


Abstract
The renal outer medullary potassium channel (ROMK) is expressed in the kidney tubule and critically regulates sodium and potassium balance. The physiological functions of other inward rectifying K+ (Kir) channels expressed in the nephron, such as Kir7.1, are less well understood in part due to the lack of selective pharmacological probes targeting inward rectifiers. In an effort to identify Kir channel probes, we performed a fluorescence-based, high-throughput screen (HTS) of 126,009 small molecules for modulators of ROMK function. Several antagonists were identified in the screen. One compound, termed VU590, inhibits ROMK with submicromolar affinity, but has no effect on Kir2.1 or Kir4.1. Low micromolar concentrations inhibit Kir7.1, making VU590 the first small-molecule inhibitor of Kir7.1. Structure-activity relationships of VU590 were defined using small-scale parallel synthesis. Electrophysiological analysis indicates that VU590 is an intracellular pore blocker. VU590 and other compounds identified by HTS will be instrumental in defining Kir channel structure, physiology, and therapeutic potential.
- ROMK, renal outer medullary K+ channel
- Kir, inward rectifying potassium
- EK, potassium equilibrium potential
- VU590, 7,13-bis(4-nitrobenzyl)-1,4,10-trioxa-7,13-diazacyclopentadecane
- HTS, high-throughput screen
- CCD, cortical collecting duct
- HEK, human embryonic kidney
- TPNQ, Tertiapin Q
- PBS, phosphate-buffered saline
- TBST, Tris-buffered saline/Tween 20
- DMSO, dimethyl sulfoxide
- DCM, dichloromethane
- Tet, tetracycline
- RMP, resting membrane potential
- CRC, concentration-response curve
- FDSS, Functional Drug Screening System 6000.
The renal outer medullary potassium (K+) channel (ROMK, Kir1.1, KCNJ1) is expressed in the kidney tubule and which it critically regulates sodium and potassium homeostasis (Hebert et al., 2005; Wang and Giebisch, 2009). In the thick ascending limb of Henle, luminal K+ recycling by ROMK supports NaCl reabsorption by the Na-K-2Cl cotransporter and loop diuretic target NKCC2, which in turn promotes osmotic water reabsorption in the distal nephron (Hebert and Andreoli, 1984; Hebert et al., 1984; Hebert, 1998). In the connecting tubule and cortical collecting duct (CCD), apical ROMK channels constitute a major pathway for K+ secretion and function to match urinary K+ excretion with dietary intake (Frindt et al., 2009; Wang and Giebisch, 2009) A growing body of genetic evidence (Simon et al., 1996; Ji et al., 2008; Tobin et al., 2008) suggests that pharmacological antagonists of ROMK could have potent diuretic effects while minimizing potentially dangerous urinary K+ loss, as seen with loop diuretics (Grobbee and Hoes, 1995; Macdonald and Struthers, 2004). However, the molecular pharmacology of ROMK, and indeed that of the entire inward rectifier family, is virtually undeveloped, precluding the assessment of ROMK's potential as a diuretic target.
At least five other members (Kir2.3, Kir4.1, Kir4.2, Kir5.1, and Kir7.1) of the Kir channel family are expressed in the nephron (Welling, 1997; Ookata et al., 2000; Hebert et al., 2005; Lachheb et al., 2008), but their physiological functions are not well understood. The newest member, Kir7.1 (KCNJ13), is expressed in several nephron segments. In principal cells of the collecting duct, Kir7.1 is proposed to mediate basolateral K+ recycling necessary for Na-K-ATPase-dependent K+ secretion (Ookata et al., 2000). However, there is no direct evidence that Kir7.1 forms functional ion channels in the renal tubule. Kir7.1 is difficult to identify in single-channel recordings because of its unusually low unitary conductance (∼50 fS) (Krapivinsky et al., 1998), and the lack of pharmacological tools does not allow one to discriminate Kir7.1 from other channels present in macroscopic current recordings. The identification of Kir7.1-targeted probes would provide important new tools with which to define the physiological functions of the channel in the nephron and other tissues.
In an effort to identify Kir channel probes, we developed and implemented a fluorescence-based assay for high-throughput screening (HTS) of chemical libraries for modulators of ROMK function. From a screen of 126,009 organic small molecules, several ROMK antagonists were identified. One compound, termed VU590, inhibits ROMK with submicromolar affinity and Kir7.1 at low micromolar concentrations, but it does not inhibit Kir2.1 or Kir4.1. The identification of VU590 and other Kir channel antagonists by HTS represents an important step toward developing the molecular pharmacology of the Kir channel family and creates new opportunities for investigating potassium transport physiology in the nephron and other tissues.
Materials and Methods
Cell Lines, Reagents, and Chemicals.
Parental tetracycline-regulated expression Human embryonic kidney (HEK)-293 cells, Dulbecco's modified Eagle's medium containing 25 mM d-glucose and 2 mM l-glutamine, and the acetoxymethyl ester form of Fluozin-2 were purchased from Invitrogen (Carlsbad, CA). HEK-293 (CRL-1573) cells used for transient transfections were purchased from American Type Culture Collection (Manassas, VA). Fetal bovine serum was from Atlanta Biologicals (Lawrenceville, GA). Tertiapin Q (TPNQ), protease inhibitor cocktail, Triton X-100, Tween 20, and salts of the highest purity available were purchased from Sigma-Aldrich (St. Louis, MO). Thallium (I) sulfate was from Alfa Aesar (Ward Hill, MA). Tetracycline HCl (Sigma), Blasticidin S HCl, and Hygromycin B (both from Invitrogen) were prepared as described previously (Fallen et al., 2009). Rabbit polyclonal ROMK antiserum was purchased from Alomone Labs (Jerusalem, Israel). Rabbit β-actin antiserum was from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase-conjugated goat anti-rabbit secondary antiserum was from Jackson ImmunoResearch Laboratories (West Grove, PA). SuperSignal West Pico chemiluminescent reagent and bicinchoninic protein quantitation kit were purchased from Pierce (Rockford, IL).
Expression Vectors.
The pcDNA5/TO expression vector carrying rat ROMK1 was created as described previously (Fallen et al., 2009). Serine 44 of ROMK1 was mutated to aspartate (S44D) to promote cell surface expression of the channel (O'Connell et al., 2005; Yoo et al., 2005). Mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The entire open-reading frame was sequenced to verify the fidelity of mutagenesis. The pIRES2-EGFP vector carrying human Kir2.1 (NM_000891.2) was provided by Dr. Alfred George (Vanderbilt University Medical Center, Nashville, TN) (Ballester et al., 2006). The pcDNA3.1 vector containing human Kir7.1 (NM_002242.2) was provided by Dr. David Clapham (Harvard Medical School, Boston, MA) (Krapivinsky et al., 1998). The pCMV6-XL4 expression vector carrying human Kir4.1 (NM_002241.2) was purchased from OriGene Technologies (Rockville, MD).
Generation of C1 Cells.
Polyclonal stable cell lines expressing ROMK-S44D were established essentially as described previously (Fallen et al., 2009). Individual clones were isolated through limiting dilution in 96-well plates. The clones were expanded, plated in duplicate 384-well assay plates, and analyzed for tetracycline (Tet)-inducible ROMK channel activity using the Tl+-flux assay described below. Of approximately 330 clones evaluated, 11 clones exhibiting the highest Tet-inducible Tl+ flux were selected for expansion from replicate plates and further characterization. One clone, termed C1, was selected for detailed electrophysiological characterization and high-throughput assay development.
Western Blot Analysis.
Cells were plated at a density of 105 cells/cm2 and cultured overnight in the absence or presence of Tet (1 μg/ml), after which they were washed three times with ice-cold phosphate-buffered saline (PBS) and solubilized in ice-cold radioimmunoprecipitation assay buffer containing 150 mM NaCl, 100 mM NaF, 50 mM Tris-HCl, pH 8.0, 35 mM sodium deoxycholate, 5 mM EDTA, 1% Triton X-100, 0.1% SDS, and 1× protease inhibitor cocktail. Cell lysates were scraped into Eppendorf tubes (Eppendorf North America, New York, NY), sonicated, rotated end-over-end for 30 min at 4°C, and then centrifuged for 10 min to pellet insoluble material. The protein concentration of cleared lysates was quantitated using bicinchoninic reagent. Cell lysates were either used immediately or frozen as single-use aliquots at −80°C for subsequent immunoblot analysis.
Lysate protein was separated electrophoretically using 10% Bis-Tris NuPAGE gels (Invitrogen) and transferred to 0.45-μm pore nitrocellulose membranes (BioRad, Hercules, CA) for immunoblot analysis. Membranes were blocked for 1 h at room temperature in Tris-buffered saline (25 mM Tris, 1.3 mM KCl, and 137 mM NaCl) containing 0.1% Tween 20 (TBST) and 5% nonfat milk. Membranes were incubated overnight at 4°C in polyclonal ROMK antiserum diluted 1:500 in milk-TBST and subsequently for 1 h at room temperature in milk-TBST containing horseradish peroxidase-conjugated secondary antiserum diluted 1:40,000. Protein bands were visualized on film using enhanced chemiluminescence detection methods. Membranes were stripped in buffer containing 100 mM 2-mercaptoethanol, 62.5 mM Tris-HCl, pH 6.7, and 2% SDS (50°C for 30 min) and reprobed with β-actin antiserum.
Cell Surface Biotinylation.
C1 cells (105 cells/cm2) cultured overnight in the absence or presence or Tet were placed on ice and washed three times in ice-cold PBS containing 1 mM MgCl2 and 0.1 mM CaCl2 (buffer A). All of the following steps were performed either on ice or at 4°C using ice-cold solutions. Membrane proteins were oxidized for 30 min with 10 mM sodium meta-periodate dissolved in buffer A, washed three times in buffer A, and rinsed with 100 mM sodium acetate, pH 5.5, containing 1 mM MgCl2 and 0.1 mM CaCl2 (buffer B). The cells were subsequently incubated with 2 mM biotin long-chain hydrazide (Pierce) dissolved in buffer B and agitated with gentle rocking for 30 min. Buffer B lacking biotin was added to nonbiotinylated samples as a control. The cells were then washed once for 5 min with quenching buffer containing 50 mM Tris-HCl in buffer A, pH 7.4, washed twice in buffer A, washed twice in divalent-free PBS, and lysed in radioimmunoprecipitation assay buffer as described above.
Biotinylated proteins were immunoprecipitated from cell lysates (30 μg in 900 μl of cell lysis buffer) with 100 μl of a 50% slurry of washed streptavidin agarose resin (Pierce). Immunoprecipitations were performed overnight at 4°C with end-over-end rotation. The next day, the resin was washed extensively (six 5-min washes with 1 ml of lysis buffer and end-over-end rotation at 4°C), pelleted by brief centrifugation and dried by aspirating the supernatant with a 27-gauge needle. Immunoprecipitated proteins were eluted at 95°C for 5 min in 1× LDS buffer (Invitrogen) and 1× reducing agent (Invitrogen) containing 50 mM DTT. Eluted biotinylated proteins were separated electrophoretically and immunoblotted for ROMK as described above.
Patch-Clamp Electrophysiology.
C1 cells were cultured in the absence or presence of Tet the night before patch clamp experiments. HEK-293 cells were transiently transfected with 0.5 to 1.0 μg of channel expression vector and 0.5 μg of pcDNA3.1-EGFP (transfection marker) using Lipofectamine 2000. Experiments were performed approximately 24 h after transfection. The day of experiments, cells were rinsed with divalent-free buffer (145 mM NaCl, 5 mM KCl, 4 mM NaHCO3, 0.3 mM Na2HP04, 0.3 mM KH2PO4, and 5.6 mM d-glucose, pH 7.4, 290 mOsmol/kg of water), dissociated by brief exposure to 0.25% Trypsin containing 1 mM EDTA, plated on poly(l-lysine)-coated round glass coverslips and allowed to recover at 37°C in a 5% CO2 incubator for at least 1 h before experiments. The coverslips were transferred to a small-volume perfusion chamber (Warner Instruments, Hamden, CT) and mounted on the stage of a Nikon Eclipse TE2000-U inverted microscope (Nikon, Tokyo, Japan).
Patch electrodes were pulled from silanized 1.5-mm outer diameter borosilicate microhematocrit tubes using a Narishige PP-830 two-stage puller. Electrode resistance ranged from 2.5 to 3.5 MΩ when filled with the following intracellular solution: 135 mM KCl, 2 mM MgCl2, 1 mM EGTA, 10 mM HEPES-free acid, and 2 mM Na2ATP (Roche Diagnostics, Indianapolis, IN), pH 7.3, 275 mOsmol/kg of water. The standard bath solution contained 135 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM glucose, and 10 mM HEPES-free acid, pH 7.4, 290 mOsmol/kg of water. Whole-cell currents were recorded under voltage-clamp conditions using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). The cell membrane potential was recorded in current clamp mode. Electrical connections to the amplifier were made using Ag/AgCl wires and 3 M KCl/agar bridges. Electrophysiological data were collected at 5 kHz and filtered at 1 kHz. Data acquisition and analysis were performed using pClamp 9.2 software (Molecular Devices).
High-Throughput Screening.
C1 cells (30,000 cells/20 μl/well were plated in clear-bottomed, black-walled 384-well plates (Greiner Bio-One, Longwood, FL) and cultured overnight in serum-free media containing Tet at 37°C in the presence of 5% CO2. The following day, a dye stock was prepared fresh by dissolving 50 μg of Fluozin-2 in 20 μl of pure, dry DMSO and mixing with 10 μl of 20% pluronic acid. The dye stock was diluted to 2 mM with assay buffer (Hanks' balanced salt solution + 20 mM HEPES, pH 7.3, adjusted to 343 mOsmol/kg of water with sucrose). A 20-μl sample was added to each well containing cells. The cells were incubated for 20 min at room temperature, followed by automated washing using the ELx405 plate washer (BioTek, Winooski, VT) primed with modified assay buffer (0.44 mM NaH2PO4, 4.17 mM NaHCO3, 137.93 mM NaCl, 0.338 mM Na2HPO4, 20 mM HEPES, and 0.25 mM K2SO4, adjusted to 343 mOsmol/kg of water with sucrose). A final aspiration step resulted in 20-μl residual volume. The plate was loaded into the Functional Drug Screening System 6000 (FDSS; Hamamatsu, Tokyo, Japan) to obtain an initial baseline image (F0) (10 images at 1 Hz; excitation, 470 ± 20 nm; emission, 540 ± 30 nm). The Bravo liquid handling platform (Velocity11, Santa Clara, CA) added 20 μl of DMSO vehicle control, TPNQ, or test compound to each plate for final concentrations of 0.1%, 2 μM, and 10 μM, respectively. During assay development, we established that Tl+ flux through ROMK was insensitive to DMSO concentrations up to 10% (data not shown). Test compounds in 40 μl of modified assay buffer were prepared by transferring 80 nl into a 384-well polypropylene plate using the ECHO 550 acoustic formatter (Labcyte, Sunnyvale, CA) from a 10 mM stock supplied by the NIH Molecular Libraries Small Molecule Repository (BioFocus DPI, San Francisco, CA). Four columns in each plate were filled with vehicle control or TPNQ-containing buffer for the determination of Z′ (Fig. 3B), whereas the remaining 20 columns were used for compound screening. The compound and cells were allowed to incubate for 20 min at room temperature, then the plate was reloaded into the FDSS, and a second baseline was acquired. The FDSS's integrated pipetter added 10 μl of thallium stimulus (125 mM sodium gluconate, 12 mM thallium sulfate, 1 mM magnesium sulfate, 1.8 mM calcium gluconate, 5 mM glucose, and 10 mM HEPES, pH 7.3, adjusted to 343 mOsmol/kg of water with sucrose), whereas imaging of the flux continued at 1 Hz for 2 min of total sampling time.
Data Analysis and Statistics.
The kinetic fluorescence values (F) from each well were divided by the initial frame of the read (F0) to give the static ratio (F/F0), which corrects for variability in cell number and dye loading. The second baseline reading was used to assess fluorescent compound artifacts, and those wells in which the reading exceeded 70% of the mean fluorescence for the plate were excluded from analysis. The slope of the static ratio from 7 to 12 s was calculated, and the value was compared with the average distribution of the remaining compound wells assumed to Gaussian. Hits were picked using a combination of Z score and B score (Malo et al., 2006) with 99.7% confidence intervals in an automated data analysis pipeline generated with Pipeline Pilot (Accelrys, San Diego, CA) and R statistics package (http://www.r-project.org). TPNQ and vehicle control wells were included on every plate and used to calculate the Z′ according to the equation below (Zhang et al., 1999). Hit compounds were reordered from Biofocus DPI as 10 mM stocks and tested in dose-response as 10-point 3-fold dilutions ranging from 30 μM to 1.5 nM. Those showing dose-dependence against induced C1 cells, and no inhibition of uninduced cells were considered compounds of interest, and powders were ordered from commercial vendors for retesting in dose-response experiments. Concentration-response curves were fitted with a single-site four-parameter logistic function using Prism version 4.0 (GraphPad Software Inc., San Diego, CA) to derive half-inhibition concentration and the Hill coefficient. 
Chemical Synthesis.
The general methods for the synthesis of VU590 analogs are depicted in Scheme 1. To an 8-ml screw-capped vial were added 2.0 equivalents of aldehyde 2, 1.0 equivalent of Kryptofix 1 (dissolved in 0.5 ml of DCM), and 4.0 equivalents of MP-BH(OAc)3. An additional 2 ml of DCM was added to each vial, and the mixtures were gently rotated for 12 h. The mixtures were filtered, concentrated under vacuum, and the crude samples were purified by either mass-directed high-performance liquid chromatography, Gilson high-performance liquid chromatography, or normal phase-column chromatography to afford analog 3. Isolated yields for analytically pure compound 3 ranged from 32 to 85%.

Synthesis of VU590 analogs.
VU590.
To an 8-ml vial were added 4-nitrobenzaldehyde (70 mg, 0.46 mmol), Kryptofix (50 mg, 0.23 mmol), and MP-BH(OAc)3 (500 mg) in DCM (3 ml). The mixture was rotated for 12 h, filtered, and concentrated. Purification of the crude oil by ISCO flash chromatography (10% MeOH/DCM containing 1% NH4OH) provided the title compound VU590 as an orange oil (39 mg, 35%). 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.8 Hz, 4H), 7.60 (d, J = 8.8 Hz, 4H), 3.79 (s, 4H), 3.65–3.62 (m, 12H), 2.86–2.79 (m, 8H); high-resolution mass spectrometry (calculated for C24H32N4O7 + H), 489.2349; found, 489.2349.
Results
C1 Cell Line Characterization.
We generated a monoclonal cell line, termed C1, expressing the S44D mutant of rat ROMK1 under the control of a Tet-inducible promoter. A Tet-regulatable system was used to avoid cytotoxic effects of constitutive overexpression of ROMK (Nadeau et al., 2000) and cell line degeneration. The ROMK-S44D mutant was used to maximize cell surface expression of the channel (O'Connell et al., 2005; Yoo et al., 2005) for HTS.
As shown in Fig. 1A, ROMK-S44D protein expression was dramatically induced in C1 cells cultured overnight with Tet. Very minor channel expression was observed in uninduced C1 cells after prolonged film exposure (e.g., Fig. 1A). Biotinylation experiments showed robust channel expression on the cell surface (Fig. 1B).

Tetracycline-inducible ROMK-S44D expression in C1 cells. A, Western blot analysis of ROMK expression in lysates prepared from parental tetracycline-regulated expression or C1 cells cultured overnight in the absence or presence of Tet (1 μg/ml). In C1 cells (5 μg of lysate), but not in parental cells (20 μg of lysate), Tet induced the appearance of three bands of ∼45, ∼47, and ∼60 kDa, which correspond, respectively, to unglycosylated, core-glycosylated, and mature glycosylated forms of ROMK (Fallen et al., 2009). B, Western blot analysis of biotinylated immunoprecipitates showing the mature-glycosylated form of ROMK-S44D is expressed at the cell surface of induced C1 cells (top middle) but not in uninduced cells (top left). ROMK-S44D was not observed in immunoprecipitates from cells treated without biotin (top right) but was present in whole-cell lysates (middle right). β-Actin (bottom) was used as a loading control in both blots. These results are representative of at least three independent experiments.
Consistent with these observations, Tet induced robust ROMK channel activity in C1 cells (Fig. 2A, right). The mean ± S.E.M. current amplitude at −120 mV from Tet-induced and uninduced cells was −46.4 ± 8.3 (n = 9) and −1.0 ± 0.3 pA/pF (n = 5), respectively (Fig. 2C). ROMK exhibited a linear current-voltage (I-V) relationship between −120 and 40 mV (Fig. 2C) and inward rectification at potentials greater than 60 mV (Fig. 2, A, arrows, and C), reversed near the equilibrium potential for K+ (Fig. 2C), and was highly selective for K+ over Na+ (Fig. 2, D and E). Overnight Tet treatment hyperpolarized the resting membrane potential (RMP) from −47.4 ± 2.6 (uninduced) to −74.3 ± 0.5 mV (induced) in C1 cells (n = 6–9) but had no significant (P = 0.9) effect on that of parental cells [−21.9 ± 2.8 mV (−Tet) and −22.4 ± 2.9 (+Tet); n = 6–7]. The RMP of uninduced C1 cells was significantly (P < 0.001) more negative than that of parental cells (Fig. 2D), consistent with Western blot results showing minor ROMK expression in the absence of Tet (Fig. 1A). Barium (Ba2+) and TPNQ blocked ROMK with half-inhibition concentrations (IC50) of 12 μM and 8 nM, respectively (data not shown).

Electrophysiological properties of C1 cells. A, whole-cell currents recorded from C1 cells cultured overnight in control media or media containing 1 μg/ml Tet. The cells were voltage-clamped and stepped between −120 and 120 mV in 20-mV increments for 500 ms from a holding potential of −75 mV. The arrows indicate current rectification at positive test potentials. B, mean ± S.E.M. I-V relationships for parental cells incubated with (•, n = 7) or without (○, n = 7) Tet. C, mean ± S.E.M. I-V relationships for C1 cells incubated with (n = 9) or without (n = 5) Tet. D, representative reversal potential (Erev) measurements of Tet-induced whole-cell currents in C1 cells after equimolar substitution of bath NaCl with KCl. The cell was voltage-ramped between −120 and 80 mV from a holding potential equal to the calculated Nernst potential for K+ in the presence of 5, 25, and 100 mM extracellular K+ (K +). E, mean ± S.E.M. Erev measurements recorded from five cells plotted against the log of extracellular K+ concentration ([K
+). E, mean ± S.E.M. Erev measurements recorded from five cells plotted against the log of extracellular K+ concentration ([K +]). The slope of the linear fit to this relationship was 55 mV/log mM K+, indicating the whole-cell current is highly selective for K+ over Na+ and dominated by ROMK. F, mean ± S.E.M. RMP measurements from parental and C1 cells cultured overnight in control (Con) or Tet-containing media. Tet had no significant (N.S.) effect (P = 0.9) on parental cell RMP, but significantly (**, P < 0.001 compared with C1 cells incubated without Tet) hyperpolarized the RMP of C1 cells. Note also that the RMP of C1 cells cultured with or without Tet was significantly (*, P < 0.001) more negative than that of parental cells, consistent with minor “leakage” of ROMK protein expression (Fig. 1A) in C1 cells.
+]). The slope of the linear fit to this relationship was 55 mV/log mM K+, indicating the whole-cell current is highly selective for K+ over Na+ and dominated by ROMK. F, mean ± S.E.M. RMP measurements from parental and C1 cells cultured overnight in control (Con) or Tet-containing media. Tet had no significant (N.S.) effect (P = 0.9) on parental cell RMP, but significantly (**, P < 0.001 compared with C1 cells incubated without Tet) hyperpolarized the RMP of C1 cells. Note also that the RMP of C1 cells cultured with or without Tet was significantly (*, P < 0.001) more negative than that of parental cells, consistent with minor “leakage” of ROMK protein expression (Fig. 1A) in C1 cells.
Assay Development.
We used C1 cells to develop a fluorescence-based assay of ROMK function for HTS. The assay reports flux of the K+ congener thallium (Tl+) through ROMK channels using the fluorescent dye Fluozin-2 (Weaver et al., 2004; Delpire et al., 2009; Fallen et al., 2009). Figure 3A shows representative fluorescence traces recorded from individual wells of a 384-well plate containing uninduced (−Tet) or Tet-induced cells bathed in control (+Tet) or TPNQ-containing assay buffer (Tet + TPNQ). Tl+ evoked a rapid fluorescence increase in induced cells but not in uninduced cells or induced cells pretreated with the ROMK blocker TPNQ. The slope of the Fluozin-2 fluorescence increase between 7 and 12 s after Tl+ addition was used as a measure of Tl+ flux. TPNQ dose-dependently decreased Tl+ flux in induced cells with an IC50 of 5.2 nM (Fig. 3C), indicating that the assay is sensitive and capable of reporting graded changes in ROMK activity. To assess the suitability of the assay for HTS, every other well of a 384-well plate was pretreated with control assay buffer or TPNQ-containing buffer before Tl+ addition. The rate of Tl+ flux varied little within treatments but was dramatically different between control- and TPNQ-treated wells (Fig. 3B). For this plate the calculated Z′ value, a statistical indicator of the assay's ability to correctly identify hits in a screen, was 0.68. Assays with Z′ values between 0.5 and 1.0 are considered suitable for HTS (Zhang et al., 1999).

High-throughput assay of ROMK function. A, Fluozin-2 fluorescence traces recorded from C1 cells before and after the addition of extracellular Tl+ (shaded area). Fluorescence emission was recorded from individual wells of a 384-well plate containing 30,000 C1 cells cultured in the absence (−Tet) or presence (+Tet) of Tet. Induced cells were pretreated for 20 min with control buffer or buffer containing 10 μM TPNQ to block ROMK activity. B, plot of the slope of Tl+-induced fluorescence increase recorded from individual wells of induced C1 cells treated with control buffer (C) or TPNQ (T). The Z′ statistic calculated for this plate was 0.68. C, concentration-response curve for TPNQ inhibition of ROMK. Data are means ± S.E.M. (n = 4). Fit of a four-parameter logistic function to the concentration-response curve (CRC) data yielded an IC50 of 5.2 nM with 95% confidence intervals between 3.5 and 7.6 nM.
High-Throughput Screening.
Using the Tl+ flux assay, we screened 126,009 compounds in 384-well plates at a single dose of 10 μM. DMSO at a final concentration 0.1% was used as the compound vehicle. During assay development, we established that Tl+ flux through ROMK was insensitive to DMSO concentrations up to 10% (data not shown). Four columns in each plate were filled with control or TPNQ-containing buffer for the determination of Z′ (e.g., Fig. 3B). Plates with Z′ values less than 0.45 were not included in the data analysis. The mean ± S.D. Z′ of plates passing quality control was 0.72 ± 0.08. The processed primary data resulted in 1758 compounds designated as inhibitors.
Identification of a Small-Molecule Inhibitor of ROMK and Kir7.1.
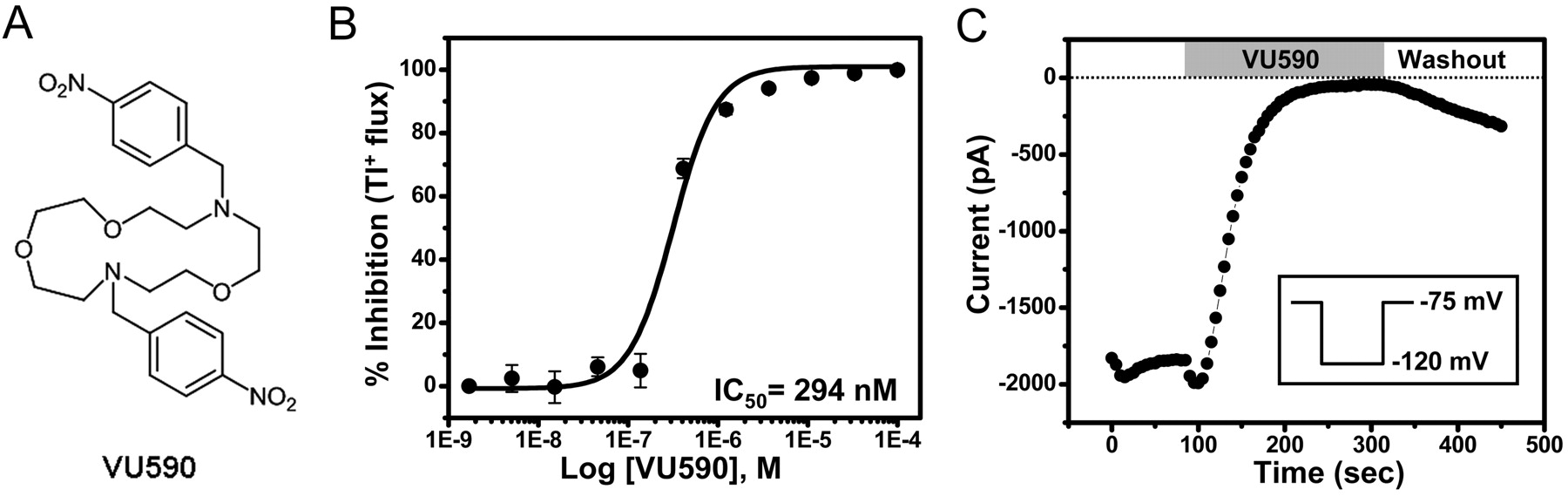
Figure 4 A shows the structure of one ROMK inhibitor identified by HTS. The HTS stock of the compound purchased from ChemDiv (San Diego, CA), which we have termed VU590, afforded an IC50 of 1.5 μM (data not shown). VU590 was resynthesized (see Materials and Methods) and characterized in detail in Tl+ flux and electrophysiological experiments. Tl+ flux was recorded from C1 cells incubated for 20 min in VU590 at concentrations between 1.69 nM and 100 μM. As shown in Fig. 4B, VU590 inhibited ROMK in a dose-dependent manner with an IC50 of 294 nM, which represents a 5-fold improvement in potency over the HTS stock. At a concentration of 10 μM, VU590 had no effect on Tl+ flux in uninduced cells (data not shown), suggesting the compound acts specifically on ROMK. To exclude the possibility that VU590 inhibits the channel indirectly through intracellular acidification (Schulte et al., 1999), we tested its effects on wild-type ROMK in whole-cell patch-clamp recordings in which intracellular pH was held constant at pH 7.35 with a well-buffered pipette solution. Figure 4C shows that VU590 abolished channel function under these conditions. These experiments confirmed that VU590 is a bona fide inhibitor of ROMK.

Small-molecule inhibitor of ROMK identified by HTS. A, structure of VU590. B, CRC for inhibition of ROMK-S44D by VU590. Values are mean ± S.E.M. Tl+ flux from four independent experiments performed on three different days. A fit of the CRC with a single-site four parameter logistic function yielded an IC50 of 294 nM and Hill coefficient of 1.8. C, electrophysiological confirmation of VU590 as a ROMK inhibitor. A cell transfected with wild-type ROMK1 was voltage-clamped in the whole-cell configuration and then stepped from a −75 mV holding potential to −120 mV (inset) for 200 ms every 5 s to evoke ROMK current. Bath addition of 10 μM VU590 led to a nearly complete inhibition of ROMK activity.
The selectivity of VU590 among members of the inward rectifier family was assessed by patch-clamp electrophysiology. Figure 5A shows representative current traces recorded from ROMK, human Kir2.1, human Kir4.1, or human Kir7.1 in the absence (black) or presence (gray) of 10 μM VU590. At this concentration, VU590 inhibited ROMK current at −120 mV by 95.3 ± 0.5% (n = 9). Consistent with Tl+ flux data (Fig. 4B), inhibition was dose-dependent. At concentrations of 1 and 3 μM, VU590 inhibited ROMK activity by 63.4 ± 1.8% (n = 5) and 78.2 ± 4.9% (n = 5), respectively (Fig. 5B). Block by submicromolar concentrations of VU590 was too slow to reliably distinguish from rundown observed occasionally during long recordings. At a concentration of 10 μM, VU590 had no significant (P > 0.05) effect on either Kir2.1 (n = 5) or Kir4.1 (n = 6). The decrease in outward current shown in the Kir2.1 current traces (Fig. 5B, top right) is due to partial inhibition of endogenous HEK-293 currents (data not shown). It is important to note that the compound inhibited Kir7.1 by 68.8 ± 6.2% (n = 10; Fig. 5B). To our knowledge, VU590 is the first small-molecule inhibitor of Kir7.1.
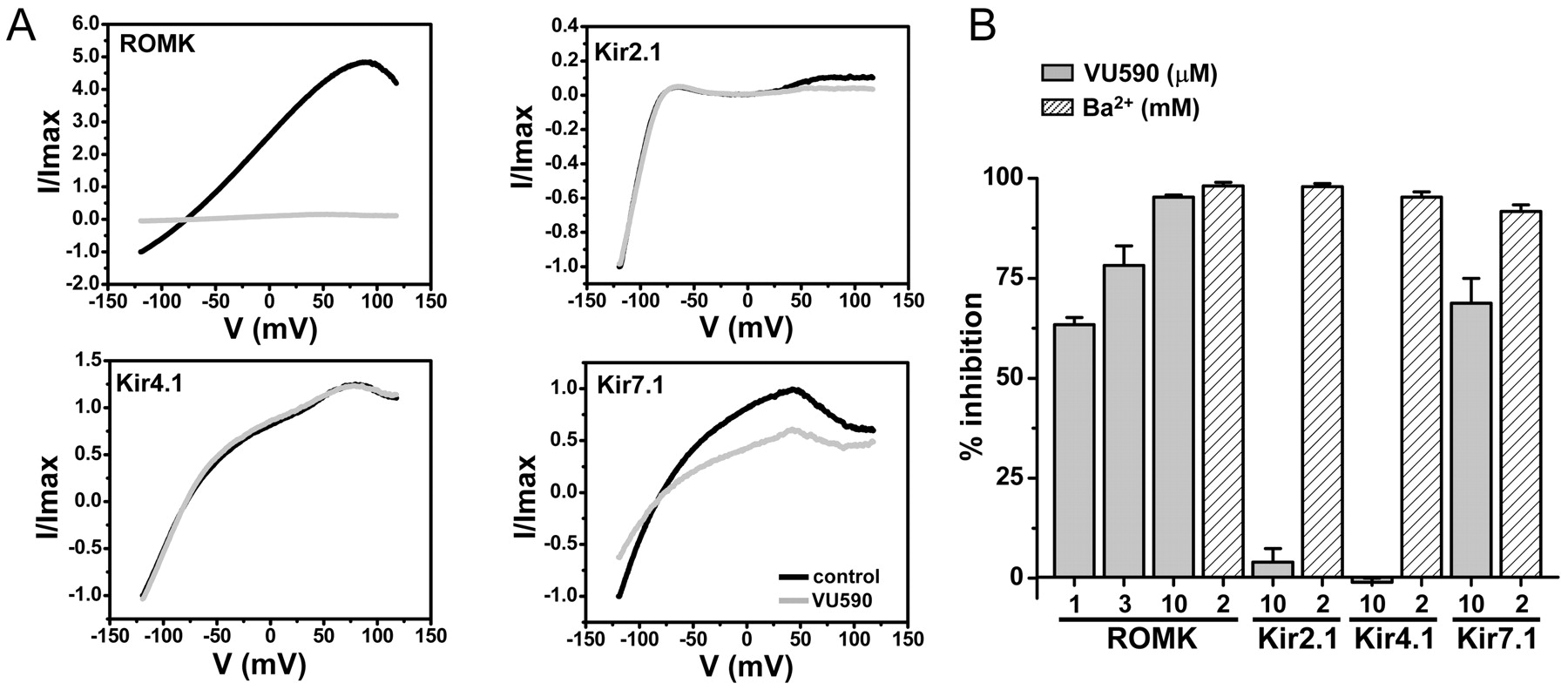
VU590 is an inhibitor of ROMK and Kir7.1. A, representative whole-cell current traces recorded from HEK-293 cells transiently transfected with wild-type rat ROMK (top left), human Kir2.1 (top right), human Kir4.1 (bottom left), or human Kir7.1 (bottom right). The cell was stepped from a holding potential of −75 to −120 mV for 200 ms (data not shown) then ramped between −120 and 120 mV at a rate of 2.4 mV/ms. Current traces were recorded before (black) and after several minutes (gray) of applying 10 μM VU590 to the bath. B, mean ± S.E.M. percentage inhibition of current recorded at −120 mV by VU590 ( ) or Ba2+ ( positive control). The concentrations of VU590 (micromolar) and Ba2+ (millimolar) used are shown below each bar.
) or Ba2+ ( positive control). The concentrations of VU590 (micromolar) and Ba2+ (millimolar) used are shown below each bar.
Synthesis and Structure-Activity Relationships of VU590 Analogs.
Lead optimization efforts directed at VU590 were then initiated using a parallel library synthesis approach wherein kryptofix 1 was treated with a diverse set of 24 aldehydes 2 to form 24 symmetrical bis-tertiary amine analogs 3 upon a reductive amination sequence with MP-B(OAc)3H. Structure-activity relationships proved shallow, yielding few active analogs (IC50 values in the 6–50 μM range). As shown in Fig. 6, moving the nitro group from the 4-position (VU590) to the 3-position (3a) resulted in a 43-fold loss in potency (IC50 = 14.2 μM) and further migration to the 2-position (3b) or a 2,4-dinitro analog (3c) led to a complete loss (IC50 > 100 μM) of ROMK inhibitory activity. Alternate functional groups for the 4-nitro moiety such as 4-Cl (3d), 4-thiomethyl (3e), and 4-methyl ester (3f) afforded weak ROMK inhibitors, with IC50 values of 13, 8.5, and 33 μM, respectively. All attempts to attenuate the basicity of VU590 by replacing the tertiary amine moieties with amides, sulfonamides, and ureas resulted in inactive compounds (IC50 values >100 μM). These data suggest VU590 is a unique ROMK inhibitor with a special balance of physiochemical properties.

Determination of VU590 SARs through combinatorial chemistry. A, structure of VU590 and areas to explore through chemical optimization. B, structure-activity relationships of analogs of VU590. C, alternate capping agents to attenuate the basicity of VU590.
VU590 Is a ROMK Channel Pore Blocker.
The rate of ROMK inhibition by 10 μM VU590 was slow (mean ± S.E.M. time constant for inhibition was 40.0 ± 0.5 s; n = 8), and washout was incomplete (e.g., Fig. 4C), suggesting that the compound might first cross the plasma membrane to reach an intracellular binding site. Furthermore, we noted a large hyperpolarizing shift in the rectification potential during the onset of inhibition (Fig. 7, A and B). Because Kir channel rectification is caused by a block of outward current by intracellular cations (Lopatin et al., 1994; Lu and MacKinnon, 1994; Wible et al., 1994; Fakler et al., 1995), this observation raised the possibility that VU590 is a cytoplasmic pore blocker. We therefore tested whether VU590 could be displaced from the pore with inwardly directed K+ ions through a mechanism known as “knockoff” (Shin and Lu, 2005). Experiments were performed by preblocking ROMK channels with 10 μM VU590 and voltage-clamping cells to potentials more negative than the K+ equilibrium potential (EK) to drive K+ ions intracellularly through the pore. In the presence of 5 mM extracellular K+ and 135 mM intracellular K+ (EK = −83 mV), block by VU590 was virtually insensitive to pulses to −120 mV. However, stronger pulses led to knockoff of VU590 that were both time- and voltage-dependent (Fig. 7, C and D). To confirm that VU590 knockoff is caused by permeant ions interacting with VU590 and not, for example, by voltage-dependent conformational changes in the compound's binding site, we tested whether increasing the electrochemical driving force on K+ would facilitate VU590 knockoff at −120 mV, a potential that was ineffective in the presence of 5 mM K+. As shown in Fig. 7, E and F, elevation of extracellular K+ to 50 mM (EK = −25 mV) readily displaced VU590 from the pore during pulses to −120 mV. The most parsimonious interpretation of these data, and the one we favor, is that VU590 is an intracellular pore blocker of ROMK.
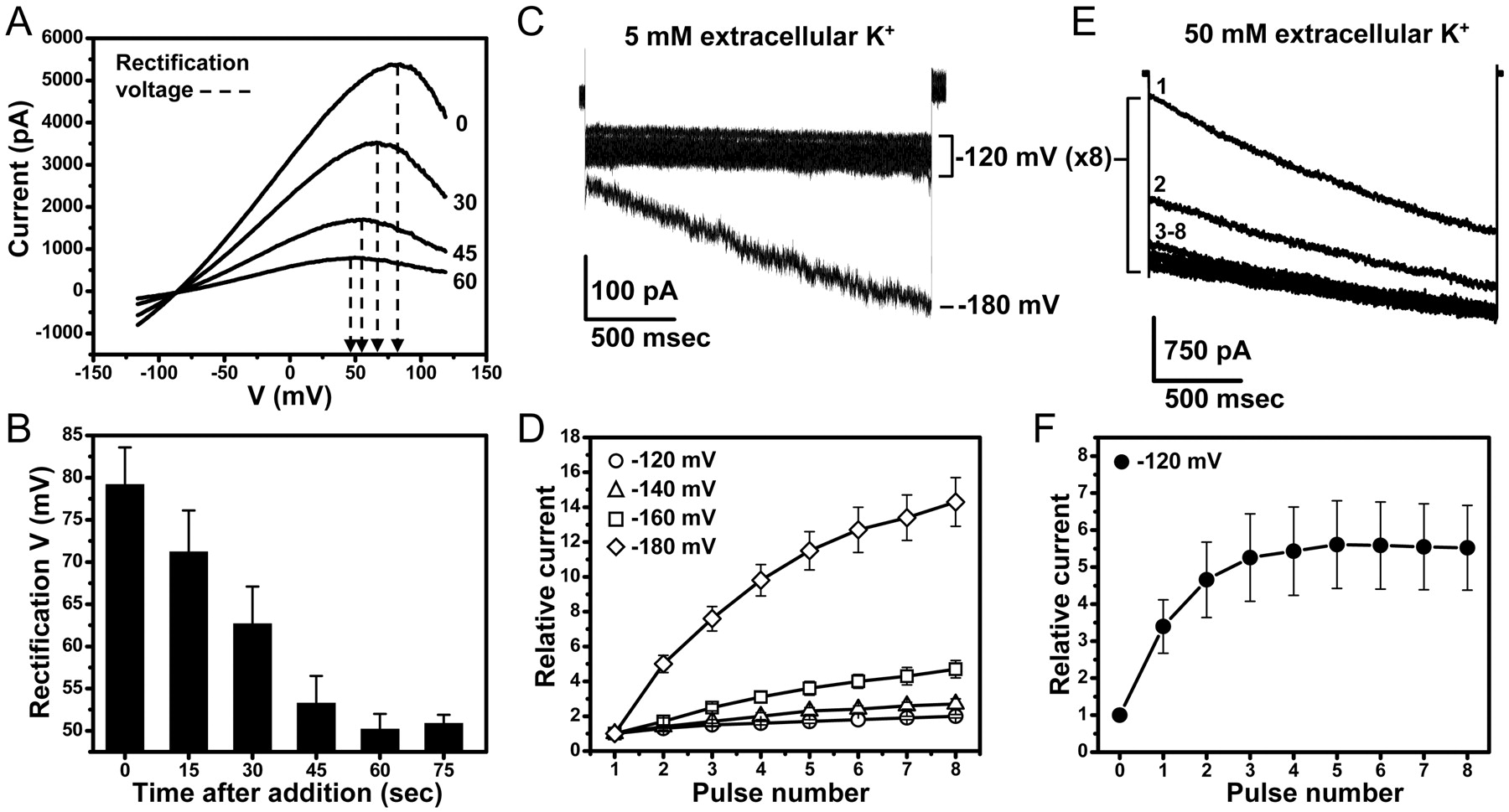
VU590 is a ROMK channel pore blocker. A, representative wild-type ROMK current traces evoked from voltage ramps between −120 and 120 mV before and during the onset of inhibition by 10 μM VU590. The time (in seconds) after compound application is shown next to each current trace. The dashed arrows are drawn at the point of current inflection to indicate the current's rectification potential. The mean ± S.E.M. rectification potentials as a function of time are shown in B. C, ROMK current traces evoked from a cell bathed in a solution containing 5 mM K+ and 10 μM VU590 after achieving maximal block. Eight successive pulses to −120 mV, separated by a 5-s interpulse interval, had virtually no effect on current amplitude, but a single step to −180 mV led to time-dependent unblock of the channel. The mean ± S.E.M. normalized current amplitude recorded at −120 mV (○, n = 5), −140 mV (▵, n = 4), −160 mV (□, n = 5), and −180 mV (⋄, n = 4) is summarized in Fig. D. E and F, elevation of extracellular K+ to 50 mM displaces VU590 from the pore. Cells were stepped to −120 mV for 2 s and then returned to a holding potential of −25 mV for 5 s between pulses (n = 6).
Discussion
In the present study, we developed a robust HTS assay for small-molecule ROMK modulators, which enabled the identification of a novel blocker of ROMK and Kir7.1 channels. The assay extends previous work using Tl+ flux to detect potassium channel and transporter activities (Weaver et al., 2004; Delpire et al., 2009; Fallen et al., 2009) and overcomes ROMK-specific technical hurdles associated with reportedly poor channel expression in mammalian cells (Brejon et al., 1999) by using an inducible expression system and point mutation (S44D) that promotes cell surface expression (O'Connell et al., 2005; Yoo et al., 2003). The identification by HTS of numerous ROMK antagonists, some of which preferentially block other Kir channels (data not shown), is a significant step toward developing the molecular pharmacology of the inward rectifier family.
Results from electrophysiological experiments indicate that VU590 blocks the ion permeation pathway of ROMK. VU590 block is relieved by hyperpolarizing pulses and increased extracellular K+ concentrations. The simplest interpretation is that these maneuvers increase the rate of blocker dissociation into the cytoplasmic compartment via ion-blocker interactions within the intracellular pore. VU590 actions on the extracellular pore with blocker “punch-through” (Kucheryavykh et al., 2007) is also conceivable, but seems less likely given the recent identification of low potency cytoplasmic Kir channel inhibitors exhibiting similar electrophysiological profiles. Tricyclic antidepressants such as nortriptyline block Kir4.1 channels with IC50 values in the 20 to 100 μM range (Furutani et al., 2009), whereas the antimalarial agent chloroquine inhibits Kir2.1 with an IC50 of ∼10 μM (Rodríguez-Menchaca et al., 2008). Block by both compounds is relieved by membrane hyperpolarization and elevated extracellular K+ concentration. Mutagenesis experiments have shown these agents block the cytoplasmic pore of the channels. We are currently using mutagenesis and molecular modeling to define the binding site of VU590 within ROMK and Kir7.1. It is noteworthy that nortriptyline and chloroquine exhibit no appreciable activities toward ROMK (Furutani et al., 2009; J.S. Denton, unpublished observations) and that VU590 has no effects on Kir2.1 or Kir4.1. These observations suggest that Kir channels possess selective drug binding sites within the cytoplasmic pore that can be targeted with organic small molecules.
Although our understanding of Kir channel structure-function relationships has advanced considerably with the determination of Kir channel X-ray structures, the physiology of some inward rectifiers remains poorly understood due in part to the lack of pharmacological tools to manipulate Kir channel activity. Kir7.1 is widely expressed in brain, retina, intestine, and kidney (Krapivinsky et al., 1998), but little is known of its function. The identification of disease-causing mutations in KCNJ13, the gene encoding Kir7.1, in a patient with Snowflake vitreoretinal degeneration suggests the channel plays a key role in retinal development and/or physiology (Hejtmancik et al., 2008). The putative role of Kir7.1 in modulating retinal pigment epithelial function can now be tested directly with VU590 because ROMK is not expressed in these cells (Yang et al., 2008). In the nephron, Kir7.1 is expressed on the basolateral surface of the distal convoluted tubule and CCD, suggesting it may play a role in regulating basolateral potassium transport and in turn sodium reabsorption (Ookata et al., 2000). Although ROMK and Kir7.1 are coexpressed in these nephron segments, particularly the CCD, it may be possible to dissect their relative roles using a combination of VU590 and TPNQ.
In contrast to Kir7.1, the functional role of ROMK in the regulation of renal sodium and potassium transport has been studied extensively. Electrophysiological studies of renal tubular potassium currents in wild-type and ROMK knock-out mice have established that ROMK underlies a major apical potassium conductance in the TAL, CNT, and CCD (Lu et al., 2002; Frindt et al., 2009). Functionally, ROMK activity supports sodium and potassium reabsorption in the TAL and potassium secretion in the CNT and CCD. ROMK antagonists could conceivably provide robust natriuresis and diuresis by acting at the TAL but do so with minimal kaliuresis by inhibiting potassium secretion at the CCD. Although appealing, there is currently no direct evidence to support this notion. The diuretic and natriuretic efficacy of ROMK seems relatively ensured given the severe salt-wasting phenotype of ROMK knockout mice (Lorenz et al., 2002; Lu et al., 2002) and Bartter syndrome patients carrying homozygous loss-of-function mutations in ROMK (Simon et al., 1996). However, the ability to limit urinary potassium wasting depends critically on the ability to inhibit distal potassium secretion in the face of high urinary flow rates caused by proximal inhibition of sodium and water reabsorption. The extent to which ROMK mediates K+ secretion in the CCD during high flow states versus other apical K+ channels, particularly calcium-activated BK channels (Liu et al., 2007), is not well understood. If ROMK plays a comparatively minor role in CCD potassium secretion during high flow states, urinary potassium wasting may not be mitigated. Indeed, Bartter syndrome patients with complete loss of ROMK function are in most cases hypokalemic, albeit to a lesser extent than patients with loss of NKCC2 function (Simon et al., 1996). It should be kept in mind, however, that global changes in kidney gene expression, development, and function caused by the complete loss of function of a gene may not accurately reflect the physiological changes that occur after short-term administration of an antagonist of the gene product. Ultimately, only ROMK-selective inhibitors studied in vivo can provide proof-of-principle evidence needed to drive clinical drug discovery efforts. We anticipate that small-molecule antagonists of ROMK identified by HTS will enable future studies to fill this important knowledge gap.
Acknowledgments
We thank Drs. Chun Jiang (Georgia State University), Alfred George (Vanderbilt University Medical Center), and David Clapham (Harvard Medical School), respectively, for providing the rat ROMK1, human Kir2.1, and human Kir7.1 expression vectors.
Footnotes
-
This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant 1R21-NS57041-1]; the National Institutes of Health National Institute of Mental Health Molecular Libraries Screening Centers Network [Grants U54-MH074427, 1U54-MH084659-01]; and a National Kidney Foundation Postdoctoral Fellowship grant.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.059840
-
ABBREVIATIONS:
-
L.M.L. and G.B. contributed equally to this work.
- Received July 27, 2009.
- Accepted August 25, 2009.
- © 2009 The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}