


Abstract
The synthetic cannabinoid WIN55,212-2 (WIN) is widely used as a pharmacological tool to study the biologic activity of cannabinoid receptors. In contrast to many other cannabinoid agonists, however, WIN also causes broad effects outside of neurons, such as reducing inflammatory responses, causing cell cycle arrest, and reducing general protein expression. How exactly WIN causes these broad effects is not known. Here we show that WIN partially disrupts the Golgi apparatus at nanomolar concentrations and fully disperses the Golgi apparatus in neuronal and non-neuronal cells at micromolar concentrations. WIN55,212-3, the enantiomer of WIN; JWH-018, a related alkylindole; or 2-arachidonoylglycerol, an endocannabinoid, did not cause Golgi disruption, suggesting that the effect was specific to the chirality of WIN. WIN treatment also perturbed the microtubule network. Importantly, WIN disrupted the Golgi in primary cortical neurons derived from mice where cannabinoid receptor-1 (CB1) was genetically knocked out, indicating that the effects were independent of CB1 signaling. The Golgi dispersion could not be explained by WIN’s action on peroxisome proliferator-activated receptors. Our results show that WIN can disrupt the Golgi apparatus independent of CB1 in cultured cells. These effects could contribute to the unique physiologic effects that WIN exhibits in neuronal behavior, as well as its role as an antiproliferative and anti-inflammatory agent.
SIGNIFICANCE STATEMENT The synthetic cannabinoid WIN55,212-2 (WIN), widely used to investigate the cannabinoid system, also shows unique broader effects at cellular and organismal levels compared to endogenous cannabinoids. Our study shows that WIN can disrupt the Golgi apparatus and the microtubule network in multiple cell types, independent of cannabinoid receptors. These results could explain how WIN reduces surface levels of proteins and contributes to the unique physiological effects observed with WIN.
Introduction
Cannabinoid ligands are versatile pharmacological agents. Cannabinoids primarily activate the cannabinoid receptor-1 (CB1), a G protein-coupled receptor that plays a vital role in modulating neurotransmission in the central nervous system (CNS) and in the peripheral nerves. In the CNS, CB1’s activation is part of a highly coordinated retrograde signaling mechanism (Castillo et al., 2012; Ohno-Shosaku and Kano, 2014), which suppresses neuronal depolarization as highlighted by numerous electrophysiological recordings (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). Cannabinoid agonists are valuable therapeutics for addressing drug addiction, neuropathic pain, epilepsy, and various psychiatric disorders (Jetly et al., 2015; Smith et al., 2015; O’Connell et al., 2017; Mücke et al., 2018; Segura et al., 2019).
The synthetic cannabinoid WIN55,212-2 (WIN) is a high-affinity CB1 agonist that is widely used to investigate cannabinoid physiology. WIN’s increased potency compared with the endogenous cannabinoids 2-arachidonoylglycerol (2AG) and anandamide [AEA; N-(2-hydroxyethyl)-5Z,8Z,11Z,14Z-eicosatetraenamide], which are partial agonists for CB1 (Pertwee, 2005), often makes it a preferred compound used to study cannabinoid pharmacology in cells and animals (Sim-Selley and Martin, 2002; Martini et al., 2010; Chen et al., 2021). However, WIN is a particularly interesting cannabinoid agonist in this regard, as it shows unique pharmacological and physiologic outcomes. These outcomes could be driven by WIN’s unique chemical properties, including stereoisomer specificity (Emery et al., 2014). One notable physiologic difference between WIN and other cannabinoids is observed in cancer pharmacology, where WIN has been shown to cause robust antiproliferative effects in oncogenic cells (Scuderi et al., 2011; Wasik et al., 2011; Emery et al., 2014; Pellerito et al., 2014; Müller et al., 2017). In addition, WIN has been proposed to have anti-inflammatory and antiproliferative effects in a variety of settings (Marchalant et al., 2007; Marchalant et al., 2008; Zhao et al., 2010; Wang et al., 2018). These broad effects have generated interest in the role of synthetic cannabinoids like WIN in physiologic systems outside of the CNS.
One overarching theme that is unique to WIN’s actions at the cellular level is a reduction in the surface expression and secretion of proteins. For neurotransmitter receptors and their accessory proteins, this reduction can have profound effects on neurotransmission (Blume et al., 2013; Perdikaris et al., 2018). WIN downregulates the surface expression of CB1 and GABAA subunits (Deshpande et al., 2011). This reduction might be partly due to a decrease in total protein expression via mRNA downregulation (Perdikaris et al., 2018; Tan and Cao, 2018). Similarly, in non-neuronal cells, WIN reduces the generation of inflammatory mediators in non-neuronal cells (Lowin et al., 2016), suggesting that the effects of WIN on surface expression and secretion of proteins could be general. However, whether WIN generally regulates protein trafficking and whether WIN’s effects on surface expression of proteins are via CB1 activation are not fully known.
In this study, we used high-resolution fluorescence microscopy to explore the effects of WIN on components of the trafficking machinery in neuronal and non-neuronal cells. We found that WIN (but not its enantiomer WIN55,212-3, the related alkylindole JWH-018, or the endocannabinoid 2AG) partially disrupts the Golgi apparatus at nanomolar concentrations and completely disrupts the Golgi apparatus in both neuronal and non-neuronal cells at micromolar concentrations. WIN treatment disrupted the Golgi in neurons obtained from CB1 knockout (CB1-KO) mice, suggesting that this effect is CB1-independent. Our findings provide a potential mechanism by which WIN can reduce levels of select surface proteins and produce a broad range of physiologic effects independent of CB1.
Materials and Methods
Cell Culture and Reagents
Experiments performed in human embryonic kidney 293 (HEK 293) cells (American Tissue Culture Collection, Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose (Thermo Fisher Scientific) + 10% fetal bovine serum (FBS; GIBCO). E18 striatal neurons were obtained from BrainBits, LLC and cultured for 2 weeks following the recommended protocol. Cortical CB1-KO neurons were obtained from P0 mice and cultured for 2 weeks following the BrainBits, LLC recommended protocol. Cells in the laboratory were tested routinely for mycoplasma contamination. CB1 knockout mice were described in Ledent et al. (1999). SNAP-Cell 647-SiR (300 nM; New England Biolabs) was used to label SNAP-CB1 in HEK293 cells. Drugs used for treatment conditions were obtained and prepared as follows: 2AG was purchased from Tocris Bioscience and prepared as a 10 mM stock with ethanol. WIN, GW6471, and GW9662 were purchased from Sigma-Aldrich (St. Louis, MO) and prepared as 10mM stocks with DMSO.
Experimental Protocols
HEK293 cells were transfected with SNAP-CB1 and GPP130-GFP using Effectene (QIAGEN) according to the provided manufacture protocols. Stable cell lines were generated using selection with geneticin (Invitrogen). Cell viability was assessed by labeling cells with Trypan Blue stain and manually counting the cells labeled using a counting chamber. Protein secretion was estimated by washing cells with PBS, incubating cells in PBS for 1 hour, and estimating the amount of protein in the supernatant by using a Pierce BCA Protein Assay (Thermo Fisher Scientific). Surface delivery of CB1 was estimated using a sequential labeling protocol. HEK cells expressing SNAP-CB1 were labeled with impermeable SNAP-Cell 488 (300 nM; New England Biolabs) to saturate the surface CB1. They were then incubated with impermeable SNAP-Cell 647 (100 nM; New England Biolabs) for 1 hour in the presence or absence of WIN. The ratio of 647/488 was used to measure the relative amount of new CB1 delivered to the surface.
Fixed-Cell Immunofluorescence
After each drug treatment, cells were fixed with 4% paraformaldehyde (P6148; Sigma-Aldrich) for 15 minutes and processed for immunofluorescence as described recently (Kunselman et al., 2021). After fixation, antibody labeling was performed as follows:
Anti-GPP130 (1:1000; provided by Adam Linstedt, Carnegie Mellon University, Pittsburgh, PA)
Anti-TGN46 (1:1000; ab50595; Abcam)
Anti-TGN38 (1:1000; T9826; Sigma-Aldrich)
Anti-GM130 (1:1000; PA5-95727; Thermo Fisher Scientific)
Anti-α-tubulin (1:1000; ab185031; Abcam)
Anti-MAP2 (1:1000; ab5392; Abcam)
Confocal images were taken on a Nikon Eclipse Ti inverted microscope using a 20×/0.75 numerical aperture (NA), 60×/1.49 NA, or 100×/1.49 NA objective. The images were acquired with an iXon+ 897 electron-multiplying charge-coupled device camera using Andor IQ software (Andor).
Image Analysis Protocols
All images were analyzed via ImageJ. We generated custom ImageJ macros for standardized quantification of GPP130 and α-tubulin in HEK cells, trans-Golgi network (TGN) marker TGN38 in rat striatal neurons (BrainBits, LLC), and GM130 in mouse CB1-KO cortical neurons (Ledent et al., 1999). Briefly, images were thresholded using a specific value, and the area and the fluorescence of the objects identified above threshold were used for data analysis. The parameters for analysis were kept identical between the control and the experimental conditions.
Statistical Analysis
This exploratory study was designed to address the effect of WIN on membrane trafficking. Sample sizes (number of cells or fields to be analyzed) were determined before the experiment was performed, and the P values reported are descriptive. All analysis was performed on GraphPad Prism 8 (2019). We determined before viewing the data that each group would be compared with the respective control conditions. Data were analyzed by D’Agostino-Pearson test for normal distribution. For normally distributed data, statistical significance for analysis comparing two experimental conditions was derived from unpaired t tests, and for three or more conditions by ordinary one-way ANOVAs and multiple comparisons by a Dunnett’s test. For data not normally distributed, statistical significance for analysis comparing two experimental conditions was derived from Mann-Whitney tests, and for three or more conditions by Kruskal-Wallis test with multiple comparisons by a Dunn’s test. All analyses are reported. For each graph, n denotes the number of cells analyzed for each condition for single-cell analyses, or the number of fields analyzed for each condition for conditions that test penetrance of phenotype. Statistical significance is denoted as follows: ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.001.
Results
WIN Disrupts the Golgi Apparatus
We first tested the effect of WIN in CB1-expressing HEK293 cells. HEK293 cells stably expressing SNAP-tagged CB1 were treated with 10 μM of either WIN or 2AG for 1 hour and labeled with a cell-permeable SNAP label to detect CB1. In control cells, CB1 was present on the cell surface and on intracellular compartments that roughly overlapped with the Golgi apparatus, detected by the cis-Golgi marker GPP130 (Fig. 1A). This suggests that CB1 could localize to the Golgi in steady state conditions in these cells. This intracellular CB1 localization is consistent with previous reports (Leterrier et al., 2004; Grimsey et al., 2010), as is also observed for other G protein-coupled receptors such as the δ-opioid receptor in neuronal cells (Shiwarski et al., 2017). This intracellular localization did not change when cells were treated with the endocannabinoid 2AG. In contrast, this intracellular pool of CB1 became more dispersed across the cytoplasm in cells treated with WIN (Fig. 1A).

WIN disrupts the Golgi apparatus. (A) HEK293 cells imaged with confocal microscopy showing SNAP-CB1 and GPP130 labeling after 1-hour drug treatment conditions. The Golgi is intact in control and 2AG [10 μM] conditions, whereas GPP130 labeling shows a robust dispersal phenotype for cells in the WIN [10 μM] condition. Similarly, we observe a dramatic redistribution of intracellular CB1 after WIN treatment, which we did not observe with 2AG (yellow arrows). Scale bar = 5 μm. (B) GPP130 labeling covers a larger surface area after WIN treatment as the Golgi is dispersed throughout the cell, and (C) the fluorescence intensity of GPP130 correspondingly decreases as the antibody label becomes less concentrated within the defined Golgi area (**P < 0.01; ***P < 0.001; ****P < 0.0001; n ≥ 9). (D) Confocal images shown at 20× magnification used for high-throughput analysis of Golgi disruption. As previously shown, Golgi dispersal is observed after 1 hour of WIN treatment, and the mask generated from antibody labeling was used to identify Golgi objects as defined by predetermined area and fluorescence constraints. Scale bar = 10 μm. Fewer objects were detected in 5 μM and 10 μM treatment conditions for both (E) cis-Golgi compartments and (F) trans-Golgi compartments. Circles represent biologic replicate means (ns, not significant; n = 3 averaged biologic replicates). (G) Representative confocal images showing GPP130 labeling and 3D object identification. Using our analysis paradigms, only one 3D object is detected (white) when the Golgi is intact, and multiple 3D objects are detected when the Golgi is fragmented. In this example, two objects are detected (white and yellow). Scale bar = 2.5 μm. (H) Golgi fragmentation, represented as a percentage of fragmented Golgi detected out of the total number of Golgi, shows that 1-hour WIN treatment at nanomolar concentrations causes Golgi fragmentation but not complete disruption.
This finding raised the question as to whether the dispersed localization induced by WIN was due to specific redistribution of CB1 or whether this was due to general dispersal of the Golgi. To test this, we imaged the behavior of the general Golgi marker GPP130. GPP130 appeared fully dispersed across the cytoplasm upon WIN treatment compared with the discrete staining seen in untreated cells or cells treated with 2AG (Fig. 1A). To quantify dispersal of Golgi compartments, we reasoned that dispersal would result in redistribution of fluorescence over a larger area and, correspondingly, that the fluorescence in each area would be reduced. Therefore, we measured the fraction of total cell area covered by the Golgi. In control and 2AG conditions, the Golgi covered roughly 20% of the cell’s total area. In contrast, in WIN-treated cells, the Golgi covered roughly 60% of the cell’s total area (Fig. 1B). We next measured the average fluorescence of the Golgi marker GPP130 in the three conditions. In WIN-treated cells, the Golgi fluorescence decreased almost 3-fold compared with that of control and 2AG-treated cells (Fig. 1C). This decrease roughly corresponded to the 3-fold increase in area. The reciprocal changes in area and fluorescence indicate that the Golgi was dispersed in cells after WIN treatment.
We next developed an automated analysis paradigm using area and fluorescence constraints to detect Golgi dispersion in cells imaged at 20× magnification (Fig. 1D; Supplemental Fig. 1). Although this assay was of lower resolution and therefore lower sensitivity, this allowed us to analyze dispersion in a higher throughput and objective manner. Using this assay, we observed that WIN dispersed the Golgi at concentrations ≥5 μM (Fig. 1E). Because GPP130 is a cis-Golgi protein, we next used TGN46 to examine the effect of WIN on the TGN. We found that the TGN46 was also redistributed at ≥5 μM concentrations of WIN treatment (Fig. 1F).
We performed high-resolution confocal imaging to examine if WIN produced a partial effect at lower concentrations in cells treated with concentrations of WIN ranging from 50 nM to 400 nM. (Fig. 1G). Our results show that WIN can fragment the Golgi in concentrations as low as 50 nM (Fig. 1H). The Golgi fragmentation we observed was not an indirect effect of WIN changing cell viability, as a 3-hour WIN [10 μM] treatment did not affect cell viability as measured by Trypan Blue (Supplemental Fig. 2).
WIN, but Not Related Alkylindoles, Disrupts the Golgi Apparatus Rapidly and Reversibly
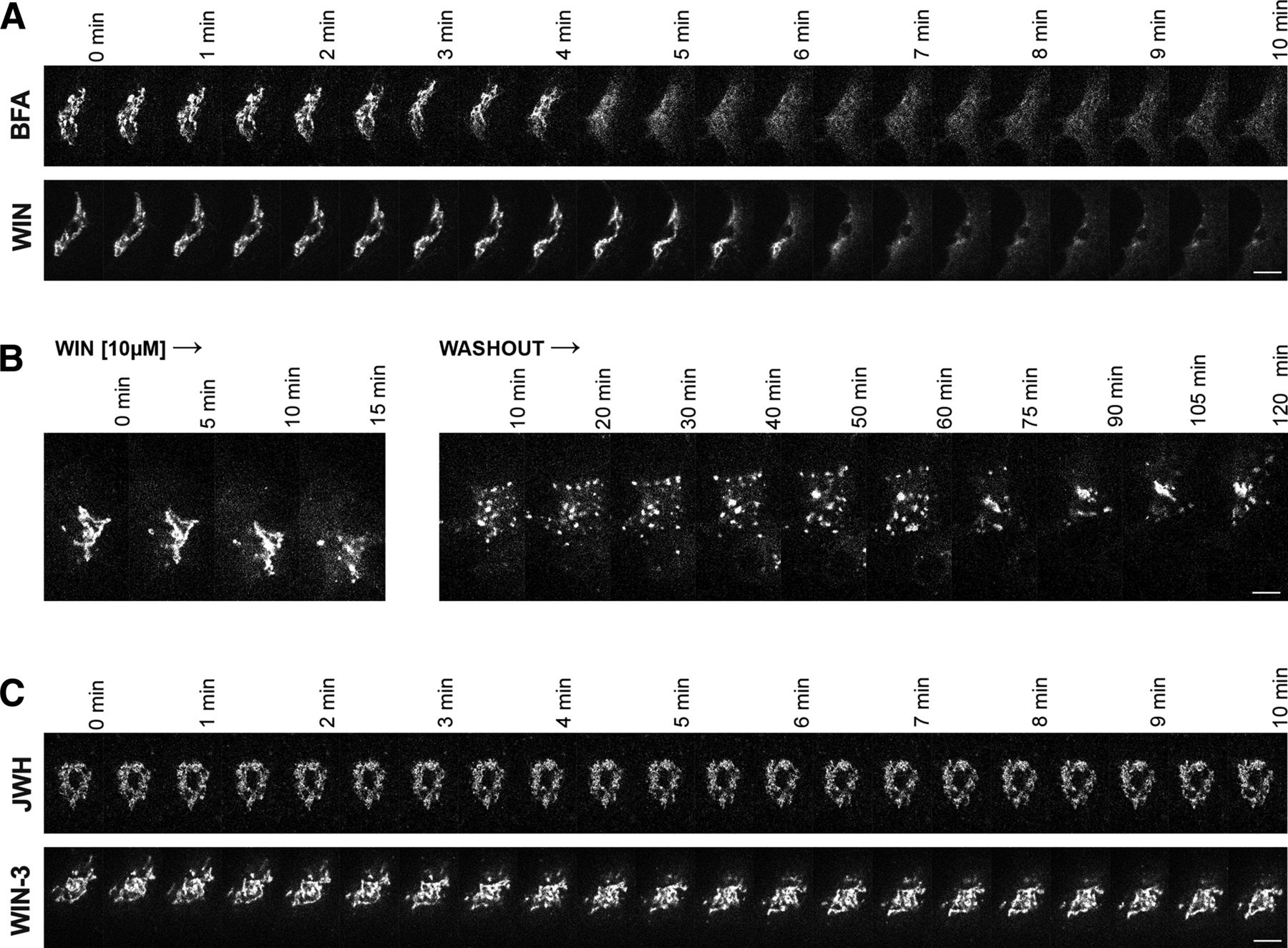
We next compared the time-course of WIN-mediated Golgi disruption to that of brefeldin-A (BFA), a fungal metabolite that has been extensively used as a standard to study Golgi collapse and reassembly. HEK293 cells expressing a GPP130-GFP were imaged live every 30 seconds for 10 minutes after treatment with 10 μM WIN or 5 μg/ml BFA as described (Donaldson et al., 1990). WIN caused Golgi disruption within 7 minutes of treatment, very similar to BFA (Fig. 2A). To test if WIN-mediated disruption was reversible, we treated cells with 10 μM WIN, washed out the agonist, and imaged GPP130-GFP in the cells for 2 hours after washout. Over this period, the dispersed Golgi fragments reassembled and formed a discrete Golgi structure in the cell center between 90 minutes and 120 minutes after washout (Fig. 2B), comparable to the kinetics of reassembly after BFA (Langhans et al., 2007).

WIN-mediated Golgi disruption is comparable to BFA. (A) Live-cell imaging of HEK293 cells transfected with GPP130-GFP. BFA and WIN disrupt the Golgi at roughly similar time scales. (B) Live-cell GPP130-GFP imaging shows that the Golgi partially reassembled within 2 hours of washout after 15 minutes of WIN [10 μM] treatment. (C) The synthetic cannabinoids JWH [10 μM] and WIN-3 [10 μM] do not cause Golgi disruption at similar time scales. Scale bar = 2.5 μm in all images.
To explore the region of WIN that was involved in Golgi disruption, we tested two other synthetic cannabinoids that shared chemical properties with WIN: the clinically relevant naphthoylindole compound JWH-018 (JWH) and the enantiomer of WIN, WIN55,212-3 (WIN-3), which differs in chirality at the morpholine group. We imaged HEK293 cells expressing a GPP130-GFP every 30 seconds for 10 minutes after JWH [10 μM] and WIN-3 [10 μM] treatment. Neither compound caused Golgi disruption, suggesting that the disruption is specific to the stereochemical structure of WIN (Fig. 2C).
WIN Disrupts Microtubules
Because Golgi structure and an intact microtubule architecture in the cell are highly interdependent, we investigated whether WIN disrupted the microtubule structure in addition to dispersing the Golgi. In HEK293 cells treated with 10 μM WIN for 1 hour, the characteristic filament network of the microtubule cytoskeleton was visually disrupted at a similar timepoint as Golgi dispersion (Fig. 3A). We next attempted to quantify this microtubule disruption using image analysis. The intricacy of microtubule filaments added a substantial degree of difficulty when processing 2D images. Therefore, we enhanced the network by using ImageJ for standardized tubule segmentation across all sample images as previously described (Kalkofen et al., 2015). We then measured the fluorescence intensity of the defined network and found a significant decrease in WIN-treated cells compared with control conditions (Fig. 3B). Our findings are further supported by a strong correlation between Golgi and microtubule disruption. We found that microtubule fluorescence decreased as Golgi fluorescence decreased (Fig. 3C), which represents a coinciding disruption of both cellular structures after WIN treatment.

WIN disrupts microtubule structure. (A) HEK293 cells imaged with confocal microscopy showing SNAP-CB1 and α-tubulin labeling after 1-hour drug treatment conditions. As shown in Fig. 1, we observe a dramatic redistribution of intracellular SNAP-CB1 after WIN [10 μM] treatment. Changes in microtubule organization are also observed, where the tubular network is less defined after WIN [10 μM] treatment. α-Tubulin labeling was used to generate a microtubule mask to quantify structural differences between treatment conditions. Scale bar = 5 μm. (B) The fluorescence intensity of α-tubulin labeling is significantly decreased after WIN treatment (**P < 0.01; n = 17 for control and 20 for WIN). (C) Scatter plot showing the matched fluorescence intensity of α-tubulin and GPP130 values. Two distinct populations are observed, where circles in the control condition (white) have higher microtubule and Golgi fluorescence values than circles in the WIN [10 μM] condition (blue).
WIN Disrupts Golgi in Primary Cultured Neurons
To test whether WIN could disrupt Golgi in multiple cell types, including physiologically relevant neurons, we treated embryonic striatal neurons with WIN and assessed Golgi disruption. Consistent with our results in HEK293 cells, we saw robust dispersal of the Golgi in neurons treated with saturating concentrations [10 μM] of WIN (Fig. 4A). When Golgi dispersal was quantified by measuring area and fluorescence, the Golgi spread across a larger surface area after WIN [10 μM] treatment (Fig. 4B), and the detected TGN38 fluorescence decreased as the surface area increased (Fig. 4C). A characteristic feature of the mammalian Golgi is that it is located at the microtubule organizing center in the center of the cell body. Therefore, as an orthogonal and higher sensitivity method to quantify neuronal Golgi disruption and localization, we measured the distribution of fluorescence intensity as a function of the distance from the cell center by generating radial profiles of antibody fluorescence across a projected 2D field (Fig. 4D). The workflow for generating radial profile plots is provided in Supplemental Fig. 3. In control and WIN [1 μM] treatment conditions, the Golgi fluorescence showed a clear peak at shorter radial lengths, indicating that Golgi fluorescence was concentrated near the cell center, consistent with an intact Golgi apparatus (Fig. 4E). When neurons were treated with 5 μM WIN, the Golgi fluorescence was more evenly dispersed across all radial lengths of the neuronal cell body (Fig. 4E). Interestingly, the fluorescence was overall lower across all radii in WIN-treated neurons, as observed by lower total fluorescence estimated as areas under the curve for radial plot values (Fig. 4F). This difference suggests that the Golgi is redistributed beyond the neuronal cell body into dendrites, as the estimates of radial profiles are restricted to the neuronal body. These results show that WIN causes Golgi disruption in multiple cell types, including physiologically relevant striatal neurons.

WIN disrupts Golgi in primary cultured neurons. (A) Striatal neurons imaged with confocal microscopy and labeled to detect TGN38 after 1-hour control and WIN treatment. As observed in HEK293 cells, Golgi is dispersed in WIN [10 μM]-treated neurons but not in control neurons. Scale bar = 2.5 μm. By thresholding the TGN38 signal, we quantified differences in area coverage and average fluorescence. (B) TGN38 labeling covers a larger surface area after WIN [10 μM] treatment, and (C) the subsequent fluorescence intensity of TGN38 signal is significantly decreased (**P < 0.01; ****P < 0.0001; n ≥ 12). (D) Schematic depicting Golgi dispersal in neurons and the radial profile quantification approach used to measure TGN38 fluorescence. Fluorescence intensity is concentrated at shorter radial lengths (top panel) when the Golgi is intact, whereas in conditions where the Golgi is disrupted (bottom panel), the fluorescence intensity becomes more evenly distributed across all radial positions. (E) Radial plots showing TGN38 fluorescence across radial position at varying concentrations of WIN treatment. Control and WIN [1 μM]-treated neurons show peaks in fluorescence intensity at shorter radial lengths, whereas WIN [5 μM]-treated neurons do not show this robust peak in fluorescence. Error bars represent mean ± 95% confidence interval (CI) (n ≥ 9). (F) TGN38 labeling does not cover a larger surface area after 2AG treatment, and (G) the subsequent fluorescence intensity of TGN38 signal is not significantly decreased (ns, not significant; n ≥ 9). (H) This is recapitulated with our radial plots, where control and 2AG treatment conditions both show peaks in fluorescence intensity at shorter radial lengths. Error bars represent mean ± 95% CI (n ≥ 8).
As a control, we treated embryonic striatal neurons with 2AG and measured Golgi stability using a similar approach as above. No significant differences in Golgi area (Fig. 4F) or fluorescence (Fig. 4G) were detected. Additionally, in both control and 2AG treatment conditions, the Golgi signal was concentrated near the nucleus, as measured through our radial profile quantification approach (Fig. 4H). These results indicate that WIN, but not 2AG, disrupts the Golgi apparatus in neurons.
Golgi Disruption by WIN Is Independent of CB1
In both HEK293 cells and embryonic striatal neurons, WIN treatment at ∼5 μM concentrations resulted in Golgi disruption despite variations in cellular CB1 expression. This raised the possibility that the effects of WIN on the Golgi are independent of CB1 receptor expression. To directly test this, we measured Golgi disruption by WIN in cortical neurons cultured from mice in which CB1 was genetically knocked out (CB1-KO) (Ledent et al., 1999) to determine if CB1 was required for WIN-mediated organelle disruption. Neuronal identity of cultured cells was confirmed using the neuronal marker microtubule-associated protein 2 (MAP2). Upon treatment with 10 μM WIN, we observed that GM130, a cis-Golgi marker, was dispersed in CB1-KO neurons as well as neurons cultured from wild-type littermates (Fig. 5A). To quantify Golgi disruption in CB1-KO neurons, we measured the fraction of total cell area covered by GM130 and the fluorescence intensity of detected antibody signal as in Fig. 1. Golgi coverage in the neuronal cell body significantly increased when the Golgi was disrupted (Fig. 5B), and the fluorescence intensity of detected antibody signal decreased as area increased (Fig. 5C). Both of these were comparable to the effects that we observed in HEK293 cells. Golgi dispersal was also evident when radial profile plots were used to quantify dispersion. The radial plots show a fluorescence peak at shorter radial lengths in control neurons but not in neurons treated with 10 μM WIN for 20 minutes or 1 hour (Fig. 5D). Together, our results show that WIN can cause Golgi dispersion in multiple cell types, independent of CB1 expression.

WIN-mediated Golgi disruption is CB1-independent. (A) Cortical neurons of CB1-KO mice and wild-type littermates imaged with confocal microscopy. The Golgi is identified with the cisternal-Golgi antibody GM130, and neurons are identified with MAP2. As shown in HEK293 cells, the Golgi is intact for neurons in the control group, whereas GM130 labeling shows a dispersal phenotype for neurons in the WIN [10 μM] condition. Scale bar = 2.5 μm. (B) GM130 labeling covers a larger surface area after WIN treatment, and (C) the subsequent fluorescence intensity of GM130 is significantly decreased (*P < 0.05; n ≥ 9). (D) Radial plots showing GM130 fluorescence across radial position at varying incubation times of WIN [10 μM] treatment. Neurons in the control group show peaks in fluorescence intensity at shorter radial positions, whereas neurons treated with 10 μM WIN for 20 minutes and 1 hour do not show this peak. Error bars represent mean ± 95% CI (n ≥ 10).
Golgi Disruption Is Independent of Peroxisome Proliferator-Activated Receptors
Because WIN-mediated Golgi dispersal was independent of the primary receptor CB1, we examined whether WIN acted via its main other known effectors: the nuclear peroxisome proliferator-activated receptors alpha and gamma (PPARα and PPAR γ) (O’Sullivan, 2016). To test if PPAR was required for Golgi disruption, we cotreated HEK293 cells with WIN and either a PPARα inhibitor (10 μM GW6471) or a PPARγ inhibitor (10 μM GW9662) for 1 hour. Inhibition of neither PPARα or PPARγ blocked Golgi disruption completely when HEK293 cells were treated with WIN, suggesting that PPAR activation was not required for WIN-mediated Golgi disruption. However, GW9662 showed a partial protective effect in comparison with control when HEK293 cells were treated with 5 μM WIN (Fig. 6A). To assess the penetrance of the phenotype of Golgi disruption, we determined the fraction of fields in which the majority (>50%) of HEK293 cells within the field displayed Golgi disruption after WIN treatment. The Golgi was intact for all fields of HEK293 cells in control conditions. In contrast, Golgi disruption was observed upon treatment with 5 μM WIN and 10 μM WIN in all cells, irrespective of whether PPAR was inhibited (Fig. 6B). The quantitation confirmed that 10 μM GW9662 produced a partial protective effect in HEK293 cells treated with 5 μM WIN, suggesting that PPARγ, although not required, might play a contributory role in WIN-mediated disruption of the Golgi.

WIN-mediated Golgi disruption does not require PPAR activity. (A) HEK293 cells imaged with confocal microscopy showing GPP130 labeling after 1-hour treatment conditions. The Golgi is intact in control, GW6471 [10 μM], and GW9662 [10 μM] conditions, whereas GPP130 labeling shows a robust dispersal phenotype after WIN treatment at both 5 μM and 10 μM. Although cotreatment with 5 μM WIN and 10 μM GW9662 collapses the Golgi apparatus, the phenotype is less pronounced than what is observed after treatment with WIN alone. Scale bar = 5 μm. (B) Stacked bar graph representation of Golgi stability as a percentage of fields exhibiting intact cis-Golgi compartments (n = 30 for each).
Discussion
In this study, we show that WIN can disrupt the Golgi apparatus across multiple cell types, including primary neurons, with partial disruptions starting at nanomolar concentrations and complete disruption at micromolar concentrations. These general effects of WIN are independent of CB1 expression and are effected through an as-yet undiscovered pathway that could be partially regulated by PPARs.
Our results provide a new context for the use of WIN in experimental and behavioral contexts. The divergent effects observed with WIN have often been considered to be due to the unique ways that it might activate CB1 compared with endocannabinoids. This could certainly be true, and part of WIN’s unique effects could be explained by differential activation of CB1. For example, WIN exhibits weakened β-arrestin signaling compared with 2AG (Flores-Otero et al., 2014). However, WIN can also elicit CB1-independent effects. WIN can halt proliferation in oncogenic cells (Emery et al., 2014; Pellerito et al., 2014; Müller et al., 2017) independent of CB1 signaling (Scuderi et al., 2011). Additionally, WIN shows anti-inflammatory and antinociceptive properties that have also been identified as CB1-independent (Price et al., 2004). One point of consideration is that WIN caused complete Golgi disruption at micromolar concentrations of WIN. However, even at these concentrations, the effect we observed is still specific to WIN, as similar concentrations of JWH-018 or WIN55,212-3 did not cause Golgi disruption. Interestingly, WIN-mediated downregulation of the expression of proteins involved in cell growth and survival was also observed only at micromolar concentrations of WIN (Sreevalsan and Safe, 2013) similar to concentrations that can completely disrupt the Golgi apparatus, as we report in this study.
WIN could, however, affect Golgi function even at nanomolar concentrations, even without causing complete collapse of the Golgi. In this context, one clear strength of our quantitative imaging approach is that we can detect small changes in the Golgi architecture, which might be missed by traditional biochemical approaches, with high sensitivity. Because the Golgi apparatus is a processing station for many post-translational modifications, including glycosylation, small changes in the Golgi architecture can cause differences in protein processing without changing trafficking rates overall (Puthenveedu et al., 2006). To the best of our knowledge, the exact concentrations of WIN in different brain regions after administration in mice are not known. In mice injected i.p. with 2.5 mg/kg of the synthetic cannabinoid JWH-018, the serum concentration can reach 250 nM (Malyshevskaya et al., 2017). Our results suggest that WIN causes partial fragmentation of the Golgi starting at 50 nM (Fig. 1), even without large changes in protein secretion (Supplemental Fig. 2B). The disruption of the Golgi and associated changes in processing of select proteins could contribute to the wide-ranging physiologic effects of WIN.
Our results suggest that the disruption of the Golgi apparatus is specific to the stereochemistry of WIN. WIN belongs to the aminoalkylindole family of cannabinoids. Some alkylindoles can destabilize microtubules and promote cell death through direct microtubule binding (Cherry et al., 2016). Moieties linked to the nitrogen of pyrrole rings are the structural determinants of alkylindole-microtubule interactions, where bulkier moieties prevent microtubule binding (Fung et al., 2017). However, it is unlikely that the effect of WIN on the Golgi is through microtubule disruption by alkylindoles. First, the interplay between microtubules and cytoskeleton is bidirectional, as microtubules are required for a discrete Golgi ribbon to exist at the cell center and as the Golgi can nucleate microtubules (Wu and Akhmanova, 2017). Second, the dispersal and kinetics of Golgi disruption that we observed match BFA and are faster than reported for microtubule disruption. Third, neither JWH-018 nor WIN55,212-3, which share the alkylindole group, caused Golgi disruption. Rather, the chirality of the morpholine group seems to be important for WIN, as WIN55,212-3 does not disrupt the Golgi. The exact mechanism by which the morpholine group disrupts the Golgi apparatus is not known. It is possible that this group binds to an unknown effector. WIN also contains an oxazine group that has been suggested to have antitumor properties, and it is possible that the morpholine group might sterically control the interactions of the oxazine group with its effectors.
Our results indicate that Golgi disruption does not require CB1 activation, as we observed similar effects in HEK293 cells not expressing exogenous CB1 and in primary cells derived from CB1 knockout mice. One potential CB1-independent target of WIN is the PPAR family of nuclear receptors. WIN can activate PPARα and PPARγ (O’Sullivan, 2016) independent of CB1. Both PPARs can regulate lipid metabolism and could contribute to disruption of the Golgi apparatus. However, our results suggest that the effect of WINs cannot be fully explained by activation of PPARs. First, WIN disrupts the Golgi at relatively fast timescales, which makes it unlikely that a transcriptional regulatory mechanism is the primary mediator. Second, blocking PPAR activation was not sufficient to block Golgi disruption (Fig. 6). PPARγ inhibition caused a partial reduction of WIN-mediated disruption of the Golgi. Interestingly, PPARγ inhibition on its own can downregulate tubulin expression and inhibit cell growth (Schaefer, 2007), but the timescale required to observed downstream effects of nuclear receptors is much longer than what is sufficient for observing WIN’s effect on cellular structures. WIN could target one of many proteins that have been identified as being critical for maintaining the integrity of the Golgi apparatus. These include trafficking proteins such as the coatomer protein I complex (COPI), which is disrupted by the fungal metabolite BFA (Donaldson et al., 1990; Orci et al., 1991). However, unlike BFA, which preferentially disrupts the stacked Golgi, WIN also disrupts the trans-Golgi network as marked by TGN38 at micromolar concentrations. Future studies that identify a target for WIN will provide us with a better understanding of how WIN regulates Golgi structure and might provide insights into the regulation of Golgi structure and trafficking in general.
Irrespective of the mechanism, our results highlight a broad effect of WIN on the Golgi apparatus that is likely to confound interpretations of its effects on cannabinoid pharmacology. These effects could contribute to WIN’s role as an antiproliferative and anti-inflammatory agent by disrupting a key organelle that is central to membrane trafficking and protein processing. This study therefore generates new considerations for interpreting the unique physiologic effects of WIN compared with other cannabinoid agonists.
Acknowledgments
The authors thank Taylor Craig and Dr. Kevin Jones for assistance with preparing neuronal cultures derived from CB1-KO mice, Dr. Stephanie Crilly and Dr. Zara Weinberg for valuable discussions that helped advance this study, Dr. Kenneth Mackie (Indiana University at Bloomington) for providing the mice and parent CB1 constructs, and Dr. Adam Linstedt (Carnegie Mellon University) for providing essential reagents.
Authorship Contributions
Participated in research design: Lott, Jutkiewicz, Puthenveedu.
Conducted experiments: Lott.
Contributed new reagents or analytic tools: Lott, Jutkiewicz, Puthenveedu.
Performed data analysis: Lott.
Wrote or contributed to the writing of the manuscript: Lott, Puthenveedu.
Footnotes
- Received July 29, 2021.
- Accepted February 22, 2022.
This work was supported by National Institutes of Health National Institute for General Medical Sciences [Grant T32-GM007767] (to J.L.) and [Grant GM117425] (to M.A.P.) and by the National Science Foundation [Grant 1935926] (to M.A.P.).
No author has an actual or perceived conflict of interest with the contents of this article.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- 2AG
- 2-arachidonoylglycerol (5Z,8Z,11Z,14Z)-5,8,11,14-eicosatetraenoic acid, 2-hydroxy-1-(hydroxymethyl)ethyl ester)
- BFA
- brefeldin A (1,6,7,8,9,11aβ,12,13,14,14αa-decahydro-1β,13α-dihydroxy-6β-methyl-4H-cyclopent(f)oxacyclotridecin-4-one)
- CB1
- cannabinoid receptor-1
- CB1-KO
- cannabinoid receptor-1 knockout
- CI
- confidence interval
- CNS
- central nervous system
- HEK
- human embryonic kidney
- JWH
- JWH-018 (1-naphthalenyl(1-pentyl-1H-indol-3-yl)-methanone)
- MAP
- microtubule-associated protein
- NA
- numerical aperture
- PPAR
- peroxisome proliferator-activated receptor
- TGN
- trans-Golgi network
- WIN
- WIN55,212-2 (R-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate)
- WIN-3
- WIN55,212-3 ([(3S)-2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenyl-methanone, methanesulfonate)
- Copyright © 2022 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}