


Visual Overview

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease that is accompanied by memory decline and cognitive dysfunction. Aggregated amyloid β formation and accumulation may be one of the underlying mechanisms of the pathophysiology of AD. Therefore, compounds that can inhibit amyloid β aggregation may be useful for treatment. Based on this hypothesis, we screened plant compounds used in Kampo medicine for chemical chaperone activity and identified that alkannin had this property. Further analysis indicated that alkannin could inhibit amyloid β aggregation. Importantly, we also found that alkannin inhibited amyloid β aggregation after aggregates had already formed. Through the analysis of circular dichroism spectra, alkannin was found to inhibit β-sheet structure formation, which is an aggregation-prone toxic structure. Furthermore, alkannin attenuated amyloid β-induced neuronal cell death in PC12 cells, ameliorated amyloid β aggregation in the AD model of Caenorhabditis elegans (C. elegans), and inhibited chemotaxis observed in AD C. elegans, suggesting that alkannin could potentially inhibit neurodegeneration in vivo. Overall, these results suggest that alkannin may have novel pharmacological properties for inhibiting amyloid β aggregation and neuronal cell death in AD.
SIGNIFICANCE STATEMENT Aggregated amyloid β formation and accumulation is one of the underlying mechanisms of the pathophysiology of Alzheimer’s disease. We found that alkannin had chemical chaperone activity, which can inhibit β-sheet structure formation of amyloid β and its aggregation, neuronal cell death, and Alzheimer’s disease phenotype in C. elegans. Overall, alkannin may have novel pharmacological properties for inhibiting amyloid β aggregation and neuronal cell death in Alzheimer’s disease.
Introduction
Amyloid β protein is produced by the cleavage of amyloid precursor protein (APP), a type I transmembrane glycoprotein. Amyloid precursor protein is cleaved by two enzymes, β-secretase and γ-secretase, which produce amyloid β protein (Selkoe, 2011). Amyloid β protein is prone to aggregation, and while monomers of amyloid β protein do not induce neuronal cell death, the aggregation of amyloid β protein can (Pike et al., 1993; Lorenzo and Yankner, 1994). It has been hypothesized that the pathophysiology of Alzheimer’s disease (AD) involves the accumulation of aggregated amyloid β protein in the brain, possibly in senile plaque, which is observed in the AD brain. In several mouse models, the induced accumulation of amyloid β protein has been found to cause neurodegeneration (Götz et al., 2018). Furthermore, evidence suggests that aggregated amyloid β protein may act as a transmitter (Meyer-Luehmann et al., 2006; Jaunmuktane et al., 2015). Overall, these reports suggest that aggregated amyloid β protein accumulation and transmission may play a key role in the pathogenesis of AD.
The inhibition of amyloid β protein aggregation may therefore be a key strategy for the treatment of AD. Several potential methods for reducing the accumulation of aggregated amyloid β proteins have been presented. One such method is the use of chemical chaperones (Cohen and Kelly, 2003). Chemical chaperones are compounds of low molecular weight that can inhibit protein aggregation (Perlmutter, 2002). Several reports have indicated the use of chemical chaperones in protein aggregation-mediated diseases such as cystic fibrosis (Hanrahan et al., 2013), diabetes (Ozcan et al., 2006), and epilepsy (Yokoi et al., 2015). Previous studies have also reported that endoplasmic reticulum stress is linked to leptin resistance in obesity, which can be ameliorated by chemical chaperones (Hosoi et al., 2008; Ozcan et al., 2009; Hosoi et al., 2014). Interestingly, the amyloid-binding dye thioflavin T (ThT) has been found to extend the lifespan and reduce β-amyloid-associated toxicity in Caenorhabditis elegans (C. elegans) (Alavez et al., 2011). Furthermore, Congo red, a histologic dye, has been shown to inhibit the fibril formation activity of amyloid β protein and thus reduce associated toxicity against neuronal cells (Lorenzo and Yankner, 1994). However, ThT and Congo red are mainly used to stain aggregated β-amyloid for diagnosis and may not be usable in medicine. Therefore, in the present study, we aimed to identify compounds that could ameliorate amyloid β protein aggregation from the library of plant compounds used in Kampo medicine, a traditional Chinese medicine. We reasoned that, since the compounds derived from Kampo medicine have been used for hundreds of years, they would be safe. Based on this screening, we found that alkannin has chemical chaperone activity and can thereby attenuate amyloid β protein aggregation and ameliorate AD pathology. In the present study, we therefore investigated the pharmacological effects of alkannin on amyloid β aggregation and neuronal cell death.
Materials and Methods
Materials
The human amyloid β1-42 peptide was obtained from the Peptide Institute (Osaka, Japan). The library of plant compounds was obtained from the Institute of Natural Medicine, University of Toyama. Each compound was dissolved at 10 mM in DMSO and stored at −20°C until use.
Measurement of Chaperone Activity
Chaperone activity measurements were performed as previously described (Huang et al., 2000; Li et al., 2001; Kubota et al., 2006). The assay was based on measuring α-lactalbumin (α-LA) aggregation, which was analyzed by measuring optimal density at 488 nm to detect turbidity using a VERSAmax microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Preparation of Amyloid β
Aβ1-42 was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at 1 mg/mL and sonicated in an ice-cooled water bath for 4 minutes. The samples were then aliquoted in a tube, dried under vacuum, and stored at −20°C until use.
Measurement of Amyloid β Aggregation Using Thioflavin T
Amyloid β aggregation was measured based on previous reports, with slight modifications (Feng et al., 2009). HFIP-treated Aβ1-42 was dissolved in DMSO at 1 mg/mL and then diluted to 10 μM with phosphate-buffered saline. The compounds were added to the Aβ1-42 solution and incubated at 37°C for the indicated times. After incubation, 180 μL of 5 μM ThT solution was added to 20 μL of the Aβ1-42 solution and the fluorescence of ex 450 nm/em and 480 nm was measured. The ThT solution (5 μM) was prepared using 50 mM phosphate buffer (pH 6.5).
Dot Blotting
Amyloid β aggregation was measured based on previous reports, with slight modifications (Izuo et al., 2012). HFIP-treated Aβ1-42 was dissolved in DMSO at 1 μM and then diluted to 20 μM with phosphate-buffered saline. Alkannin was added to the solution to a final concentration of 10 μM and incubated at 37°C for 8 hours. Control samples were treated with DMSO to a final concentration of 0.1%. The samples were then applied to nitrocellulose membranes, i.e., 2μL for 6E10 and 5μL for 11A1. After 10 minutes, the membranes were blocked by 2.5% skim milk in Tris-buffered saline containing 0.1% Tween-20. Then the membranes were probed with one of the primary antibodies, anti-Aβ1–17 (6E10, 1 μM) or anti-E22P-Aβ10-35 (11A1, 1 μM) overnight at 4°C followed by incubation with the secondary antibody (1 hour at room temperature). The intensities of the dots were quantified using Image J software.
Cell Culture
PC12 cells were maintained in an RPMI medium with 5% (v/v) heat-inactivated fetal calf serum, 10% (v/v) heat-inactivated horse serum, 100 units/mL penicillin G, and 100 mg/mL streptomycin. Cultured cells were maintained at 37°C in 5% CO2/95% air. PC12 cells were cultured on poly D-lysine-coated plates for the assay. Three days before Aβ1-42 stimulation, the cultured medium was replaced with a neurobasal medium containing 2% B-27 supplement and 0.5 mM L-glutamine. On the day of Aβ1-42 stimulation, the cultured medium was replaced with a neurobasal medium containing 2% B-27 supplement without antioxidant and 0.5 mM L-glutamine. For Aβ1-42 stimulation, amyloid β1-42 peptide was dissolved in 0.02% NH3 (200 μM) and added to the PC12 cells to obtain the final concentration indicated in the Results section.
Measurement of Cell Death Using Lactate Dehydrogenase Assay
Cell death was analyzed by measuring the amount of lactate dehydrogenase (LDH) released into the medium. The LDH assay was performed using a cytotoxicity detection kit (Roche Molecular Biochemical, Basel, Switzerland) according to the manufacturer’s instructions. LDH activity was measured at an optimal density of 492 nm. The percentage of cell death was calculated by measuring the ratio of LDH activity in the culture medium to the total LDH activity [i.e., (extracellular LDH)/(extracellular LDH + cellular LDH)].
Measurement of Amyloid β Secondary Structure by Analyzing Circular Dichroism Spectra
A 200 µM Aβ1-42 stock solution (in 0.02% NH3) was dissolved in a 50 mM potassium phosphate buffer to obtain a final concentration of 10 μM of Aβ1-42. Concurrently, alkannin was added to the solution to a final concentration of 10 μM. Control samples were treated with DMSO to a final concentration of 0.1%. The samples were then incubated at 37°C for 24 hours to form the secondary structure of Aβ1-42. The circular dichroism (CD) spectra were measured using a spectropolarimeter J720 (JASCO, Japan).
Measurement of Amyloid β Fibrils Using Scanning Electron Microscope
HFIP-treated Aβ1-42 was dissolved in DMSO, sonicated, and then diluted to 10 μM with phosphate-buffered saline. The samples were then incubated at 37°C for 48 hours. A scanning electron microscope analysis was performed at the Hiroshima University of Imaging Platform, Japan.
Maintenance of C. Elegans
The N2 control and CL2122, CL2355, and CL4176 worm strains were maintained on nematode growth medium plates with OP50 as a food source. These strains were obtained from the Caenorhabditis Genetics Center. For the experiments, the age of the C. elegans was synchronized by bleaching. Samples were then plated and OP50 bacteria were added after the eggs had hatched. They were then grown at 16° to 25°C until adulthood. 5-Fluoro-2’-deoxyuridine was added 72 hours after synchronization.
Chemotaxis Assay of C. Elegans
A chemotaxis assay of C. elegans was performed 144 hours after synchronization. Following several washes with M9 buffer, C. elegans was plated at the center of the nematode growth medium plate. For the assay plate, 1 μL of 1% benzaldehyde (dissolved in ethanol) and 0.5 μL of 1M sodium azide were added to the attractant side of the plate. Similarly, 1 μL of ethanol and 0.5 μL of 1M sodium azide were added on the opposite side of the plate for the control. The plate was then incubated at 23°C in a dark room for 4 hours, after which the amount of C. elegans that had moved was counted and the chemotaxis index was calculated using the following formula: chemotaxis index (%) = [(number of attractant side − number of control side)/number of C. elegans] × 100.
Staining of Amyloid β in C. Elegans
After hatching, CL4176 cells were incubated at 16°C for 36 hours, treated with alkannin (3 µM), and then incubated at 25°C for another 36 hours. After incubation, the worms were washed with M9 buffer and transferred to microfuge tubes. The worms were then fixed with 4% paraformaldehyde in M9 buffer (pH 7.4), incubated at 4°C for 24 hours, and subsequently permeabilized with 5% 2-mercaptoethanol, 1% Triton X-100, and 125 mM Tris (pH 7.4), at 37°C for 24 hours. After washing twice with M9 buffer, the worms were stained with 0.125% ThT (in 50% ethanol) for 2 minutes. The worms were then sequentially washed with 50%, 70%, 90%, 70%, and 50% ethanol for 2 minutes each. Finally, they were washed with M9 buffer containing 1% Triton X-100, and fluorescence images were acquired using a microscope (Keyence, BZ9000). For the data analysis, we used nontreatment AD model C. elegans (CL4176 strain) sample as control, and wild-type, DMSO control-, or alkannin-treated samples were divided by nontreatment AD sample (wild-type, DMSO control, or alkannin AD/nontreatment AD). For the data analysis, we used non-treatment AD model C. elegans (CL4176 strain) sample as a control. Fluorescence intensity of each sample (wild-type N2 strain, DMSO-treated or Alkannin-treated AD model) was divided by non-treatment AD sample (wild-type, DMSO control or Alkannin AD / non-treatment AD).
Statistics
The results are expressed as the mean ± SD. Statistical analyses were performed using the paired t test, Tukey-Kramer, or Dunnett’s test. We considered the data to be statistically significant when P values were lower than 0.05.
Results
Identification of Candidate Compound from Screening Assay
A compound that could inhibit Aβ aggregation may be a candidate drug for the treatment of AD. To identify such a compound, we first performed a screening assay. We selected compounds derived from traditional Chinese medicine to create a library for the screening assay. The list of compounds used in the present study is shown in Supplementary Fig. 1. We performed two different types of screening assays: a measurement of chemical chaperone activity and a measurement of amyloid β aggregation. The chaperone activity measurement was based on α-LA aggregation (Huang et al., 2000; Li et al., 2001; Kubota et al., 2006). We incubated the cells with or without each compound for 6 hours at 37°C and measured the chemical chaperone activity. Using this assay, we identified three compounds (alkannin, coptisine chloride, and shikonin) that had chemical chaperone activity (Fig. 1A). Next, we screened amyloid β aggregation using a ThT assay (LeVine, 1993). We incubated the cells with or without each compound for 7 hours at 37°C and measured amyloid β aggregation using the ThT assay. From this assay, we found that five compounds, (alkannin, barbaloin, (z)-ligustilide, luteolin, and shikonin) had inhibitory effects on the aggregation of amyloid β (Fig. 1B). Based on these two assays, we found that only alkannin and shikonin had both chemical chaperone activity and inhibitory effects on amyloid β aggregation. Alkannin and shikonin are enantiomers of each other. In the present study, we focused on alkannin and analyzed the effect of alkannin on AD pathology in more detail.

Screening assay for the identification of candidate compounds used in Kampo medicine. (A) Screening of candidate compounds to determine which have chemical chaperone activity. The relative intensity of α-LA aggregation was analyzed by measuring optimal density at 488 nm to detect turbidity after incubation at 37°C for 7 hours. Mean ± SD, n = 3. (B) Screening of candidate compounds that could inhibit amyloid β aggregation. The relative intensity of amyloid β aggregation using ThT fluorescence intensity measured at an excitation wavelength of 450 nm and emission wavelength of 480 nm after incubating at 37°C for 6 hours. Mean ± SD, n = 3. Candidate compounds were as follows: #5: alkannin, #15: barbaloin, #29: coptisine chloride, #65: (Z)-ligustilide, #69: luteolin, #91: shikonin. A list of all the compounds tested is shown in Supplementary Fig. 1.
Alkannin Inhibits Aβ Aggregation
To confirm the results of the screening assay, we performed a time-course analysis of Aβ aggregation using the ThT assay. Amyloid β1-42 (Aβ1-42) (10 μM) was incubated with or without alkannin (10 μM) for 0, 6, 12, 24, and 48 hours at 37°C, and Aβ1-42 aggregation was measured. Aβ1-42 alone increased aggregation in a time-dependent manner (Fig. 2B). However, the cotreatment of Aβ1-42 and alkannin dramatically attenuated Aβ1-42 aggregation at 6, 12, 24, and 48 hours (Fig. 2B). Alkannin alone did not affect the ThT-induced o.d. signal, suggesting that alkannin may not directly affect the action of ThT. These results suggest that alkannin inhibits Aβ1-42 aggregation without secondary effects.

Alkannin inhibits the aggregation of amyloid β. (A) Chemical structure of alkannin. (B) A time-course analysis of the effect of alkannin on ThT fluorescence intensity was measured at 0, 6, 12, 24, and 48 hours after incubating Aβ1-42 (10 µM) and alkannin (10 µM). Aβ1-42 alone dramatically increased ThT fluorescence intensity whereas Aβ1-42 + alkannin did not. Alkannin alone did not affect ThT fluorescence intensity. Mean ± SD, n = 3. (C) Alkannin inhibited preformed amyloid β aggregates. Amyloid β was incubated for 6 hours to form amyloid β aggregates in the absence of alkannin, after which the amyloid β aggregates were incubated with alkannin for the additional times and ThT fluorescence was measured. *P < 0.05, ***P < 0.001 versus cont (DMSO). Tukey-Kramer. Mean ± SD, n = 5. (D) Alkannin inhibited preformed amyloid β aggregates. Aβ was incubated for 24 hours to form amyloid β aggregates in the absence of alkannin, after which the amyloid β aggregates were incubated with alkannin for the additional times. ThT fluorescence was measured after incubation. *P < 0.05, **P < 0.01 versus control (DMSO). Tukey-Kramer. Mean ± SD, n = 5. (E) A time-course analysis of the effect of alkannin and shikonin on ThT fluorescence. The intensity was measured at 0, 6, 12, and 24 hours after incubating Aβ1-42 (10 µM) and alkannin (10 µM) or shikonin (10 µM). Sikonin similarly attenuated ThT fluorescence intensity compared with alkannin. Mean ± SD, n = 4.
To determine whether alkannin could ameliorate preformed amyloid β aggregation, we treated amyloid β aggregates with alkannin. To achieve this, we preincubated Aβ1-42 for 6 or 24 hours and observed a dramatic increase in Aβ1-42 aggregation (Fig. 2C-D). After preincubating Aβ1-42 for 6 or 24 hours, we then added alkannin and further incubated for 6 to 42 hours and analyzed Aβ1-42 aggregation. To compare the effect of shikonin versus alkannin on Aβ1-42 aggregation, we next analyzed the effect of shikonin and alkannin on Aβ1-42 aggregation at 6, 12, and 24 hours. As shown in Fig. 2E, we observed similar inhibitory action against Aβ1-42 aggregation (Fig. 2E). From these experiments, we found that Aβ1-42 aggregation was attenuated by alkannin (Fig. 2). These results indicate that alkannin can also ameliorate preformed Aβ aggregates, suggesting that alkannin may be beneficial for the treatment and prevention of AD.
Alkannin Inhibits the Formation of the β-sheet Secondary Structure of Amyloid β
After finding that alkannin inhibited amyloid β aggregation, we next analyzed whether alkannin affected the secondary structure of amyloid β. To analyze this, we performed CD spectral analysis. In general, the secondary structure of amyloid β is randomly coiled when it is in monomer formation, whereas it changes to a β-sheet structure during multimer formation and aggregation. We analyzed the CD spectra before and after incubation with Aβ1-42 (10 μM) at 37°C for 24 hours. After incubating Aβ1-42 for 24 hours, we observed the typical CD spectra of the β-sheet structure (Fig. 3B panel of 24 hours). We found that these typical CD spectra were ameliorated by 10 μM of alkannin treatment (Fig. 3B). In contrast, the CD spectra were not affected by alkannin treatment alone, suggesting that alkannin itself does not affect the CD spectra (Fig. 3AB). These results suggest that alkannin may affect conformational structural changes in amyloid β, thereby inhibiting the β-sheet secondary structure of amyloid β.

Alkannin attenuates the β-sheet structure and fibril formation of amyloid β. Aβ1-42 (10 μM) was incubated with or without alkannin (10 μM) for 24 hours at 37°C and CD spectra were analyzed. CD spectra of amyloid β and alkannin at (A) 0 hour and (B) 24 hours after incubation. Alkannin was found to attenuate the typical CD spectra signal of β-sheet structure formation of amyloid β. Mean ± SD, n = 4. (C) Alkannin attenuated amyloid β fibril formation. Scanning electron micrograph analysis of Aβ1-42 (10 μM) alone or Aβ1-42 + alkannin (10 μM) after incubation at 37°C for 48 hours.
Alkannin Moderately Attenuated Amyloid β Fibril Formation
Since we found that alkannin could inhibit the β-sheet secondary structure of amyloid β and subsequent formation of aggregation, we presumed that alkannin could also attenuate amyloid β fibril formation. To analyze this, we performed an electron microscopy analysis. Aβ1-42 (10 μM) was incubated for 48 hours at 37°C with or without alkannin (10 μM), and amyloid β fibril formation was analyzed. Amyloid β fibril formation was detected by electron microscopy analysis, which showed the width of the fibril formed was moderately reduced by alkannin (Fig. 3C). These results indicate that alkannin may also moderately inhibit amyloid β fibril formation.
Alkannin Inhibited Toxic Conformer of Amyloid β
As we found that alkannin can inhibit aggregation of amyloid β, we next investigated whether alkannin can inhibit toxic conformer of amyloid β. To this end, we had done dot blotting of amyloid β using 11A1 antibody, which can specifically bind with toxic conformer of amyloid β (Izuo et al., 2012). Amyloid β1-42 was incubated for 8 hours at 37°C with or without alkannin. We observed increase in dot blotting signal of 11A1 antibody, when incubated for 8 hours (Fig. 4). On the other hand, we observed attenuation of the signal when we treated with alkannin. No difference was observed when we used anti-Aβ1-17 antibody (6E10) antibody (Fig. 4). These results suggest that alkannin may inhibit toxic conformer of amyloid β.

Alkannin attenuated formation of toxic conformer of amyloid β. (A) Amyloid β1-42 was incubated for 8 hours with or without alkannin and analyzed toxic conformer of amyloid β using specific antibody 11A1. We observed inhibition of formation of the toxic conformer of amyloid by alkannin. On the other hand, similar signal was detected when we used anti-Aβ1-17 antibody (6E10) antibody. (B) Densitometric analysis of the amyloid β dot blot analysis. Mean ± SD, n = 5. Tukey-Kramer *p < 0.05 versus control 8 hours.
Alkannin has a Neuroprotective Effect on Amyloid β-induced Cell Death
Aggregated amyloid β is toxic to neurons (Pike et al., 1993; Lorenzo and Yankner, 1994). Since we found that alkannin could reduce amyloid β aggregation, we next investigated whether alkannin had a neuroprotective effect on amyloid β-induced cell death. To test this, we treated PC12 neuronal cells with alkannin and analyzed Aβ1-42-induced cell death. PC12 cells were treated with alkannin (0.1–1 μM) and Aβ1-42 (5 μM) for 48 hours, and cell death was analyzed by measuring LDH. We observed that Aβ1-42 caused cell death from 13.0% to 25.6%, and this effect was reduced to 17.8% by alkannin treatment (Fig. 4). An enantiomer of alkannin, shikonin has been reported to protect against amyloid β-induced cell death (Tong et al., 2018). Therefore, to compare the neuroprotective action of these compounds, we also analyzed the effect of shikonin on Aβ1-42 (5 μM)-induced cell death. As shown in Fig. 5, shikonin similarly inhibited Aβ1-42 (5 μM)-induced cell death (Fig. 5). These results suggest alkannin can have a protective effect on amyloid β-induced neuronal cell death.

Alkannin protects amyloid β-induced neuronal cell death. PC12 cells were treated with amyloid β (5 µM) and alkannin (0.1, 0.3, and 1 µM) or shikonin (0.1, 0.3, and 1 µM). The percentage of cell death was measured by LDH assay after 48 hours of incubation. Alkannin was found to attenuate amyloid β-induced neuronal cell death. Mean ± SD, n = 5. Dunnett’s test compared with amyloid β-treatment alone was done. Aβ versus Aβ + alkannin (P=0.14). Aβ versus Aβ + shikonin (P = 0.11).
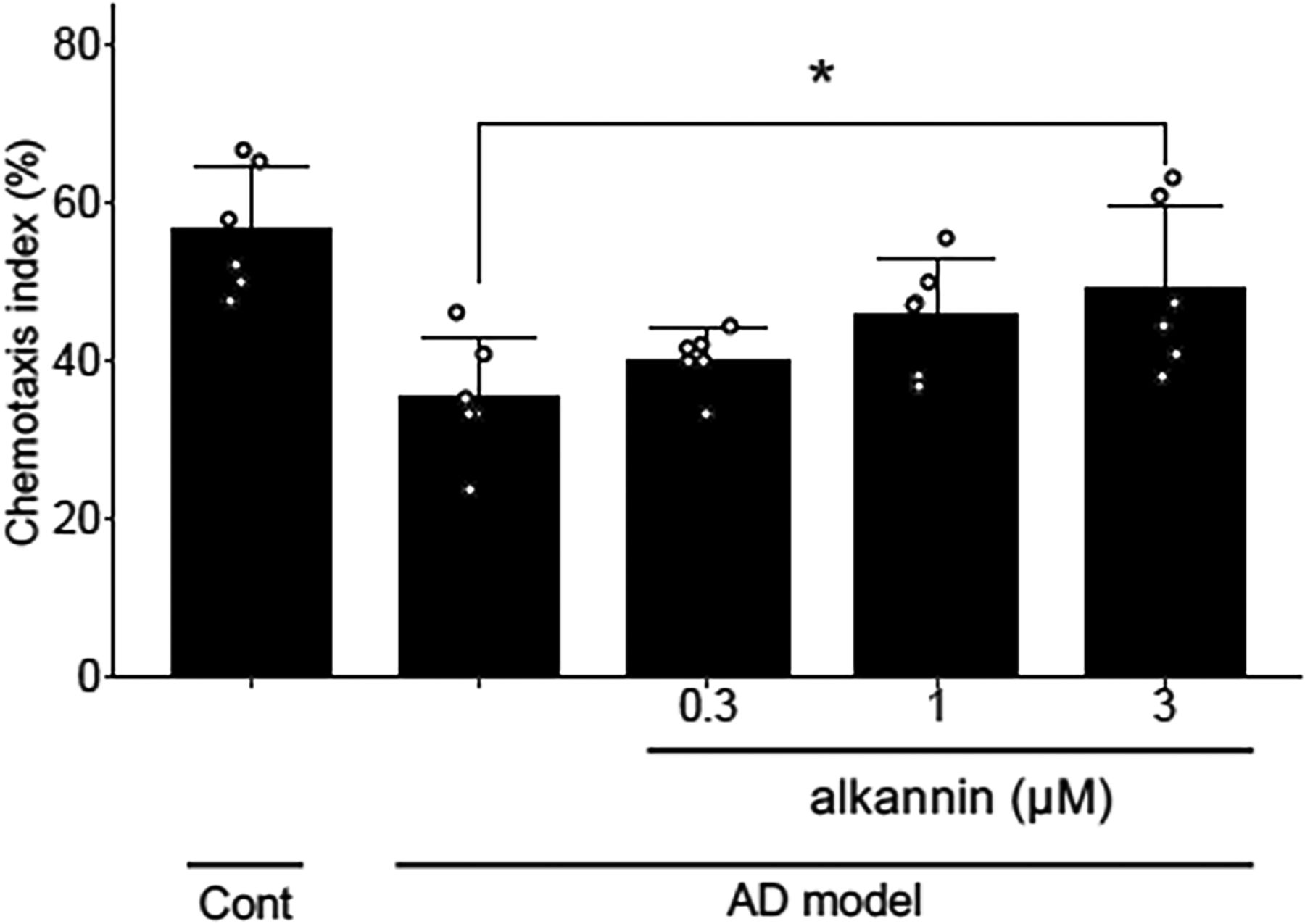
Alkannin Attenuates Chemotaxis in an AD C. Elegans Model
Since we found that alkannin attenuates amyloid β-induced neuronal cell death, we next investigated whether alkannin could ameliorate AD pathology in vivo. In the present study, we used an AD model of C. elegans. The worm strain CL2355 has a transgene that expresses human amyloid β peptide, which shows defects in chemotaxis associated with learning (Wu et al., 2006). The worm has a temperature-sensitive transgene, which expresses human amyloid β peptide when the culture conditions are shifted from 16°C to 25°C. Therefore, CL2355 was maintained at 16°C during synchronization and changed to 25°C 36 hours after the synchronization. A control strain of C. elegans (CL2122) was maintained at 25°C. The basal level of the chemotaxis index (CI) of CL2122 was approximately 60%. On the other hand, the CI of the AD C. elegans (CL2355) was approximately 35% (Fig. 6), indicating an AD phenotype. We next investigated the effect of alkannin on the AD phenotype observed in CL2355. Alkannin (0.3, 1, and 3 μM) was added 36 hours after synchronization and chemotaxis was analyzed 72 hours after drug application. We observed that the chemotaxis index was improved by alkannin in CL2355, the AD model of C. elegans (Fig. 6), while alkannin did not affect the chemotaxis index in the wild-type control C. elegans (CL2122) (Supplementary Fig. 2), suggesting that the effect of alkannin was specific to the AD model of C. elegans. These results suggest that alkannin can ameliorate AD pathology.

Alkannin ameliorates AD and amyloid β aggregation in C. elegans. (A) Alkannin ameliorates chemotaxis index of AD in C. elegans. Treatment of CL2355 (AD model of C. elegans) with alkannin increased CI. CL2122 (normal control of C. elegans) was incubated at 25°C. CL2355 was incubated at 16°C initially; however, 36 hours after hatching, the temperature was increased to 25°C. Concurrently, CL2355 was treated with alkannin (0.3, 1, 3 µM). At 72 hours after hatching, all the worms were treated with 5-fluoro-2’-deoxyuridine (0.6 mM) for sterilization. The chemotaxis assay was performed 144 hours after hatching. CL2355 showed a reduced CI, which indicated AD, while treatment with alkannin showed a concentration-dependent increase in CI, which reached significance at 3 µM. Mean ± SD, n = 6. *P < 0.05. Dunnett’s test compared with AD model worms was performed.
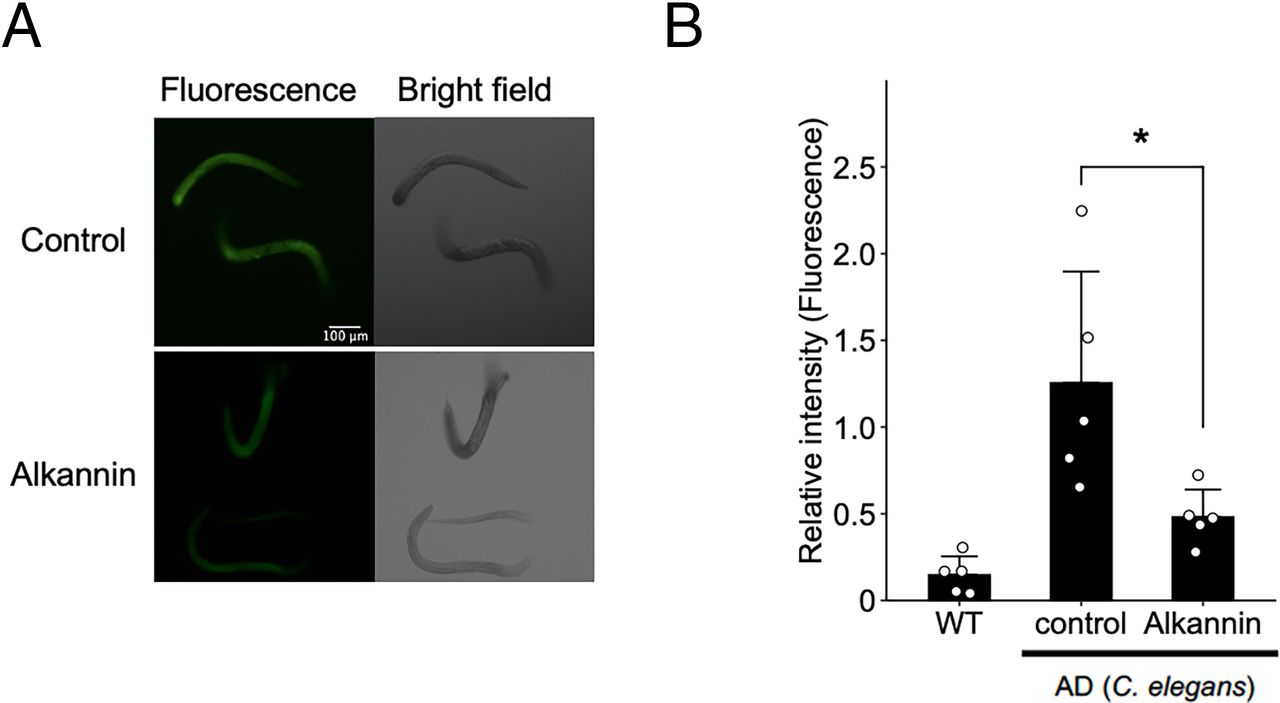
Alkannin Attenuates Amyloid β Aggregation in C. Elegans In Vivo
Since we found that alkannin could attenuate AD pathology in C. elegans, we next investigated whether alkannin could ameliorate amyloid β aggregation in vivo. To achieve this, we treated an AD model of C. elegans (CL4176) with alkannin and analyzed amyloid β aggregation by measuring ThT-induced fluorescence. CL4176 is a temperature-sensitive Aβ1-42 transgene transgenic worm. In wild-type control worms (N2), which do not express human Aβ1-42, the ThT fluorescence signal was almost undetectable (Fig. 6). In contrast, we detected a dramatic 7.9-fold increase in ThT fluorescence in CL4176, the AD model worms (Fig. 7). These results suggest that ThT can specifically detect aggregated Aβ1-42 in CL4176 worms. We next investigated whether Aβ1-42 aggregation in CL4176 was attenuated by alkannin. As shown in Fig. 7, we observed a decrease in the ThT fluorescence signal in alkannin-treated AD worms. These results suggest that alkannin can attenuate amyloid β aggregation in vivo.

Reduced amyloid β aggregation in C. elegans treated with alkannin. CL4176 (AD model of C. elegans) was incubated at 16°C for 36 hours. The worms were then treated with 3 µM alkannin and incubated at 25°C for 36 hours. After staining with thioflavin T, fluorescence images were acquired. (A) Fluorescence images of C. elegans. (B) Fluorescence intensity of C. elegans. Alkannin reduced aggregated form of amyloid β in the AD model of C. elegans. Mean ± SD, n = 5. *P < 0.05. Dunnett’s test.
Discussion
Several efforts have been made to identify compounds that can ameliorate aggregation of amyloid β peptide (Giorgetti et al., 2018; Pagano et al., 2020). Among these compounds, dye, such as Congo red, has been reported to ameliorate aggregation of amyloid β peptide. Alkannin is also a natural dye used for food coloring. However, currently, the relation between Congo red and alkannin against amyloid β aggregation are unknown. Alkannin is isolated from the roots of Alkanna tinctoria, a borage family compound, and has been used as a medicine for centuries (Papageorgiou, 1978). Alkannin has been reported to have antitumor activity (Zhang et al., 2018) and wound-healing properties (Papageorgiou, 1978; Papageorgiou et al., 1999). However, the molecular mechanisms underlying its action on wound-healing properties are not well understood. Induction of protein synthesis and protein folding is required during wound healing (Schürmann et al., 2014; Bachar-Wikstrom et al., 2021). When unfolded proteins accumulate, endoplasmic reticulum stress is activated (Walter and Ron, 2011). Interestingly, endoplasmic reticulum stress may be involved in wound healing, and the activation of protein folding by the chemical chaperone 4-PBA improved it (Schürmann et al., 2014; Bachar-Wikstrom et al., 2021). In the present study, we found that alkannin has chemical chaperone activity (Fig. 1A). Therefore, one of the mechanisms by which alkannin may contribute to wound healing may be mediated through this chemical chaperone activity.
In the present study, we found that alkannin may have a pharmacological mechanism of action for inhibiting formation of β sheet structure of amyloid β, thereby inhibit amyloid β aggregation and AD pathology. We performed ThT fluorescence assay for the measurement of amyloid β aggregation. However, we could not deny the possibility that alkannin may replace ThT bound to the amyloid β aggregates/fibrils, which may be the limitation of this assay. Therefore, we performed a different assay to confirm the results. We did CD spectra analysis and found that alkannin reduced formation of β-sheet formation of amyloid β. As the formation of β-sheet structure is an essential step for the formation of aggregates, we think alkannin may indeed reduce aggregate of amyloid β.
It is reported that aggregated amyloid β is toxic to neurons (Pike et al., 1993; Lorenzo and Yankner, 1994). Therefore, we assume that amyloid β-induced neuronal cell death of the present condition may be mediated through aggregated amyloid β. As we observed that alkannin reduced amyloid β aggregation, it is possible that inhibitory action of alkannin on amyloid β-induced cell death may be due to its inhibitory action against amyloid β aggregation. As we also observed that alkannin reduced toxic conformer of amyloid β, it is possible that alkannin’s effect in inhibiting neuronal cell death may be due to the inhibitory effect against formation of toxic conformer of amyloid β. Akannin has been reported to suppress nuclear factor-κB signaling in lipopolysaccharide-induced lung injury (Li et al., 2019). Amyloid β-induced inflammation will be observed in brain glial cells, such as microglia or astrocytes (Kaur et al., 2019). Therefore, in addition to neuronal cells, alkannin may also affect glial inflammatory condition elicited by amyloid β. Amyloid β peptide accumulates at extracellular space (Marksteiner and Humpel, 2008) and forms aggregate. As we found that alkannin can inhibit aggregation of amyloid β peptide, it is possible that alkannin inhibits aggregation of amyloid β at extracellular space. On the other hand, amyloid β peptide was also suggested to accumulate at the intracellular level (LaFerla et al., 2007). It is possible that alkannin may penetrate through plasma membrane and affect amyloid β peptide aggregation as shikonin does (Tong et al., 2018).
To analyze the observed effects in vivo, we performed chemotaxis assay in the transgenic C. elegans model of AD and found that alkannin recovered its chemotaxis. Considering this model, although the change in chemotaxis is a known phenotype, the causal relationship between reduced chemotaxis and amyloid β aggregation was not established. Therefore, we could not conclude that reduced chemotaxis was due to the reduced aggregation of amyloid β. On the other hand, using ThT analysis, we found that amyloid β was aggregated in this model and the aggregation was reduced by alkannin (Fig. 6). Therefore, it is possible that reduced aggregation of amyloid β by alkannin may contribute to the recovery of the chemotaxis in this model. Future analysis may be required to investigate these in vivo effects.
AD is a progressive neurodegenerative disease that is active many years before the symptoms of dementia appear. Therefore, drugs that are safe and can be administered over a long period are required. Alkannin has been used as a food additive in New Zealand and Australia (Awuchi et al., 2020), suggesting that the compound is safe to consume. Moreover, alkannin has been used for many decades as a Kampo medicine. Therefore, alkannin may be a safe drug candidate for the treatment of AD. However, future analyses are required to investigate these possibilities.
In the present study, through a screening assay, we found that the compound alkannin could inhibit amyloid β aggregation. Further analyses revealed that alkannin has a neuroprotective effect against AD. Thus, the results support the hypothesis that amyloid β is actively involved in AD, and this compound may be useful for the treatment of AD.
Acknowledgments
Scanning electron microscope analysis was performed at the Hiroshima University of Imaging Platform supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. The authors thank Dr. Misako Imai-Takemoto for her kind support in the analysis of the electron microscope.
Authorship Contributions
Participated in research design: Hosoi, Tohda, Nomura, Ozawa.
Conducted experiments: Yazawa, Imada, Tawara.
Performed data analysis: Yazawa, Imada, Tawara.
Wrote or contributed to the writing of the manuscript: Hosoi.
Footnotes
- Received December 1, 2021.
- Accepted January 4, 2023.
This work was supported by a Grant-in-Aid for the Cooperative Research Project from the Institute of Natural Medicine, University of Toyama in 2015 and 2016, JSPS KAKENHI [18KT0072], and the Takeda Science Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
↵1These authors contributed equally to this study.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- α-LA
- α-lactalbumin
- Aβ
- amyloid β
- Aβ1-42
- Amyloid β1-42
- AD
- Alzheimer’s disease
- CD
- circular dichroism
- C. elegans
- Caenorhabditis elegans
- CI
- chemotaxis index
- HFIP
- 1,1,1,3,3,3-hexafluoro-2-propanol
- LDH
- lactate dehydrogenase
- ThT
- thioflavin T
- Copyright © 2023 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}