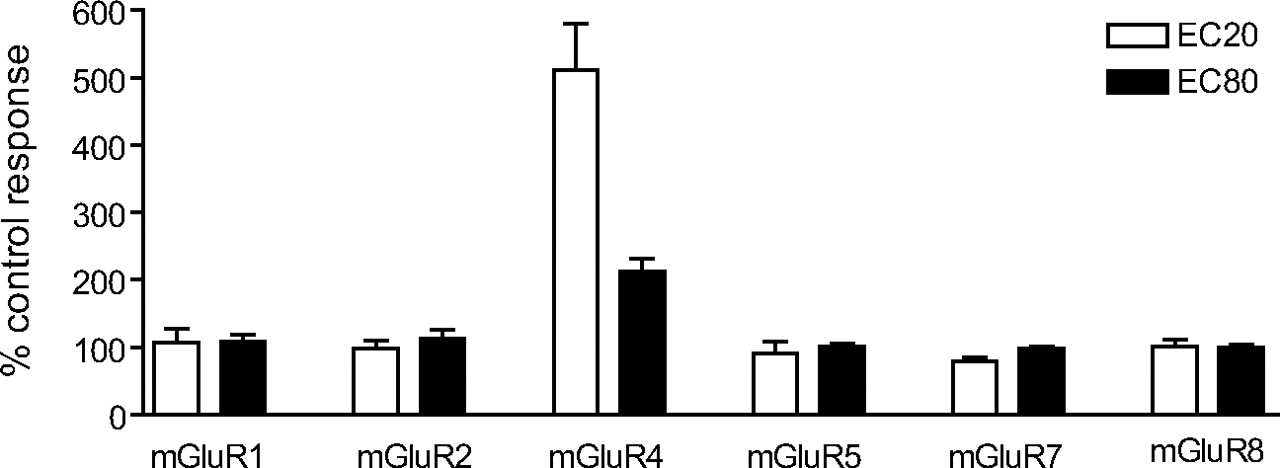
Novel mGluR4 PAM Was Selective for mGluR4 Relative to Other mGluR Subtypes. For compounds to be useful as tools for the study of mGluR4, they must be selective for this receptor. As mentioned above, PHCCC is an antagonist at mGluR1 (Annoura et al., 1996), potentially limiting its utility as an mGluR4 PAM. This is especially important in interpreting effects of PHCCC in rodent models of PD, because mGluR1 has physiological effects in basal ganglia nuclei which suggest that mGluR1 antagonists could have antiparkinsonian activity (Conn et al., 2005). We examined the selectivity profile of VU0003423 by determining its activity at mGluR subtypes 1, 2, 4, 5, 7, and 8. We first examined the ability of the compound to potentiate other mGluR responses by examining their effects on the response induced by an EC20 concentration of agonist. Data were normalized to the response for each receptor obtained in the absence of VU0003423. As can be seen in Fig. 5, 30 μM VU0003423 did not potentiate responses at any of the other mGluR subtypes. Potential antagonism of the other mGluRs was assessed by examining the ability of a 30 μM concentration of each compound to affect the response to an EC80 concentration of agonist. VU0003423 did not affect the EC80 concentration responses of the other mGluRs, indicating that this compound is selective among these mGluRs as an mGluR4 PAM.
Separation of VU0003423 into Cis and Trans Regioisomers. The compounds in this cluster, including VU0003423, have unknown stereochemistry. To determine whether one regioisomer preferentially exhibits mGluR4 PAM activity, we synthesized both the cis (VU0155041) and trans (VU0155040) regioisomers of VU0003423 and evaluated them for potency and efficacy at human and rat mGluR4. These studies revealed that at both human and rat receptors, the cis regioisomer of VU0003423 (VU0155041) was similar in potency to the lead compound (798 ± 58 nM at human mGluR4 and 693 ± 140 nM at rat mGluR4). On the other hand, the concentration-response curve for the trans regioisomer (VU0155040) did not plateau at the maximum concentration tested (Fig. 6, A and B). Fold-shift experiments at 30 μM concentration of each compound also showed that the cis regioisomer was more effective at this concentration on both human and rat mGluR4 (Fig. 6, C and D). Further resolution by preparative chiral liquid chromatography of the pure cis-regioisomer into the two single cis-enantiomers revealed that both the (1R, 2S) and (1S, 2R) enantiomers were of equal potency and efficacy (data not shown).

VU0003423 was effective in shifting the glutamate concentration-response to the left in two independent assays of mGluR4 function. Various concentrations of PHCCC (A and B) and VU0003423 (C and D) were applied to either human mGluR4/Gqi5 (A and C) or rat mGluR4/GIRK cells (B and D) approximately 2.5 min before the addition of increasing concentrations of glutamate, and responses were measured as described under Materials and Methods. Each compound resulted in progressive leftward shifts of the glutamate concentration-response curve; shifts induced in the presence of a 30 μM concentration of each compound for the calcium assay were the following (mean ± S.E.M.): PHCCC: 6.7 ± 0.8-fold; VU0003423, 6.5 ± 1.0-fold; and for the GIRK assay: PHCCC, 13.7 ± 1.6-fold; VU0003423, 7.7 ± 0.6-fold.
Partial Agonist Activity of VU0155041 as Revealed Using the GIRK-Mediated Thallium Flux Assay. As shown in Figs. 4 and 6, we observed that VU0003423, as well as the resolved regioisomers VU0155040 and VU0155041, induced concentration-dependent shifts in the baseline when examined in fold-shift experiments using the thallium flux assay. In calcium assay experiments, we also observed that compounds related to the VU0003423 scaffold induced a weak response when added alone. For instance, Fig. 7A is a calcium trace generated in the presence of 10 μM ChemBridge compound 7307507 (VU0003423) from the original HTS; a small calcium response can be observed upon the addition of the compound in the absence of glutamate. These effects were not observed with PHCCC (Fig. 2B). This suggested that these new compounds might possess some intrinsic agonist activity at mGluR4. When increasing concentrations of PHCCC and VU0155041 were added to rat mGluR4/GIRK cells in the absence of glutamate (Fig. 7B), VU0155041 induced a response that reached approximately 45% of the maximal glutamate response. Preincubation of rat mGluR4/GIRK cells with the orthosteric antagonist LY341495 resulted in a complete blockade of the response elicited by an EC80 concentration of glutamate with no effect on the response of an EC80 concentration of VU0155041 (Fig. 7C). These results are similar to the inability of orthosteric antagonists to block an allosteric agonist response of the mGluR5 PAM CDPPB on mGluR5 (Kinney et al., 2005) and indicate that the effect of the compound alone is not due to potentiation of “endogenous” glutamate, which might be remaining from the cell culture media before the assay. This suggests that VU0155041 is a partial agonist of mGluR4 that activates the receptor by interacting with a site that is distinct from the glutamate binding site.
Discovery of the allosteric agonist activity of VU0155041 provides an exciting advance and suggests that it is possible to develop both pure allosteric potentiators and compounds with allosteric agonist activity at this receptor. It was interesting to find that VU0155041 had robust agonist activity, whereas PHCCC did not exhibit this property in either assay. It is possible that these compounds act at distinct sites on mGluR4, which may allow interactions between the mGluR4 PAMs. To begin to address this question, we assessed the ability of PHCCC to potentiate the agonist activity of VU0155041. PHCCC (30 μM) was applied before the application of increasing concentrations of VU0155041. It is interesting that PHCCC was unable to potentiate or antagonize the agonist response induced by VU0155041 (Fig. 7D). Although further studies are needed to determine the binding sites of these compounds, these data suggest that PHCCC and VU0155041 may interact at distinct sites on mGluR4.
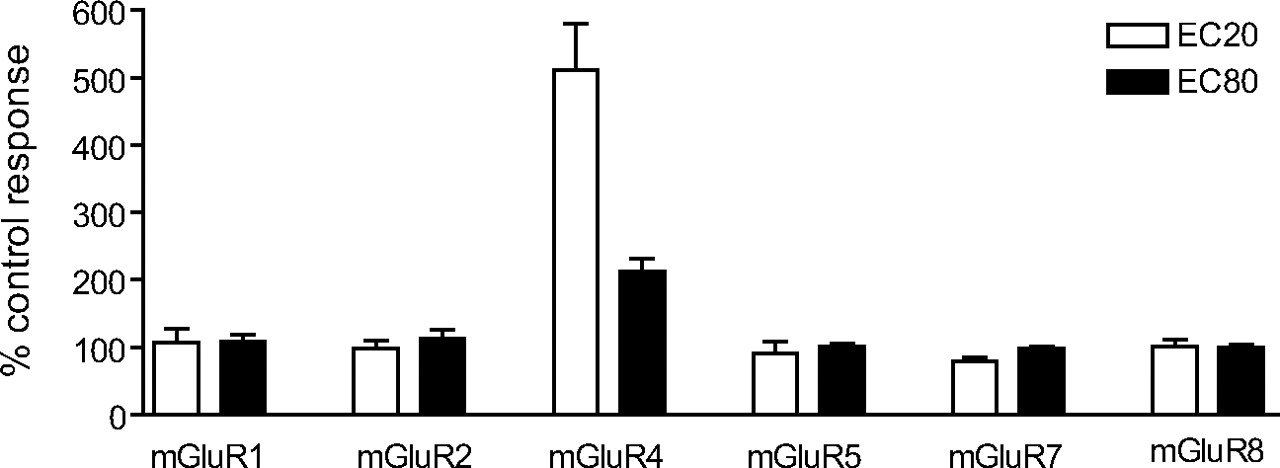
VU0003423 selectively potentiated the response of mGluR4 to glutamate compared with other mGluRs. A 30 μM final concentration of VU0003423, followed by an EC20 or EC80 concentration of agonist, was applied to the following cell lines to test for selectivity among the mGluRs: rat mGluR1/BHK, human mGluR2/CHO, human mGluR4/Gqi5, rat mGluR5/HEK, rat mGluR7/GIRK, and rat mGluR8/GIRK. Assays for each receptor were carried out as described under Materials and Methods. For mGluR1, 2, 4, 5, and 8, the agonist used was glutamate, whereas the agonist for mGluR7 was l-AP4. Data were normalized to the corresponding response observed in the absence of VU0003423. Results represent the mean ± S.E.M. of three to four independent experiments performed in triplicate or quadruplicate.

The cis regioisomer (VU0155041) of VU0003423 was more potent and efficacious than the trans regioisomer (VU0155040) in two assays of mGluR4 function. The cis (VU0155041) and trans (VU0155040) regioisomers of VU0003423 were synthesized as described in Supplementary Materials and Methods. A and B, potencies of VU0155041 and VU0155040 were determined by adding increasing concentrations of each compound to cells, followed after 2.5 min by a submaximal (EC20) concentration of glutamate. At human mGluR4 (A), potencies were the following: VU0155041, 798 ± 58 nM; and VU0155040, >10 μM. At rat mGluR4 (B), potencies were the following: VU0155041, 693 ± 140 nM; and VU0155040, >10 μM. C and D, a30 μM final concentration of compound, followed after 2.5 min by increasing concentrations of glutamate, was applied and responses were measured. At human mGluR4 (C), VU0155041 and VU0155040 caused 6.4 ± 0.7-fold and 3.0 ± 0.3-fold leftward shifts in the glutamate concentration-response curve, respectively. At rat mGluR4 (D), VU0155041 and VU0155040 caused 4.7 ± 0.4- and 2.5 ± 0.5-fold leftward shifts in the glutamate concentration-response curve, respectively. Results represent the mean ± S.E.M. of three to four independent experiments performed in triplicate.
VU0155041 Exhibited Selectivity for mGluR4 Relative to 67 Different Targets and Did Not Affect the Function of Striatal NMDA Receptors. As discussed above, mGluR4 has been postulated to be a target for novel therapeutic agents used for the treatment of PD. Identification of novel structures with PAM activity at mGluR4 now allows us to further address the role of this receptor in experimentally induced movement phenotypes characteristic of PD. Previous studies have shown that icv administration of the general group III agonist l-AP4, as well as the mGluR4 PAM/mGluR1 antagonist PHCCC, are effective in reversing reserpine-induced akinesia, a preclinical model of PD (Marino et al., 2003; Valenti et al., 2003). The development of alternate mGluR4 PAMs with better selectivity for mGluR4 has now allowed us to further test the hypothesis that compounds with the ability to increase mGluR4 activity can have beneficial effects on movement in these models. Although we had confirmed the selectivity of our lead PAM relative to other mGluR subtypes, before conducting animal studies we sought to determine whether this compound might exhibit ancillary activity at other GPCRs, transporters, or ion channels that could complicate implementation or interpretation of in vivo experiments. VU0155041 was evaluated at MDS-Pharma Services for effects on radioligand binding at 67 different targets, including GPCRs, ion channels, and transporters. As shown in Supplemental Table 2, VU0155041 had no effect on binding at any target examined.

VU0155041 exhibited partial agonist activity at a site on rat mGluR4 that is distinct from the glutamate and PHCCC binding sites. A, fluorescence trace of compound VU0003423 from the primary HTS revealing an increase in calcium mobilization upon the addition of a 10 μM concentration of compound. B, increasing concentrations of PHCCC or VU0155041 were added to rat mGluR4/GIRK cells directly in thallium buffer, and responses were measured. Maximal agonist responses observed at 30 μM, expressed as a percentage of the maximal glutamate response, were the following: VU0155041, 41.6 ± 5.3; and PHCCC, -2.3 ± 2.0. The EC50 value for the partial agonist activity of VU0155041 was 2.5 ± 0.5 μM. C, increasing concentrations of LY341495 were added to rat mGluR4/GIRK cells, followed 2.5 min later by an EC80 concentration of glutamate or VU0155041. LY341495 inhibited the glutamate response with an IC50 value of 14.5 ± 4.3 μM; LY341495 failed to inhibit the VU0155041 response. Data were normalized to the percentage of the relevant EC80 agonist response. Results represent the mean ± S.E.M. of three to five independent experiments performed in triplicate or quadruplicate. D, a 30 μM final concentration of PHCCC was added to rat mGluR4/GIRK cells, followed after 2.5 min by various concentrations of VU0155041. PHCCC did not significantly alter the potency or efficacy of VU0155041. The EC50 values for VU0155041 in the absence and presence of PHCCC were 2.3 ± 0.5 and 1.1 ± 0.1 μM, respectively (P = 0.082, unpaired t test). Results represent the mean ± S.E.M. of at least three independent experiments performed in quadruplicate.
Despite the lack of binding activity at a large number of targets, it is possible that VU0155041 could have functional effects at other targets that could be responsible for, or at least confounding to, the interpretation that mGluR4 PAMs have antiparkinsonian activity. In particular, antagonism of the N-methyl-d-aspartate receptor (NMDAR)-NR2B subtype has been shown to reverse haloperidol-induced catalepsy (Liverton et al., 2007). The mGluR5 negative allosteric modulator 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP) is a weak mGluR4 PAM and blocks NMDA receptors containing NR2B subunits (Lea and Faden, 2006). To verify that VU0155041 did not antagonize NMDA receptors, we tested the ability of this mGluR4 PAM to functionally block NMDA receptor currents in striatal medium spiny neurons. These results were compared with the ability of a known NR2B antagonist, Ro25-6981 (Fischer et al., 1997), to regulate NMDA currents in these same neurons. As can be seen in Fig. 8, 10 μM VU0155041 did not affect NMDA receptor currents in striatal medium spiny neurons, whereas 1 μM Ro25-6981 induced significant blockade. These results indicate that VU0155041 does not antagonize NMDA receptor activity and that direct NMDA receptor antagonism should not confound interpretation of the effects of VU0155041 in models of PD.
VU0155041 Had Antiparkinsonian Effects in Preclinical Rodent Models of PD. It is encouraging that in addition to improvements in potency and selectivity over mGluR4 PAMs described previously, VU0155041 was also found to be soluble in an aqueous vehicle. In contrast, PHCCC is only soluble in vehicles containing high concentrations of DMSO or other relatively toxic vehicles; these vehicles can cause tissue damage when injected icv and can compromise the blood-brain barrier when a compound is administered systemically. Because of the tolerance of VU0155041 for a more toxicology-friendly vehicle, the effects of this compound were compared with the general group III agonist l-AP4, because the vehicles used for their preparation were both aqueous (see Materials and Methods).

VU0155041 had no effect on NMDA receptor-mediated currents in striatal medium spiny neurons. A, averaged traces of NMDA receptor currents evoked by pressure-ejection of 1 mM NMDA before and during application of VU0155041 (10 μM) (top). Time course of normalized amplitude of NMDA receptor currents before and after application of VU0155041 from five cells (bottom). VU0155041 had no significant effect on the amplitude of NMDA receptor currents (106.3 ± 5.8% of control, n = 5; p > 0.3, Student t test). B, averaged traces of NMDA receptor currents before and during application of Ro25-6981 (1 μM) (top). Time course of normalized amplitude of NMDAR currents before and after application of Ro25-6981 (bottom). Ro25-6981 significantly inhibits the peak amplitude of NMDAR currents (53.1 ± 8.5% of control, n = 5; p < 0.003, Student t test). Each trace in A and B is an average of three. Data are presented as mean ± S.E.M.
l-AP4 and VU0155041 were first assessed for their ability to decrease haloperidol-induced catalepsy. As demonstrated previously, l-AP4 significantly decreased haloperidol-induced catalepsy in a dose-dependent manner (Fig. 9). It is encouraging that VU0155041, at doses of 31 and 92 nmol, was also able to significantly decrease the cataleptic effects of haloperidol, and the effects of the compound were still present 30 min after infusion.
Reversal of reserpine-induced akinesia is another preclinical model of PD used to assess the activity of compounds for antiparkinsonian effects, although the effects are much more difficult to reverse compared with the catalepsy model. Infusion of 300 and 1000 nmol icv doses of l-AP4 into ventricles of animals that had been pretreated for 2 h with reserpine induced significant reversals of akinesia (Fig. 10). Infusion of a 316 nmol icv dose of VU0155041 also resulted in a significant reversal of akinesia. VU0155041 represents only the second unique chemical scaffold with mGluR4 PAM activity that shows efficacy in rodent models of PD, further validating the role of this receptor as a therapeutic target in PD. It is exciting that the improvements in potency, selectivity, and solubility of VU0155041 compared with PHCCC suggest that this compound will serve as a valuable research tool to continue to define the role of mGluR4 in normal physiology and to further explore the therapeutic potential of mGluR4 in PD and other disease states.
Discussion
Parkinson's disease is a chronic neurodegenerative disorder resulting from the loss of dopaminergic neurons in the substantia nigra. Although dopamine-replacement therapies are useful early in the disease, these treatments generally lose their efficacy as the disease progresses. In addition, dopamine replacement does not correct the underlying disease process, and neurons continue to degenerate over time. Treatment options that bypass the dopamine system are currently being explored in the hope that therapies that do not rely on intact dopamine neurotransmission will remain effective late in the disease and slow disease progression. Patients who have undergone surgical intervention to diminish or normalize patterns of output through the basal ganglia, for example by implanting electrodes to induce high-frequency stimulation in the subthalamic nucleus or basal ganglia output nuclei (Garcia et al., 2005), often respond with a dramatic reduction in PD motor symptoms. The beneficial effects of surgical intervention suggest that pharmacological approaches that achieve effects similar to those of surgical procedures could provide new avenues for PD treatment.
Building on observations of surgical patients, mGluR4 has been shown to be a candidate GPCR that might be exploited for therapeutic benefit in PD. mGluR4 is expressed at the inhibitory synapse projecting from the striatum to the external segment of the globus pallidus (GPe, striatopallidal synapse) within the indirect pathway of the basal ganglia. The indirect pathway has been shown to be overactive in PD patients because of a loss of modulatory input from the dopaminergic neurons of the substantia nigra (Conn et al., 2005), resulting in an abnormally high inhibitory tone at the level of the GPe and disinhibition of the subthalamic nucleus. Activation of mGluR4 at the striatopallidal synapse reduces the abnormally high release of GABA that occurs at this synapse in PD, balancing GPe output (Valenti et al., 2003).
Behavioral studies provide further support for mGluR4 activation in PD. Valenti et al. (2003) showed that icv injection of the group III agonist l-AP4 could reverse reserpine-induced akinesia. To further define the roles of the group III mGluRs in modulating basal ganglia function, several studies have examined the effects of infusion of l-AP4 and another group III-preferring agonist, (1S, 3R, 4S)-1-aminocyclopentane-1,3,4-tricarboxylic acid into specific regions of the basal ganglia such as the GPe and SNr (MacInnes et al., 2004; Konieczny et al., 2007; Lopez et al., 2007; Sibille et al., 2007). It is interesting that infusion of these agonists into the GPe elicited marked improvement in motor symptoms in several preclinical PD models. Infusion of these compounds into the SNr, however, has been shown to enhance the akinesia produced by lesioning and did not reverse haloperidol-induced catalepsy (Lopez et al., 2007); however, it should be noted that there is controversy in this regard because other studies suggest that group III mGluR agonist infusion into the SNr can reverse motor defects in certain models (MacInnes et al., 2004; Konieczny et al., 2007). In the study by Lopez et al. (2007), the effect of these agonists in the SNr was mimicked by a selective agonist of mGluR8, (S)-3,4-dicarboxyphenylglycine. These observations suggest that mGluR4 is responsible for mediating beneficial effects on motor function in PD models via function at the striatopallidal synapse and that a potential therapeutic strategy that should be explored for PD is selective activation of mGluR4.

VU0155041 reversed catalepsy induced by the dopamine D2 receptor antagonist haloperidol in rats. Rats were treated with 1.5 mg/kg haloperidol as described under Materials and Methods. After 2 h, animals were infused icv with the indicated doses of either l-AP4 or VU0155041, and catalepsy was measured 15, 30, and 60 min after injection [15 min (□) and 30 min (▪) results shown]. Experiments represent data obtained for six rats per group. *, P < 0.05, Dunnett's comparison with vehicle group.

VU0155041 reversed reserpine-induced akinesia in rats. Rats were treated with 5 mg/kg reserpine as described under Materials and Methods. Two hours later, animals were infused with the indicated doses of either l-AP4 or VU0155041, and a reversal of akinesia, monitored via locomotor activity, was measured. Data are plotted as baseline locomotor activity (□) and then the activity measured 15 min after compound infusion (▪). Experiments represent data obtained for seven rats per group. *, P < 0.05, Dunnett's comparison with vehicle group.
Because of the high conservation of endogenous ligand binding sites, strategies have emerged for activation of GPCRs that exploit the ability of small molecules to bind to sites on the receptor that are distinct from the orthosteric neurotransmitter binding site and potentiate the effects of the endogenous agonist. For mGluR4, three PAMs have been described: PHCCC, MPEP, and SIB-1893 (Maj et al., 2003; Marino et al., 2003; Mathiesen et al., 2003). MPEP and SIB-1893 are weak mGluR4 PAMs and potent mGluR5 antagonists; as such, they are not useful as selective mGluR4 PAMs. In addition, as mentioned previously, MPEP has activity in antagonizing NMDA receptor currents (Lea and Faden, 2006). PHCCC has greater efficacy as an mGluR4 PAM and has been shown to demonstrate efficacy in a rodent model of PD (Marino et al., 2003). However, the low potency and poor solubility of PHCCC, along with its activity as an mGluR1 antagonist (Annoura et al., 1996), complicates the use of this compound.
Discovery of novel mGluR4 PAMs in the current studies provides a major advance in demonstrating that robust mGluR4 PAM activity can be achieved with diverse chemical scaffolds. Furthermore, these novel mGluR4 PAMs include multiple compounds that are structurally related, suggesting that it may be possible to develop clear structure-activity relationships for mGluR4 potentiation. Finally, the lead compound described here provides major advances in that it exhibits submicromolar potency, is highly selective for mGluR4 relative to other mGluR subtypes and other targets including NMDA receptors, and has improved physicochemical properties in terms of solubility. The finding that icv administration of VU0155041 is efficacious in two rodent models of PD provides exciting new support for mGluR4 activation as a novel strategy for treatment of PD. A critical role for mGluR4 in mediating this antiparkinsonian action is strengthened when taken together with previous studies demonstrating that antiparkinsonian effects occur with l-AP4 and PHCCC, which are structurally distinct from VU0155041. Furthermore, the high selectivity of VU0155041 for mGluR4 suggests that the antiparkinsonian activities of these compounds are unlikely to be due to an off-target activity that is shared by the structurally and mechanistically distinct mGluR4 activators.
The studies here also provide important new insights into the molecular pharmacology of allosteric modulators of mGluR4. It is interesting that both PHCCC and VU0155041 affected the concentration-response curves of glutamate differently in the two assays used for assessing mGluR4 activity. Both compounds increased the maximum response seen in the calcium assay but not the maximum response obtained using the thallium flux method. There are many possible explanations for this observation, including differences in signal amplification between the two pathways, possible species differences, or difference in receptor expression. Unfortunately, it is difficult to accurately measure the expression of mGluR4 in cell lines because of the lack of a high-affinity radiolabeled antagonist for the receptor (Wright et al., 2000); we should note, however, that the potency of glutamate in the two mGluR4 cell lines is similar (5.7 ± 0.7 μM, calcium assay versus 9.2 ± 1.2 μM, thallium flux/GIRK assay; data obtained from Fig. 4), suggesting that there may not be dramatic differences in receptor reserve between the two systems. Another possible explanation for the ability of compounds to potentiate maximal responses in the calcium assay may involve distinctions in the coupling efficiency of mGluR4 to Gqi5 versus Gi/o. This might manifest in an inability of glutamate alone to achieve the maximal response possible within the Gqi5-mediated signaling pathway; the presence of a PAM might stabilize the receptor or enhance its function in such a way as to permit the efficacy and the potency of glutamate to be enhanced.
An additional distinction between VU0155041 and PHCCC was revealed by the ability of VU0155041 to activate mGluR4 in the absence of glutamate. When added alone in the GIRK assay, the concentration-dependent response induced by VU0155041 reached approximately 45% of the maximal glutamate response and was not affected by preincubation with the orthosteric antagonist LY341495. This suggests that the activity of VU0155041 is not due to a potentiation of glutamate present in the assay system and further suggests that the agonist activity of VU0155041 is not mediated by actions on the orthosteric LY341495 binding site. Together, these data suggest that VU0155041 possesses allosteric agonist activity, which further differentiates the compound from PHCCC.
It is interesting that PHCCC was unable to affect the concentration-response curve of VU0155041. These data suggest that PHCCC and VU0155041 are not likely to act at a single allosteric site. Consistent with this, we have provided compelling evidence that there are multiple allosteric sites on both mGluR1 and mGluR5 and have identified PAMs that act on at least two sites on each of these mGluR subtypes (O'Brien et al., 2004; Hemstapat et al., 2006; Chen et al., 2008). Further studies using mutagenesis strategies will be needed to explore the binding pockets of these new compounds to determine how many binding sites might exist on mGluR4 that could be exploited for allosteric modulation.
In summary, we have identified a series of novel compounds with mGluR4 PAM activity. These compounds are active on both the rat and human receptors and function as PAMs in two different assays of mGluR4 function. The lead compound, VU0003423, is more potent than PHCCC and selective for mGluR4 among the mGluRs. The cis-regioisomer of VU0003423, VU0155041, does not exhibit binding activity at a large number of off-target receptors, transporters, and ion channels; does not inhibit NMDA receptor functional activity; is soluble in an aqueous vehicle; and exhibits activity in two different rodent behavioral models of PD. These compounds represent breakthrough new tools for the study of the role of mGluR4 in normal brain function and in pathophysiological states such as PD.
Footnotes
-
This work was supported by National Institutes of Health grants NS053536 (to C.M.N.), NS051342, NS031373, MH074953, and NS048334 (to P.J.C.), the Dystonia Research Foundation (to Z.X.), the Alpha Foundation (to P.J.C.), and the Michael J. Fox Foundation (to P.J.C.).
-
C.M.N. and K.A.J. contributed equally to this work.
-
ABBREVIATIONS: mGluR, metabotropic glutamate receptor; GPCR, G protein-coupled receptor; HTS, high-throughput screening; GIRK, G-protein-regulated inwardly rectifying potassium channel; HEK, human embryonic kidney; DMSO, dimethyl sulfoxide; PHCCC, N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide; MPEP, 2-methyl-6-(phenylethynyl)pyridine hydrochloride; LY341495, (2S)-2-amino-2-[(1S,2S)-2-carboxyclycloprop-1-yl]-3-(xanth-9-yl) propanoic acid; Ro25-6981, (αR,βS)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidinepropanol; NMDA, N-methyl-d-asparate; VU0003423, 2-(3,5-dichlorophenylcarbamoyl)cyclohexanecarboxylic acid; VU0155040, (±)-trans-2-(3,5-dichlorophenylcarbamoyl)cyclohexanecarboxylic acid; VU0155041, (±)-cis-2-(3,5-dicholorphenylcarbamoyl)-cyclohexanecarboxylic acid; l-AP4, l-(+)-amino-4-phosphonobutyric acid; TVC, third ventricle cannulated; PD, Parkinson's disease; PAM, positive allosteric modulator; icv, intracerebroventricular; CHO, Chinese hamster ovary; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; HEK, human embryonic kidney; FDSS, Functional Drug Screening System; BHK, baby hamster kidney; GTPγS, guanosine 5′-O-(3-thio)triphosphate; ACSF, artificial cerebrospinal fluid; NMDAR, N-methyl-d-aspartate receptor; GPe, globus pallidus external segment; SNr, substantia nigra pars reticulata; QX-314, N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide; SIB-1893, 2-methyl-6-(2-phenylethenyl)pyridine.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Received June 6, 2008.
- Accepted July 29, 2008.

- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}