


In the article above [To K, Zhao Y, Jiang H, Hu K, Wang M, Wu J, Lee C, Yokom DW, Stratford AL, Klinge U, Mertens PR, Chen CS, Bally M, Yapp D, and Dunn SE (2007) Mol Pharmacol 72:641–652], an error was introduced by mistake when preliminary data were prepared for a poster presentation at a Keystone Symposia meeting in 2007 [To K, Zhao Y, Jiang H, Wu J, Stratford AL, and Dunn SE (2007) Phosphoinositide-dependent kinase-1 inhibitor, OSU-03012, prevents Y-box binding protein-1 (YB-1) from inducing epidermal growth factor receptor (EGFR) expression in basal-like breast cancer cells. Keystone Symposia on Molecular Targets for Cancer; 2007 March 18–23; Whistler, BC, Canada]. Subsequently, during the writing of the manuscript, the authors inadvertently neglected to replace the control subjects with a different set that was intended for submission to Molecular Pharmacology. A.S. authored two articles simultaneously describing experiments with SUM 149 cells but with two different sets of control subjects: one intended for publication in Molecular Pharmacology and the other intended for publication in Breast Cancer Research. The control subjects are shown below.

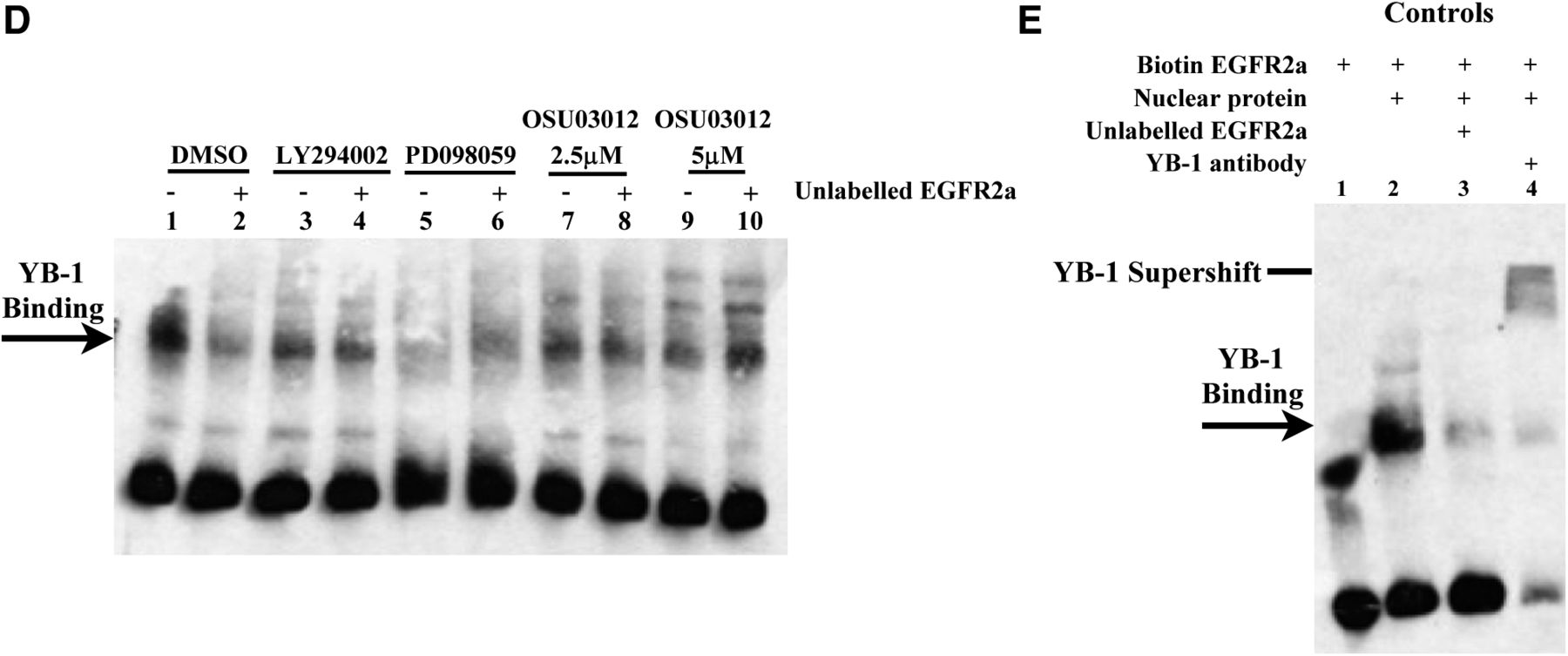
The same control blot showing the YB-1 supershift was inadvertently included in both publications. Herein, we have submitted the intended data now shown in Fig. 3E, which reiterates that the protein to which EGFR2a loci bind is YB-1 because its binding was inhibited with a YB-1 antibody causing a mobility supershift. This mirrors the data that were submitted inadvertently as a control subject. However, the conclusions are the same.
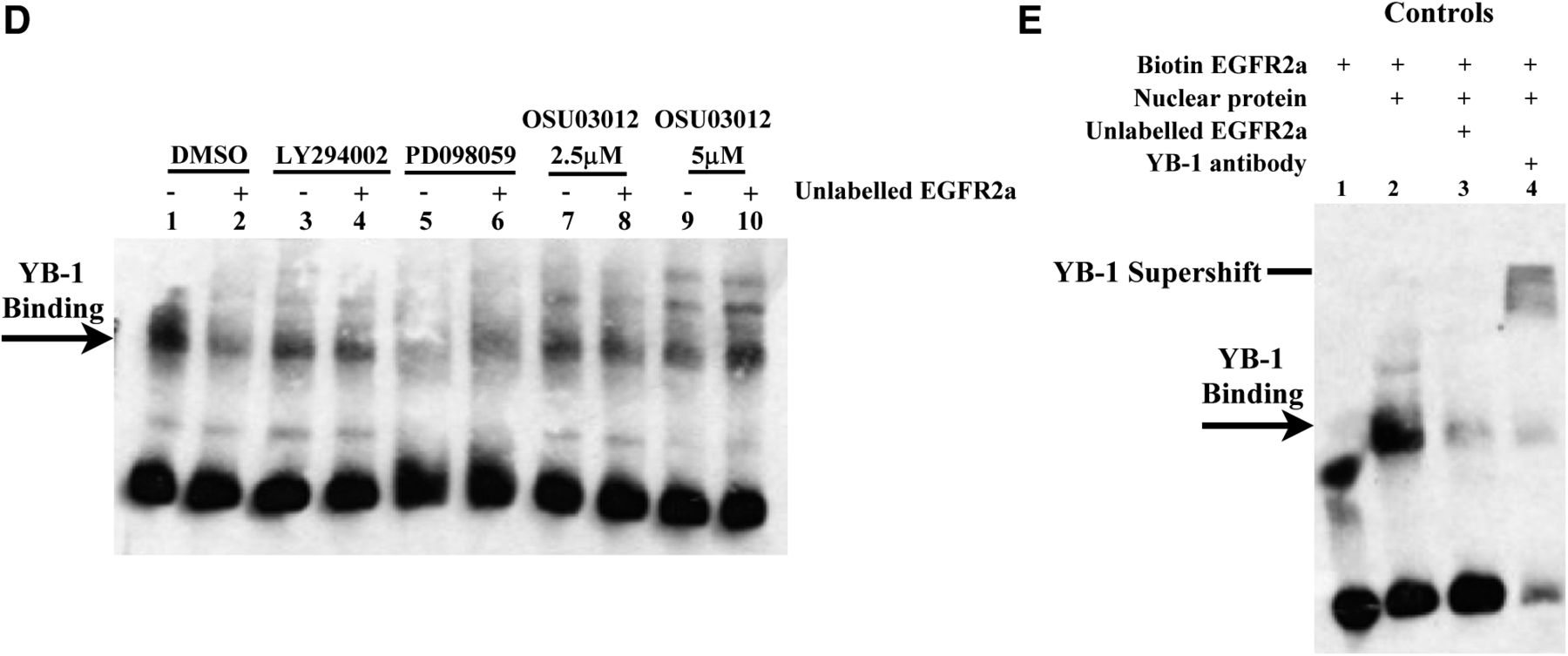
The new control subjects (Fig. 3E) do not change the interpretation of the original findings in any way; they simply validate the original control subject that was mistakenly provided. These control subjects support the main message of Fig. 3D, which was that OSU-O3012 inhibits YB-1 from binding to the EGFR promoter in nuclear extracts isolated from SUM 149 cells. These data collectively support the original ChIP data that also show that treating SUM 149 cells with OSU-O3012 suppresses YB-1 from binding to the EGFR2a loci (Fig. 3D).
Corrected Fig. 3D, bottom right: In addition to ChIP analysis, electrophoretic mobility shift assays were performed to confirm YB-1 binding to the EGFR promoter. Nuclear proteins were isolated from SUM 149 cells, and electrophoretic mobility shift assays were performed in which the biotin-labeled oligonucleotide was designed against the 2a binding site of the EGFR promoter. When SUM149 cells were treated with DMSO, YB-1 bound to the EGFR oligo EGFR (lane 1), which was inhibited with competitive unlabeled oligonucleotide (lane 2). Cells treated with LY294002 (10 μM) (lane 3), PD098059 (20 μM) (lane 4), or OSU-03012 (2.5 and 5 μM) (lanes 7 and 9) for 72 hours had decreased binding to the EGFR2a probe compared with the DMSO control (lane 1). The decreased binding by the signal transduction inhibitors was similar to that observed in the presence of an excess of unlabeled EGFR2a probe (lanes 2, 4, 6, 8, and 10). Thus, blocking cellular signaling through PI3K with LY294002 or MAPK with PD098059 suppressed YB-1’s ability to bind to the EGFR promoter. Because the PI3K and MAPK pathways impinge on PDK-1, these data further support the observation that the PDK-1 inhibitor OSU-O3012 blocks YB-1’s ability to bind to the EGFR promoter.
New Fig. 3E. Control subjects were included to show that YB-1 binding to the EGFR promoter can be supershifted with an YB-1 antibody. As a negative control, no binding was observed in the absence of nuclear extract (lane 1). When nuclear proteins were isolated from SUM 149 cells and incubated with biotin-labeled EGFR2a oligonucleotide, binding occurred (lane 2), which could be inhibited with either unlabeled probe (lane 3) or supershifted with an antibody to YB-1 (lane 4).
The authors regret this error and any inconvenience it may have caused.
- Copyright © 2014 The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}