


Abstract
Melatonin (N-acetyl-5-methoxytryptamine) is an indoleamine secreted by the pineal gland during the dark phase of the photoperiod. Its main function is the synchronization of different body rhythms with the dark-light cycle. Research on melatonin has significantly advanced since its discovery and we now know that it has considerable significance in various physiological processes, including immunity, aging, and reproduction. Moreover, in recent years evidence of the pharmacological possibilities of melatonin has increased. Indoleamine, on the other hand, has antidepressant-like effects in rodents, which may be mediated by the activation of calcium-calmodulin–dependent kinase II (CaMKII) and are also related to the regulation of neuroplasticity processes, including neurogenesis, synaptic maintenance, and long-term potentiation. Remarkably, patients with major depression show decreased levels of circulating melatonin in plasma. This review presents evidence of the antidepressant-like effects of melatonin in preclinical models and the participation of CaMKII in these actions. CaMKII’s role in cognition and memory processes, which are altered in depressive states, are part of the review, and the effects of melatonin in these processes are also reviewed. Furthermore, participation of CaMKII on structural and synaptic plasticity and the effects of melatonin are also described. Finally, the advantages of using melatonin in combination with other antidepressants such as ketamine for neuroplasticity are described. Evidence supports that CaMKII is activated by melatonin and downstream melatonin receptors and may be the common effector in the synergistic effects of melatonin with other antidepressants.
SIGNIFICANCE STATEMENT This review compiled evidence supporting that melatonin causes antidepressant-like effects in mice through calmodulin kinase II stimulation of downstream melatonin receptors as well as the participation of this enzyme in neuroplasticity, memory, and cognition. Finally, we describe evidence about the effectiveness of antidepressant-like effects of melatonin in combination with ketamine.
Introduction
Melatonin (N-acetyl-5-methoxytryptamine) is an evolutionarily conserved molecule present in plants and the animal kingdom (Reiter et al., 2010). Research on this molecule has significantly advanced since Aaron B. Lerner isolated the indoleamine from more than 250,000 bovine pineal glands (Lerner et al., 1958). Its chemical structure characterization and the discovery of its endogenous biosynthesis in the pineal gland as a molecule derived from tryptophan was performed by Julius Axelrod, shedding new light on central nervous system (CNS) control in circadian cycles (Axelrod and Weissbach, 1960).
Initially, melatonin was thought to regulate only the sleep-wake cycle; however, further research confirmed the complexity of its functions as a synchronizer of physiological functions with the environmental photoperiod and seasonal changes (Cipolla-Neto and Amaral, 2018). Melatonin is produced during the dark phase of the photoperiod, signaling darkness to the body (Zisapel, 2018). During the night, melatonin reduces body temperature and alertness and induces sleep (Arendt and Skene, 2005). Additionally, melatonin exerts long-lasting effects, priming the body for the upcoming seasons (Cipolla-Neto and Amaral, 2018).
Circadian regulation caused by melatonin influences immunity, aging, and reproduction and decreases disease vulnerability (Zisapel, 2018; Samanta, 2020, 2021, 2022). Research has also explored its potential benefits in bone health and oral health (Maria and Witt-Enderby, 2014; Kim et al., 2022).
Melatonin can exert its effects by activation of two protein receptors coupled to Gi/o proteins, melatonin receptors 1 and 2 (MT1 and MT2). These molecules have high affinity for melatonin binding and are located in the plasma membrane (Reppert, 1997; Dubocovich et al., 2010).
Thanks to its amphiphilic nature, it can also cross through the plasma membranes and bind to intracellular molecules such as calmodulin (CaM) (Benítez-King et al., 1996; Romero et al., 1998; Benítez-King, 2000).
The use of melatonin for therapeutical purposes has been extensively explored in neuropsychiatric diseases such as depression, Alzheimer’s and Parkinson’s diseases. Individuals with depression exhibit a specifically increased number of MT1 melatonin receptors (Wu et al., 2013). Melatonin also modulates the mitochondrial function in the hippocampus in animal models of Alzheimer’s disease, improving cognition and memory functions (Chen et al., 2020; Labban et al., 2021; Sano et al., 2022).
Additionally, dysregulation of the circadian cycle has been reported as a common factor in mood disorders such as bipolar, seasonal depression, and major depressive disorder. In all of these, there are variations in the plasma levels of melatonin or some of its synthesis metabolites (Dmitrzak-Weglarz and Reszka, 2017). Therefore, the endogenous levels of melatonin have been proposed as a potential biomarker in the diagnosis of depression, anxiety, and chronic stress (Chojnowska et al., 2021). Antidepressant-like effects in rodents are produced through stimulation of melatonin receptors; however, the signaling pathways are unknown. Here we review evidence supporting that calcium-calmodulin–dependent kinase II (CaMKII) participates in the signaling pathway of melatonin to produce antidepressant effects.
Antidepressant-Like Effects of Melatonin in Rodents
Animal models have provided valuable information about the pineal gland and melatonin’s role in physiological and pathological conditions such as mood disorders. These paradigms shed light on the pleiotropic role of melatonin in the human brain and its advantages on human mental health.
Evidence about the antidepressant actions of melatonin is extensive. Melatonin induces antidepressant-like effects in a wide variety of preclinical models, including tests of learned helplessness such as the forced swimming and the tail suspension tests, models of chronic mild stress, and models of anhedonia such as the sucrose preference and cognition like the Morris’ water maze, among others (Yao et al., 2023). These antidepressant actions are associated with the acute and prolonged administration of a wide range of melatonin doses (Kennaway, 2019a,b).
Melatonin synchronizes the daily sleep-wake cycle, which is disrupted in major depression, atypical depression, and seasonal affective disorder (Silva et al., 2021). This imbalance in the sleep-wake rhythm can produce sleep disorders, leading to memory consolidation loss, which takes place during sleep (Creery et al., 2022).
Melatonin has chronobiotic properties, which have been evidenced by the dependence of its antidepressant-like effects on the timing of administration in relation to the circadian cycle in rodent models; for example in the forced swimming test, melatonin caused a robust antidepressant-like effect when administered at the time at which endogenous melatonin is at the highest levels, that is, in the dark phase of the photoperiod (Estrada-Reyes et al., 2018).
Moreover, knockout mice that have a deletion in the clock gene Bmal1 showed depressive-like behaviors, evidenced by a delay in the escape time and the number of escape failures, as well as increased escape latencies and number of escape failures in the learned helplessness paradigm (Landgraf et al., 2015). Remarkably, this gene participates in the suprachiasmatic nucleus (SCN) to maintain the circadian periodicity. In the tail suspension test, the lack of expression of the Bmal1 gene resulted in higher immobility time, whereas in anxiety tests it resulted in increased anxiety levels compared with animals without gene deletions (Landgraf et al., 2016). In addition, the lack of multiple clock genes (global knockout mice) resulted in altered behaviors associated with the multiple functions codified by these clock genes. These results indicate that disrupting the SCN rhythm produces depressive-like and anxiety-like behaviors. In contrast, no changes were observed in the hedonic behavior of rodents with these gene deletions. Such behavior was measured by the sucrose preference, spatial preference, and open field tests as well as the by the aversion to eating in a novel environment. The measures suggest that only specific aspects of depressive-like behavior are influenced by the disruption of the central clock (Kukula-Koch et al., 2021). Remarkably, the circadian oscillations in the levels of melatonin circulating in the blood, similarly to serotonin, are altered in humans with mood disorders (Tonon et al., 2021). Currently, it is not entirely clear whether or how mood disorders might affect the circadian cycle of blood circulating levels of both indoleamines. Studies about the efficacy of melatonin in the treatment of mood disorders in humans are controversial. However, the use of the melatonin analog agomelatine, an agonist of MT1 and MT2 receptors and a 5HT2C antagonist, has been successful in the treatment of depression (Plesničar, 2014). Some authors argue that agomelatine synchronizes circadian rhythms such as the sleep-awake cycle through interaction with MT1 and MT2 receptors, whereas its antidepressant effects are produced through 5HT2C antagonism (Naveed et al., 2022). More clinical studies are necessary to disclose the role of melatonin as an antidepressant in humans and to define its use in therapeutic approaches, especially the use of melatonin in combination with other antidepressants, which has been demonstrated to have an additive effect in the antidepressant-like effects in rodents (Ramírez-Rodríguez et al., 2014; Estrada-Reyes et al., 2022).
In spite of all the chronobiotic actions of melatonin, its antidepressant actions cannot be explained only by the interplay of its concentration levels with the timing of the circadian clock; melatonin has other direct actions on the CNS: some are mediated by its binding to melatonin receptors and some others are mediated by its direct interaction with intracellular molecules. The melatonin signaling pathway is, however, not fully known.
Calmodulin-Dependent Kinase II As an Effector of Antidepressant Effects
CaMKII is an enzyme that is sometimes activated by antidepressants and recognized by its participation in cognition and memory consolidation, functions that are dysregulated in depression (Adaikkan et al., 2018; Yasuda et al., 2022).
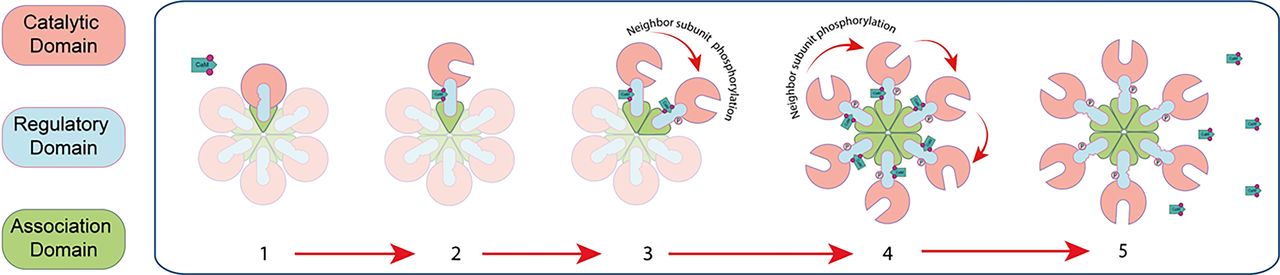
CaMKII is a holoenzyme composed of 12 subunits of 56–60 kDa that are assembled into two rings of six subunits (Zalcman et al., 2018). The subunits have a conserved amino acid sequence, with an −NH2 terminal catalytic domain, followed by a regulatory domain that contains a self-inhibitory region and a calmodulin-binding site as well as a −CO2H terminal association domain (Fig. 1). There are four isoforms of CaMKII, named α, β, γ, and δ, which differ in their subcellular localization, calcium binding kinetics, and affinity to other proteins. The α and β isoforms are predominantly expressed in the brain, whereas the γ and δ isoforms are also expressed in the heart and other tissues. The α isoform of CaMKII plays a crucial role in learning and memory; although the role of the β, γ, and δ isoforms is less understood, they have also been implicated in synaptic plasticity and memory formation. The isoforms of CaMKII are encoded by different genes and have different properties according to their subunit composition, which can be constituted by either a single type (homomeric holoenzyme) or a combination of subunits (heteromeric holoenzyme) (Lisman et al., 2012; Zalcman et al., 2018).

Schematic representation of one hexamer of the calcium-calmodulin–dependent protein kinase II. CaMKII is formed by 12 subunits arranged in two hexamers. Each subunit is constituted by three domains: catalytic domain (pink), regulatory domain (blue), and association domain (green). Upon activation by Ca2+-CaM binding, the subunit changes its conformation structure from the closed state (1) to the open state (2). The conformational change of CaMKII induces its autophosphorylation in the threonine 286 (T286) located at neighbor subunit (3); subsequently, all subunits are phosphorylated (4). Then the kinase activity becomes independent of Ca2+-CaM calcium-calmodulin (5).
CaMKII phosphorylates multiple targets, among which are the synaptic proteins. These proteins participate in both structural and functional synaptic plasticity (Colbran and Brown, 2004). For instance, phosphorylation of N-methyl-D-aspartate (NMDA) receptors and the GluR1 subunit by CaMKII regulates the placement of both receptors in the plasma membrane. Furthermore, actin phosphorylation leads to microfilament organization; EF2 phosphorylation stimulates protein synthesis, among other events. Therefore, activated CaMKII triggers a downstream signaling processes during synaptic and behavioral plasticity (Duman and Aghajanian, 2012; Adaikkan et al., 2018).
CaMKII activation in the synapses starts with a presynaptic electrical activity that causes a transient increase in the cytoplasmic calcium concentration. Calmodulin is one of the main acceptors of calcium and transduces the increases in intracellular calcium concentration through the activation of multiple target proteins, including CaMKII. Calmodulin activated by calcium (Ca2+-CaM) adopts a double dumbbell shape and binds to the calmodulin binding site of CaMKII, stimulating its activity (Rostas and Skelding, 2023). The transitory increase in intracellular calcium levels in response to each action potential at presynaptic terminals and postsynaptic dendritic spikes lasts only a few milliseconds (Llinás et al., 1995). The activation of CaMKII by Ca2+-CaM lasts from seconds to minutes, and the interaction of CaMKII with its associated proteins may persist for more than 30 minutes (Yasuda et al., 2022). Once activated by Ca2+-CaM, the kinase transfers a phosphate group from ATP to the amino acid threonine 286 (T286) of a neighbor CaMKII subunit, making the activity of the kinase independent from Ca2+-CaM (Fig. 1). CaMKII is activated within seconds after synaptic stimulation and remains active even when the cytoplasmic calcium concentration returns to its basal levels (Miller and Kennedy, 1986). All of these events participate in the consolidation of memory and cognition in the hippocampus.
Recent studies suggest that CaMKII dysfunction in the brain is associated with neuropsychiatric disorders, including depression. Alterations in protein phosphorylation have been documented in individuals diagnosed with major depressive disorder when compared with their healthy counterparts (Martins-de-Souza et al., 2012, 2014). People with major depression also show a decrease in cognitive functions associated with a reduction of the hippocampal volume (Drevets et al., 2008). This atrophy is related to a decrease in the number of neurons as well as with an impairment of synaptic plasticity (Duman and Aghajanian, 2012). Remarkably, the deletion of genes, which codify for CaMKII, or the inhibition of CaMKII’s activity blocks synaptic formation, suggesting that neurons need CaMKII to maintain their connectivity (Martin et al., 2000; Silva et al., 1992). Alterations in CaMKII also affect neurotransmitter signaling and synaptic communication (Robison, 2014). Antidepressant drugs act in the hippocampus by increasing neurogenesis and connectivity. Furthermore, evidence indicates that one of the effector molecules in the signaling pathways of antidepressants is CaMKII (Barbiero et al., 2007; Adaikkan et al., 2018).
Melatonin Receptors and Calmodulin Kinase II Participate in the Mechanism through Which Melatonin Stimulates Neurogenesis
The participation of melatonin receptors in the mechanism involved in the antidepressant effects of melatonin have been studied in detail. However, the signaling pathways and participation of CaMKII are not entirely known. Abundant evidence suggests that melatonin promotes neurogenesis and the incorporation of new neurons to brain circuits to compensate for neuronal damage and loss during pathological processes (Ramírez-Rodríguez et al., 2018, 2020).
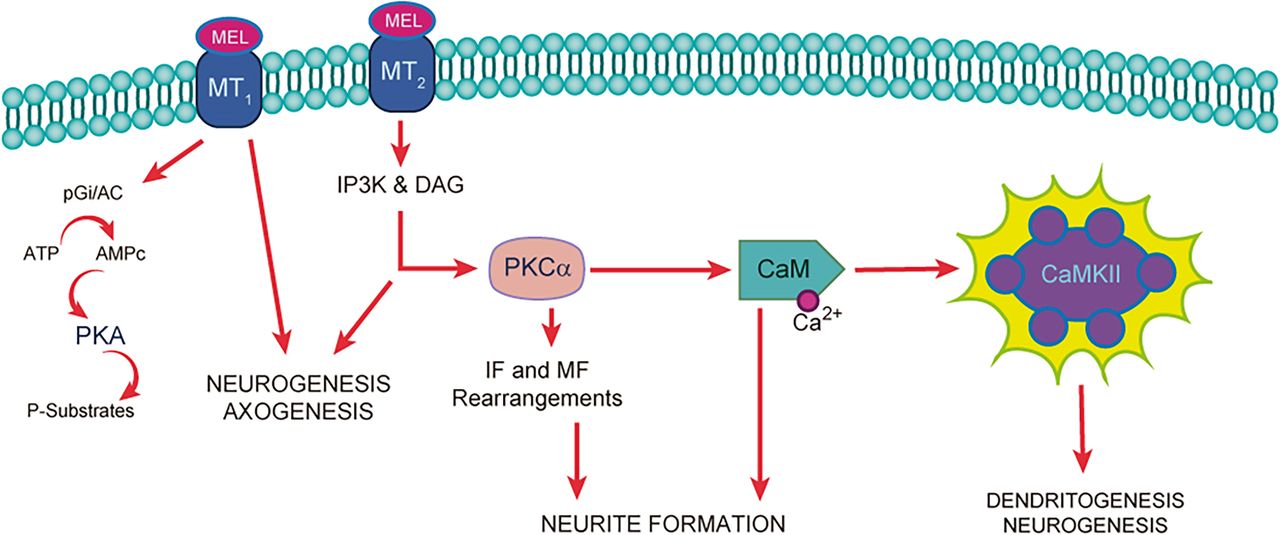
Some of the antidepressant effects of melatonin are produced through MT1 and MT2 receptor stimulation (Liu et al., 2016), and some effector proteins downstream of melatonin receptor stimulation have been described in neurogenesis, axogenesis, and dendritogenesis (Fig. 2). Experiments with cell cultures, derived from both neural precursor cells from neurogenic niches (Sotthibundhu et al., 2010, 2016,; Liu et al., 2016a) and pluripotent cells from amniotic fluid, have demonstrated that melatonin is a factor in cell survival, proliferation, and neuronal differentiation, the latter mediated by histone acetylation (Phonchai et al., 2019). Melatonin also increases the production of brain-derived neurotrophic factor (BDNF), which is involved in the neuronal differentiation of neural stem/progenitor cells. Furthermore, melatonin has been proposed as a neurotrophic (Miranda-Riestra et al., 2022) and anti-inflammatory agent (Permpoonputtana et al., 2018; Taniguti et al., 2018; Lv et al., 2020; Mansouri et al., 2022; Muñoz-Jurado et al., 2022). Further experiments using human olfactory neuronal precursors also showed that melatonin promotes neurogenesis. This neurogenic response elicited by melatonin is mediated by melatonin receptors (MT1, MT2) and the signaling pathways related to activation of mitogen-activated protein kinases (MAPK), extracellular signal–regulated kinases 1/2 (ERK1/2), phosphoinositide 3-kinase (PI3K), and protein kinase B (Akt) (Chu et al., 2016). These signaling pathways are frequently involved in melatonin receptor–related effects on cell survival, proliferation, and neuronal differentiation of neural stem/progenitor cells (Liu et al., 2016; see also Chen et al., 2020) (Fig. 2). The neurogenic effect of melatonin has also been shown in the dentate gyrus of Balb-C mice (Ramírez-Rodríguez et al., 2020). However, the antidepressant effect obtained in animals might not only occur due to the generation of new neurons but also due to the strengthening of neuronal communication or the consolidation of new neuronal circuits. In this regard, melatonin has also been shown to promote maturation processes in new postmitotic neurons, including the formation, growth, and complexity of neurites, both in cultured neuronal precursors (Galván-Arrieta et al., 2017) and in the hilus of organotypic hippocampal slice cultures (Domínguez-Alonso et al., 2012). In addition, melatonin modulates the structural plasticity of the mossy fiber projection in the hippocampus in mice (Ramírez-Rodríguez et al., 2018), and said modulation increased the number and complexity of dendrites of newborn neurons in the dentate gyrus as well as concomitantly decreased the depression-like behavior in mice submitted to the forced swimming test (Ramírez-Rodríguez et al., 2020).

Melatonin receptors’ participation in neurogenesis and neurodevelopment. Melatonin binding to MT1 and MT2 receptors activates the PGi/AC (Inhibitory G protein/Adenylate cyclase) and IP3K/DAG (Inositol triphosphate kinase/ Diacylglycerol) signaling pathways, respectively. DAG activates PKC (Protein Kinase C) that phosphorylates Ca2+-CaM, which in turn binds to CaMKII. The activation of this enzyme phosphorylates different substrates to increase neurite formation, dendritogenesis, axogenesis, and new neuron formation.
Melatonin has also neuroregenerative effects in pathological conditions. This indoleamine reversed the reduced axogenesis associated with lower levels of pGSK3β in olfactory neuronal precursors derived from a patient with schizophrenia (Galván-Arrieta et al., 2017). Prolonged treatment with melatonin improved the cognitive deficit produced after transient global cerebral ischemia by increasing the number of oligodendrocytes and restoring myelin through the activation of ERK1/2 signaling and by increasing glutamatergic synapses in the ischemic brain area (Chen et al., 2018). Melatonin also significantly reduced the deficits in spatial memory and the motor deficits caused by cortical compact impact injury in mice by preserving the integrity of cortical and hippocampal dendritic spine morphology and by reducing microgliosis and central neuroinflammation (Lin et al., 2020). Finally, melatonin also accelerated the process of motor nerve repair after different types of injury through activation of the ERK1/2 pathway downstream of the MT1 receptors (Stazi et al., 2021).
MT receptors, calmodulin, and the activation of CaMKII are involved in the neurogenic effects of melatonin because these effects are inhibited by luzindole, a MT1 and MT2 receptor antagonist, and KN-62, an inhibitor of CaMKII activity, respectively (Domínguez-Alonso et al., 2015; Estrada-Reyes et al., 2022) (Fig. 2). However, it remains unclear which effects of melatonin take place at the cell membrane or cytoplasmic level, which interactions occur downstream of the membrane receptors, and whether there are molecular interactions in the synaptic clefts. Still, CaMKII plays a crucial role in the regulation of synaptic plasticity.
Calmodulin-Dependent Kinase II Involvement in Long-Term Potentiation: Effects of Melatonin
The limbic system, particularly the hippocampus, plays a crucial role in the acquisition of new information. It allows information stored in the short-term memory to pass to the long-term memory. Remarkably, in major depression there is a significant reduction in the volume of the hippocampus (Drevets et al., 2008). Moreover, synaptic plasticity in the hippocampus is altered in major depressive disorder (MDD), and failures in mnemonic functions have been observed in animal models of depression (Pittenger and Duman, 2007) and in people suffering depression (Benson et al., 2015). This effect can be explained by changes in hippocampal synaptic plasticity modeled by long-term potentiation (LTP). Depression can downregulate synaptic proteins and growth factors required for hippocampal LTP in rodents (Citri and Malenka, 2008).
LTP participates in the consolidation of some cognitive functions such as learning and memory (Smith et al., 2003; Kerchner and Nicoll, 2008). Importantly, CaMKII plays a key role in LTP, which is altered in rodents submitted to depression models, for instance, learned hopelessness in mice (Machado et al., 2017), Whistar-Kyoto rats (Aleksandrova et al., 2020), and other models of depressive-like behavior (Batalha et al., 2013; She et al., 2015; Yang et al., 2018). LTP is a phenomenon of synaptic plasticity defined as a lasting increase in synaptic communication between two neurons and caused by high-frequency electrical stimulation. Therefore, the changes in synaptic plasticity in the hippocampus may be a plausible explanation for the memory dysfunction induced by depression (Duman and Aghajanian, 2012; Duman et al., 2016).
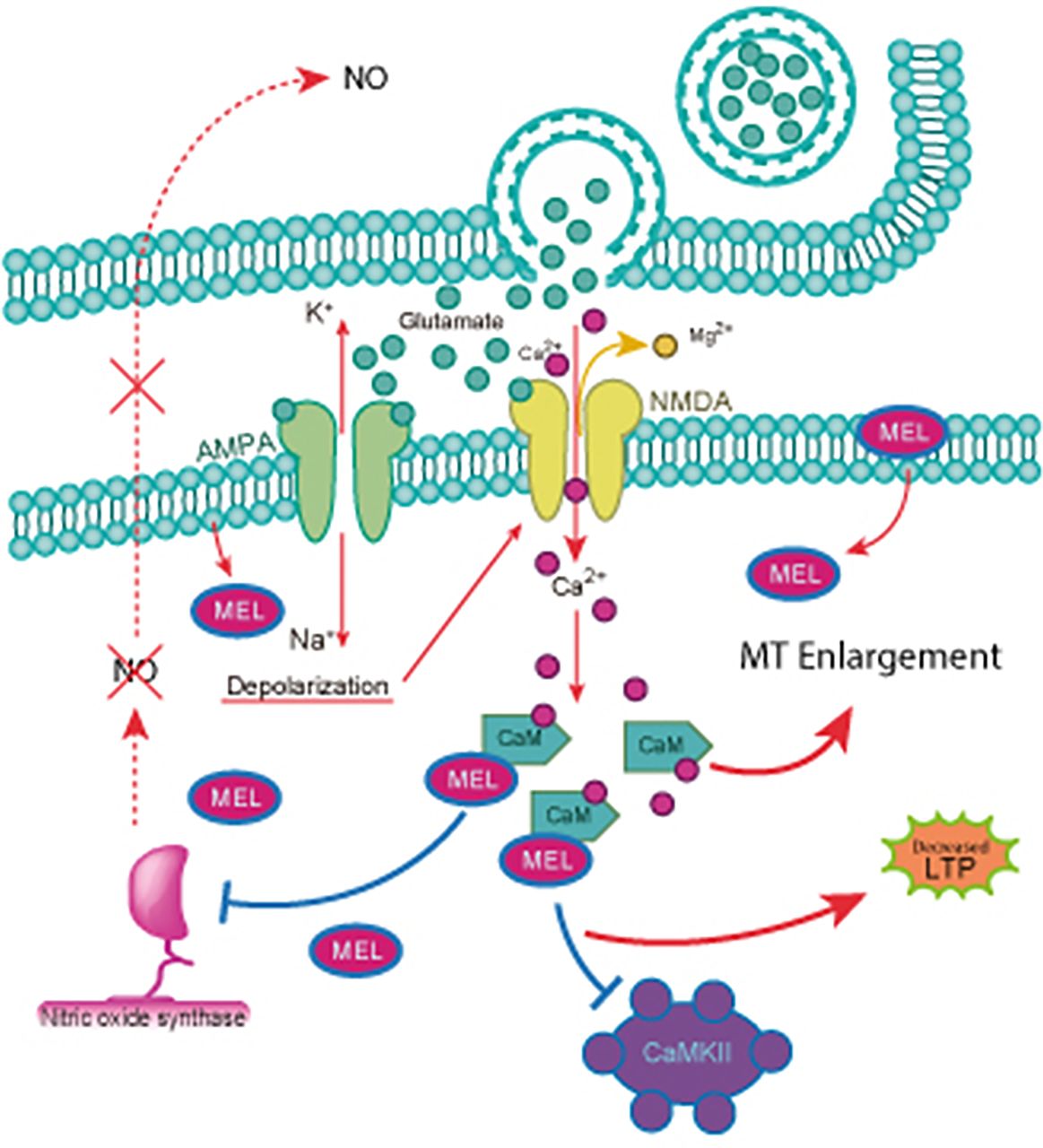
In depression disorders, there is an imbalance in the hypothalamus-pituitary-adrenal (HPA), which leads to a failure in the negative feedback of glucocorticoid secretion. Such failure produces a high level of cortisol or corticosterone and glutamate in rodents. This last one is the most studied neurotransmitter related to LTP. In the hippocampus, the CA3 Schaffer collateral axons release glutamate in response to stimulation at high frequencies. Glutamate activates postsynaptic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)-type receptors in CA1 pyramidal cells. Induction of LTP depends on the postsynaptic depolarization produced by the activation of AMPA-type receptors, allowing the influx of Ca2+ to the postsynaptic terminal and the subsequent formation of the Ca2+-CaM complex that binds and activates CaMKII. Activated CaMKII promotes the recruitment of AMPA receptors to the plasma membrane. Moreover, CaMKII phosphorylates AMPA receptors, increasing their conductance. Therefore, CaMKII activation makes the postsynaptic responses to glutamate more efficient. Furthermore, Ca2+-CaM activates nitric oxide synthase (NOS), increasing the levels of nitric oxide (NO), which diffuses to the presynaptic terminal where it increases the release of glutamate. Also, at the presynaptic level, CaMKII phosphorylates synapsin (Huttner et al., 1983; Schiebler et al., 1986; Sihra et al., 1989; Benfenati et al., 1989a,b; Jovanovic et al., 2001; Hosaka et al., 1999; Chi et al., 2003), promoting mobilization of vesicles to the active zone and further increasing glutamate release (Malenka et al., 1989; Malinow et al., 1989; Malenka and Nicoll, 1999; Collingridge et al., 2004). Altogether, these events cause a prolonged increase in the response of the CA1 neurons to stimulation of the CA3 Schaffer collaterals known as LTP, which in turn consolidates memory.
Moreover, stimulation of glutamate receptors located at the postsynaptic terminal produces a modest but statistically significant increase in CaMKII levels (Dosemeci et al., 2001). On the other hand, mutations that prevent the activation and autophosphorylation of CaMKII cause deficits in long-term synaptic plasticity and defects in learning or behavioral memory (Martin et al., 2000; Silva et al., 1992). Altogether, the evidence supports that CaMKII has an important role in the glutamate signaling pathway in LTP, which, as mentioned before, plays a key role in mnemonic functions altered in major depression. In this regard, melatonin also stimulates memory and cognition in animal models, indicating its participation in LTP regulation. In fact, melatonin regulates the electrical activity of hippocampal neurons (Zeise and Semm, 1985; Musshoff et al., 2002) and modifies the synaptic transmission among these neurons (Wan et al., 1999; Hogan et al., 2001; El-Sherif et al., 2002), which can be implicated in the antidepressant effects of the indoleamine.
Moreover, it has been described that melatonin decreases neuronal excitability by increasing the activation of G-protein–coupled inwardly rectifying potassium (GIRK) channels (Nelson et al., 1996; van den Top et al., 2001; Djebari et al., 2021) (Fig. 3), which also produce a decrease in LTP, reducing the number of NMDA channels activated by glutamate (Shibata et al., 1989; Stehle et al., 1989; Mason and Rusak, 1990; Jiang et al., 1995; van den Top et al., 2001).

Schematic representation of the mechanism by which melatonin decreases LTP through the activation of K+ channels. The outflow of K+ through these channels hyperpolarizes the membrane, preventing the activation of NMDA receptors, which need a depolarization of the membrane to conduct calcium.
Additionally, melatonin binds to CaM in the presence of Ca2+ with high affinity (1 nM) and decreases the activity of several CaM-activated enzymes like NOS with an EC50 0f 1 nM through a noncompetitive mechanism (for a review, see Pozo et al., 1994; Benítez-King et al., 2024). The decrease in NO production results in a decrease in LTP since this retrograde transmitter diffuses to the presynaptic terminal and enhances glutamate release as part of the mechanism for LTP. The direct interaction of melatonin with Ca2+-CaM also prevents the activation of CaMKII by Ca2+-CaM (Fig. 4).

Schematic representation of melatonin binding to Ca2+-CaM: participation in the antidepressant-like effects of melatonin. Melatonin crosses the plasma membrane and binds to Ca2+-CaM, preventing the activation of NOS and of CaMKII. Consequently, melatonin reduces LTP, a crucial step for the consolidation of memory, preventing the increase in presynaptic glutamate release mediated by NO and decreasing the incorporation of AMPA receptors to the membrane. Also, inhibition of Ca2+-CaM causes microtubule polymerization, a crucial step in neuritogenesis. Both LTP and neurite formation contribute to the actions of melatonin.
Remarkably, the inhibitory effects of melatonin on LTP seem contradictory in view of the effects of this indoleamine in the improvement of memory and cognition. These dual opposite effects might be explained by the different mechanisms of action of melatonin and by the opposite effects of melatonin on CaMKII activity in different microenvironments: in in vitro aqueous microenvironments melatonin increases the activity of CaMKII, whereas in a lipidic microenvironment it inhibits the activity of the kinase (Argueta et al., 2022). Furthermore, the decreased neuronal excitability produced by melatonin through the GIRK potassium channels can be a preliminary step necessary to get full responses in LTP. Thus, melatonin can decrease LTP to prepare the system for a high LTP response.
Furthermore, we found that melatonin prevents the oxidation of CaM through its binding to the hydrophobic domain of this protein, increasing the available levels of Ca2+-CaM for CaMKII activation (Benítez-King et al., 2024). In this regard, preliminary experiments showed that the amount of phospho-CaMKII (the activated form of CaMKII) is increased in the hippocampus of mice submitted to the forced swimming test where melatonin causes an antidepressant-like effect.
One explanation on the apparently contradictory effects of melatonin on CaMKII activity can be the rheostatic actions of melatonin on physiological functions. One example is the effects of melatonin a on the secretion of neurotransmitters in neuronal precursors isolated from the human olfactory epithelium. Melatonin increased the amount and the velocity of secretion when applied to olfactory neuronal precursors obtained from a healthy control subject. In contrast, in neuronal precursors obtained from patients diagnosticated with schizophrenia, which showed an increased level of secretion, melatonin decreased these parameters, bringing them to levels similar to those in cells from healthy subjects (Cercós et al., 2017). These differential effects suggest that melatonin can restore the equilibrium in basic biological processes such as secretion of biochemical mediators (Cercós et al., 2017). Similarly, the stimulation of CaMKII mediated by MT1 and MT2 receptors and the inhibition of this enzyme by melatonin binding to CaM results at final in the antidepressant effects caused by melatonin.
Other signaling pathways that are independent from CaMKII also participate in the mechanisms of action by which melatonin modulates LTP. Melatonin decreases the adenylyl cyclase–protein kinase A (PKA) pathway, reducing the incorporation of AMPA receptors to the postsynaptic membrane; through this mechanism, melatonin has inhibitory actions on hippocampal LTP (Wang et al., 2005). These inhibitory actions were prevented by luzindole, an MT1/MT2 nonselective melatonin receptor antagonist, as well as by the MT2-selective antagonist 4-phenyl-2-propionamidotetraline, indicating the involvement of melatonin receptors. Moreover, the PKA inhibitor H-89 mimicked the effects of melatonin and precluded further inhibition by melatonin (Wang et al., 2005). Additionally, forskolin, an activator of adenylyl cyclase, overcame the inhibitory effects of melatonin on LTP without affecting its magnitude. Previous work also showed that mice deficient in components of the cAMP-PKA signaling pathway also had deficits in LTP and hippocampal-dependent memory (Bourtchuladze et al., 1994; Wu et al., 1995; Abel et al., 1997).
Additive Antidepressant-Like Effects of Melatonin in Combination with Other Antidepressants: Importance of CaMKII
Melatonin has been used in combination with selective reuptake serotonin inhibitors (SSRIs) such as fluoxetine and citalopram and was found to produce additive antidepressant-like effects in mice (Ramírez-Rodríguez et al., 2014; Li et al., 2018; Filopovic et al., 2022). It is known that SSRIs affect CaMKII activity. However, it is necessary to study more deeply the participation of CaMKII in the mechanisms of antidepressant molecules used in the treatment of mood disorders (Barbiero et al., 2007; Zhuang et al., 2023). Similarly to the SSRIs, recent evidence indicates that ketamine has antidepressant-like effects in mice associated with stimulation of CaMKII activity (Adaikkan et al., 2018).
A growing body of evidence demonstrates that a single infusion of a subanesthetic-dose of ketamine [2-(2-Chlorophenyl)-2-(methylamino)cyclohexan-1-one;hydron;chloride] rapidly relieves depressive symptoms (within hours) in individuals with major depressive disorder (Zarate et al., 2006; Murrough et al., 2013a,b; Fava et al., 2020). This positive effect has, however, a negative counterpart since ketamine also produces psychomimetic effects, which have limited its clinical use as an antidepressant (Na and Kim, 2021).
Remarkably, a leading hypothesis regarding the antidepressant properties of ketamine underscores its role in neural plasticity. Moreover, antidepressant-like effects caused by ketamine has been due to activation of CaMKII (Duman and Aghajanian, 2012; Adaikkan et al., 2018).
Patients with depression often show a decrease in cognitive functions. The antidepressant effects of the NMDA receptor antagonists, like ketamine, have been classically associated with enhanced neurogenesis in the prefrontal cortex and in the hippocampus (Castrén and Rantamäki, 2010a,b; Hillhouse et al., 2015; Hillhouse and Porter, 2015). Since in patients with depression there are high levels of glutamate in plasma and in CNS structures (Hashimoto, 2009), contrary to the plasma levels of serotonin and melatonin, which are diminished, the effect of ketamine may be a consequence of a regulation of the glutamate neurotransmission system.
Additionally, ketamine has been shown to enhance hippocampal synaptic plasticity in normal animals and to partially restore the levels of LTP induction in murine models of depression (Yang et al., 2018). In Wistar-Kyoto rats, which are used as a model of depression, in vivo extracellular recordings showed a deficit in LTP, which was rescued with a single low dose of ketamine (Aleksandrova et al., 2020). Ketamine also enhances LTP in other areas of the CNS, such as the visual and auditory cortices, in individuals with mood disorders (Sumner et al., 2020) who have reduced LTP in these areas (Normann et al., 2007; Yeap et al., 2009; Ostermann et al., 2012). These studies support that ketamine has effects on LTP in humans with depression, although more work is needed to disclose the mechanisms involved. It is possible that ketamine acts by inhibiting NMDA receptors in GABAergic interneurons, decreasing their activity and thus causing a disinhibition of glutamatergic neurons that are normally inhibited by GABA.
Recently, our group found that noneffective doses of melatonin and ketamine administered in combination (acute and triple administration) produced antidepressant-like effects in mice. Animals showed a decreased immobilization time in the forced swimming test (Estrada-Reyes et al., 2018, 2021) and they didn’t show increased movements in the open field test, thus showing that this dose of ketamine does not produce negative psychomimetic effects. Importantly, these antidepressant-like effects were also associated with increased neurogenesis in the dentate gyrus of the hippocampus, and preliminary data indicates that CaMKII activity is enhanced (Estrada-Reyes et al., 2021). Since CaMKII participates in the signaling pathways downstream of both melatonin and ketamine, it is possible that this kinase is the common target in the action of this combination. It is enticing to speculate that the ketamine/melatonin combination could have a regulatory effect on LTP; however, further studies are necessary to assess this possibility.
Conclusions and Perspective
Experimental preclinical evidence indicates that melatonin can be useful in the treatment of neuropsychiatric diseases, particularly major depression. In this regard, concentrated efforts on the study of dose adjustments seem promising for the development of new pharmacological therapies for the treatment of depression that consider the use of melatonin; the mediation of melatonin’s actions by its receptors; its effector proteins or second messengers that are known to be potentiated by melatonin; and its involvement in mechanisms of neuroprotection, neuroplasticity, cognition, and memory. A solid body of evidence indicates that the mechanism of action of melatonin—by itself and in synergy with other antidepressants—involves the activation of CAMKII. Furthermore, melatonin can exert its antidepressant effects in both rodents and humans at both pharmacological and physiological doses, the first to trigger the signaling pathways involved in its antidepressant effects of melatonin and the second to reestablish endogenous levels of the hormone to synchronize the sleep-wake rhythms. This opens a field of study to investigate how manipulating CaMKII activity, expression, subcellular targets, binding partners, or metabolic pathways could provide the basis for therapeutic interventions in a variety of mood disorders (Sałaciak et al., 2021). These pathways represent vital aspects in comprehending the biological foundations of depression. Incorporating these understandings into innovative therapeutic approaches has the potential to clear a path for effective treatments that target the complex mechanisms at play in depressive disorders.
Acknowledgments
The authors thank Jorge Estrada and Itzel Aquino for English corrections.
Data Availability
This article contains no datasets generated or analyzed during the present study.
Authorship Contributions
Participated in research design: Miranda-Riestra, Benítez-King, Estrada-Reyes.
Performed data analysis: Miranda-Riestra, Oikawa-Sala, Argueta, Constantino-Jonapa, Benítez-King, Estrada-Reyes.
Wrote or contributed to the writing of the manuscript: Miranda-Riestra, Cercós, Trueta, Cruz-Garduño, Benítez-King, Estrada-Reyes.
Footnotes
- Received February 8, 2024.
- Accepted June 26, 2024.
This work was supported by Instituto Nacional de Psiquiatría Ramón de la Fuente Muñiz [Grant IC22173.0] and by CONAHCYT [Grant CBF2023-2024-457].
The authors declare that they have no conflicts of interest with the contents of this article.
↵1 A.M.-R. and M.G.C. contributed equally to this work.
Abbreviations
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- Ca2+-CaM
- calmodulin activated by calcium
- CaM
- calmodulin
- CaMKII
- calcium-calmodulin–dependent kinase II
- CNS
- central nervous system
- ERK1/2
- extracellular signal–regulated kinases 1/2
- LTP
- long-term potentiation
- MT1
- melatonin receptor 1
- MT2
- melatonin receptor 2
- NMDA
- N-methyl-D-aspartate
- NO
- nitric oxide
- NOS
- nitric oxide synthase
- PKA
- adenylyl cyclase–protein kinase A
- SSRI
- selective serotonin reuptake inhibitor
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Antidepressant-Like Effects of Melatonin in Rodents
- Calmodulin-Dependent Kinase II As an Effector of Antidepressant Effects
- Melatonin Receptors and Calmodulin Kinase II Participate in the Mechanism through Which Melatonin Stimulates Neurogenesis
- Calmodulin-Dependent Kinase II Involvement in Long-Term Potentiation: Effects of Melatonin
- Additive Antidepressant-Like Effects of Melatonin in Combination with Other Antidepressants: Importance of CaMKII
- Conclusions and Perspective
- Acknowledgments
- Data Availability
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters