


Abstract
There are now several examples of single G protein-coupled receptors to which binding of specific agonists causes differential effects on the associated signaling pathways. The dopamine D2 receptor is of special importance because the selective activation of functional pathways has been shown both in vitro and in situ. For this reason, the present work characterized a series of rigid D2 agonists in Chinese hamster ovary cells transfected with the human D2L receptor using three distinct functional endpoints: inhibition of cAMP synthesis, stimulation of mitogen-activated protein (MAP) kinase phosphorylation, and activation of G protein-coupled inwardly rectifying potassium channels (GIRKs). In this system, S-propylnorapomorphine (SNPA), R-propylnorapomorphine (RNPA), dihydrexidine (DHX), dinapsoline (DNS), and dinoxyline (DNX) all inhibited forskolin-stimulated adenylate cyclase activity to the same extent as the prototypical D2 agonist quinpirole (QP). The rank order of potency was the following: RNPA ≫ QP = DNX > SNPA > DHX = DNS. For MAP kinase phosphorylation, DHX, DNS, DNX, and RNPA had efficacy similar to QP, whereas SNPA was a partial agonist. The rank order of potency for MAP kinase phosphorylation was RNPA ≫ QP = DNX > DHX > DNS = SNPA. DNX activated GIRK channels to the same extent as QP, whereas DHX and DNS were partial agonists, and RNPA and SNPA caused no appreciable activation. These findings indicate that DHX, DNS, RNPA, and SNPA have atypical functional properties at the hD2L receptor and display different patterns of functional selectivity. We hypothesize that this functional selectivity may be a result of ligand induction of specific conformations of the D2L receptor that activate only selected signaling pathways.
The proximal event mediating cellular signaling through G protein-coupled receptors (GPCRs) is the binding of ligand, with subsequent conformational changes initiating secondary and distal events. In the past, compounds were believed to cause a single type of functional response for all effectors linked to a given receptor; hence, a compound could be labeled according to its intrinsic efficacy as full agonist, partial agonist, antagonist, or, more recently, inverse agonist. This traditional view of the receptor as operating in a digital fashion, either active or inactive, has been challenged, however, by evidence that some ligands working at a single GPCR cause markedly dissimilar degrees of activation for different effector pathways. This phenomenon that we term “functional selectivity” is vastly different from classic receptor theory. Although it is still not generally appreciated, it is probably universal. It has been demonstrated in many different receptor systems, and termed not only “functional selectivity” [dopamine D2L receptors (Lawler et al., 1999; Kilts et al., 2002; Shapiro et al., 2003; Mottola et al., 2002)] but also “agonist-directed trafficking of receptor signaling” [5-HT2A and 5-HT2c (Berg et al., 1998) and α2A-adrenergic receptors (Brink et al., 2000; Kukkonen et al., 2001)]; “functional dissociation” [μ-opioid receptors (Whistler et al., 1999)]; “biased agonism” [neuropeptide cytokine receptors (Jarpe et al., 1998)]; “biased inhibition” [A1 adenosine receptors (Kudlacek et al., 2002)]; “trafficking of receptor stimulus” [human calcitonin type 2 receptor (Watson et al., 2000)]; “differential engagement” (Manning, 2002), “discrete activation of transduction” [muscarinic cholinergic receptors (Gurwitz et al., 1994)], as just some examples. The ubiquitous nature of this mechanism has been suggested by several groups (Kenakin, 2002; Gurwitz and Haring, 2003; Hermans, 2003; Mailman and Gay, 2004).
Although dopamine D2 receptors originally were defined by their inhibitory actions on adenylate cyclase (Chern, 2000), they have subsequently been found also to regulate activation of GIRK channels, inhibit high-voltage-activated Ca2+ channels, activate the MAP kinase cascade, potentiate arachidonic acid release, and increase Na+/K+-ATPase activity (Missale et al., 1998). As GPCRs, D2 receptors mediate these downstream effectors via activation of Gαi/o and various Gβγ subunits, as well as other proteins such as G protein-coupled receptor kinases. Studies over the last decade have shown that some D2 ligands can cause functionally selective signaling both in vivo and in vitro (Lawler et al., 1999; Kilts et al., 2002; Mottola et al., 2002; Shapiro et al., 2003). In the striatum, the dopamine receptor agonist dihydrexidine (DHX) was able to inhibit adenylate cyclase activity with the same maximal intrinsic activity as quinpirole, a prototypical D2 receptor agonist. It is surprising that DHX was unable to inhibit dopamine release in the striatum or cell-firing in the substantia nigra (Mottola et al., 2002). DHX seemed to be acting as a full agonist for adenylate cyclase inhibition while acting as an antagonist at presynaptic receptor functions (i.e., dopamine release and inhibition of cell firing). Subsequent investigations in both MN9D cells and pituitary lactotrophs demonstrated that DHX was able to inhibit selectively cAMP synthesis but was a very weak partial agonist for inhibition of dopamine release or activation of GIRK channels (Kilts et al., 2002). In fact, DHX could antagonize the ability of quinpirole to inhibit dopamine release. Equally important, it was shown that S(+)-propylnorapomorphine (SNPA) elicits functional effects opposite to those of DHX. SNPA was a partial agonist for the inhibition of cAMP accumulation and a full agonist for inhibition of dopamine release, whereas R(-)-propylnorapomorphine (RNPA), like quinpirole, was able to maximally activate both functional measurements. These data strongly support the hypothesis of agonist trafficking of receptor signaling at the D2L receptor by atypical dopamine receptor agonists.
If truly universal, this phenomenon of functional selectivity/agonist trafficking has profound implications (e.g., that it is inappropriate to describe the functional interactions of many compounds with a given receptor as simply agonist, antagonist, etc.). Thus, the present study was designed to characterize the functional profile of a range of dopamine D2 receptor agonists to activate three independent functional effectors mediated by activation of cloned and heterologously expressed D2 receptors. The ability of several dopamine D2 receptor agonists to inhibit FSK-stimulated cAMP synthesis, to stimulate the phosphorylation of ERK1/2, and to activate GIRK channels was measured in CHO cells stably transfected with the hD2L receptor. Each of these functions is known to be modulated by the D2 receptor in CHO cells through activation of pertussis toxin-sensitive G proteins (Senogles, 1994; Kuzhikandathil et al., 1998; Choi et al., 1999). Inhibition of cAMP synthesis is believed to occur through a Gαi/o subunit, whereas both MAP kinase phosphorylation and GIRK channel activation are believed to be mediated by release of Gβγ subunits. In addition, these experiments provide the first thorough study of the D2 receptor functional profile of two novel dopamine receptor agonists, dinapsoline (DNS; Ghosh et al., 1996) and dinoxyline (DNX; Grubbs et al., 2004), comparing them with QP, DHX, RNPA, and SNPA.
Materials and Methods
Materials. Racemic dihydrexidine, dinapsoline, and dinoxyline were synthesized by published procedures (Brewster et al., 1995; Ghosh et al., 1996; Grubbs et al., 2004). For this series of compounds, the (+)-enantiomer is the one known to have affinity for dopamine receptors (Knoerzer et al., 1994; Sit et al., 2002). [3H]Spiperone was purchased from Amersham Biosciences Inc. (Piscataway, NJ). Quinpirole (LY171555) and domperidone were purchased from Sigma/RBI (Natick, MA). Isobutylmethylxanthine, dopamine, R(-)-propylnorapomorphine, S(+)-propylnorapomorphine, EDTA, dithiothreitol, sucrose, pepstatin A, leupeptin, PMSF, and other standard laboratory chemicals were purchased from Sigma Chemical Co. (St. Louis, MO). HEPES buffer was purchased from Research Organics (Cleveland, OH). cAMP primary antibody was obtained from Dr. Gary Brooker (George Washington University, Washington, DC), and secondary antibody (rabbit anti-goat IgG) covalently attached to magnetic beads was purchased from Advanced Magnetics (Cambridge, MA). Ham's F-12 and Opti-MEM media, penicillin, streptomycin, and geneticin (G418) were purchased from Invitrogen (Carlsbad, CA). LipofectAMINE was also purchased from Invitrogen. Primary antibody to phospho-p44/42 MAP kinase and secondary anti-rabbit HRP-conjugated antibody were purchased from Cell Signaling Technology Inc. (Beverly, MA). QP, DHX, DNS, and DNX were made up fresh for each experiment from 10 mM methanolic stocks stored at -20°C, whereas RNPA and SNPA were made up daily for each experiment from the solid salt.
Cell Maintenance. The CHO hD2L cells are a stable line that was obtained originally from Dr. Tony Sandrasagra (Aventis, Bridgewater, NJ). CHO hD2L cells were maintained in Ham's F-12 medium (Invitrogen) containing 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 500 μg/ml G418.
Membrane Harvesting. CHO hD2L cells were grown to confluence in 75-cm2 flasks. Cold phosphate-buffered saline was used to rinse the flasks and then was aspirated off. Flasks were incubated in 5 ml of lysis buffer (2 mM HEPES, 2 mM EDTA, 1 mM dithiothreitol, 1 μ g/ml pepstatin A, 0.5 μg/ml leupeptin, and 10 μg/ml PMSF, pH 7.4, with HCl) for 10 to 20 min. Cells were scraped and collected. Flasks were rinsed with another 5 ml of lysis buffer, and the buffer was added to the cell suspension. The cell suspension was homogenized with five strokes in a Wheaton glass homogenizer. The cell suspension was centrifuged at 30,000g for 20 min, and the supernatant was discarded. The pellet was resuspended in storage buffer (50 mM HEPES, 0.32 M sucrose, 1 μg/ml pepstatin A, 0.5 μg/ml leupeptin, and 10 μg/ml PMSF, pH 7.4, with NaOH) at approximately 1 mg protein/ml, homogenized as above, and aliquoted into 1-ml microcentrifuge tubes. Cell membranes were frozen and stored at -80°C until further use.
Saturation and Competition Binding. Saturation binding studies were done to evaluate the level of expression of hD2L receptors in the stable CHO cell line. Membranes were incubated with eight different concentrations of [3H]spiperone in binding buffer (50 mM HEPES, 4 mM MgCl2, and 0.1% ascorbic acid, pH 7.4, with KOH). Nonspecific binding was determined using 10 μM domperidone. Competition binding studies using 0.1 nM [3H]spiperone were carried out to determine the affinity of each test compound for the hD2L receptor in the CHO cell line. Total binding was defined as the amount of radioligand bound in the absence of any competing drug, and nonspecific binding was defined as binding in the presence of 10 μM domperidone. Eight to 10 concentrations of each test compound were used. For both saturation and competition assays, binding was initiated by the addition of tissue to the assay tube. Each condition was run in triplicate in a final volume of 500 μl. After incubation at 37°C for 15 min, tubes were filtered through a FilterMate 196 Cell Harvester (PerkinElmer Life and Analytical Sciences, Boston, MA). Plates were washed four times with ice-cold buffer. Filters were allowed to dry, and 25 μl of Packard MicroScint 20 scintillation cocktail was added to each well (PerkinElmer). Radioactivity in each sample was determined on a Packard TopCount NXT (PerkinElmer). Saturation binding data were expressed as femtomoles of receptor per milligram of protein. Competition binding data were expressed as the percentage of specific binding.
Adenylate Cyclase Assay. For measurement of dopamine receptor agonist inhibition of FSK-stimulated cAMP accumulation, cells were seeded at a density of 5 × 104 cells/well in a 24-well plate and grown for 48 h in appropriate media. Cells were preincubated for 5 min in assay media (serum-free media containing 25 mM HEPES, 500 μM isobutylmethylxanthine, and 0.1% ascorbic acid) at 37°C. Assay medium was aspirated off, and new assay media containing FSK and/or drug were added. Various concentrations of test compounds were added, and 10 μM domperidone was used to block agonist inhibition of FSK-stimulated cAMP synthesis. Plates were incubated for 15 min at 37°C. Cells were rinsed in assay media, and then the reaction was halted with 1 ml of cold 0.1 N HCl. cAMP accumulation was measured using a radioimmunoassay modified from that described previously (Harper and Brooker, 1975). Data were expressed as the percentage of maximal quinpirole inhibition of FSK-stimulated cAMP accumulation.
MAP Kinase Assay. Phosphorylated p44/p42 MAP kinase detection was carried out via Western blot, after minor modifications of a protocol published previously (Choi et al., 1999). Cells were grown in six-well plates with media containing 0.5% fetal bovine serum. Cells were exposed to various concentrations of test ligand for 5 min and then collected in SDS-polyacrylamide gel electrophoresis sample buffer. Phosphorylated p44/p42 MAP kinase was detected using a polyclonal primary antibody to phospho-p44/42 MAP kinase, followed by an HRP-conjugated secondary anti-rabbit antibody. To quantify ligand-induced stimulation of MAP kinase phosphorylation more accurately, detection of agonist-mediated MAP kinase phosphorylation was done using an enzyme-linked immunosorbent assay using minor modifications of a protocol published previously (Versteeg et al., 2000). Cells were plated at 3 × 104 cells/well in 96-well plates and were grown for 48 h. Cells were serum-starved for 4 h before drug treatment. Cells were exposed to various concentrations of agonist for 10 min and then fixed using 4% formaldehyde. Domperidone (10 μM) was used to antagonize D2 receptor-mediated phosphorylation. Detection of phosphorylated p44/42 MAP kinase was carried out using the antibodies described above. Quantification of HRP-induced o-phenylenediamine color change was measured spectrophotometrically (A490-A650) using a kinetic microplate reader (Molecular Devices, Sunnyvale, CA). Data are expressed as the percentage of the maximal stimulation of phosphorylated p44/42 MAP kinase by quinpirole after subtraction of basal stimulation.
Transfection of GIRK Channels into CHO hD2L Cells. CHO hD2L cells were plated on 15-mm coverslips in 12-well dishes and were grown for 48 h. A pcDNA3.1 plasmid encoding hKir3.2 was transiently transfected into CHO hD2L cells using LipofectAMINE by following the protocol recommended by the manufacturer. Cells were simultaneously transfected with a separate plasmid expressing green fluorescent protein, at a ratio of 3:1 (hKir3.2/green fluorescent protein). Experiments were performed 48 to 72 h after transfection.
GIRK Channel Electrophysiology. Measurement of agonist activation of GIRK channels was performed as described previously (Kuzhikandathil and Oxford, 2000). In brief, agonist-activated currents were measured in CHO hD2L cells transfected with hKir3.2 by the whole-cell patch-clamp technique using an Axopatch 200 amplifier (Axon Instruments). Currents were elicited by a hyperpolarizing step (-100 mV) from a holding potential of -50 mV, followed by a ramp voltage command (-120 to +40 mV). The cells were bathed in a standard external solution (145 mM NaCl, 5 mM KCl, 2 mM CaCl, 1 mM MgCl, 10 mM glucose, and 10 mM HEPES buffer adjusted to pH 7.4 and 295-300 mOsm). A quartz microtube was positioned to apply test compounds directly to the cell being recorded. The cells were first exposed to the standard external solution containing 30 mM KCl and then were randomly exposed to various concentrations of ligands. Every cell was exposed to 100 nM QP and one other test ligand to compare results across cells and transfection batches. The current response was normalized to the cell capacitance (pA/pF) and then expressed as the percentage of maximal quinpirole activation. It has been demonstrated previously that in untransfected CHO hD2L cells, no inwardly rectifying currents are observed (Kuzhikandathil et al., 1998).
Data and Statistical Analysis. Binding data were analyzed by nonlinear regression using a sigmoidal equation with variable slope (Prism 3.0; GraphPad Software Inc., San Diego, CA) to obtain estimates of IC50 values and Hill slopes. The IC50 values were then converted to K0.5 using the Cheng-Prusoff relationship for a bimolecular model. The functional dose-response curves were analyzed by nonlinear regression using a sigmoidal equation with variable slope (Prism 3.0) to obtain estimates for apparent potency (EC50, in nanomolars) and maximal intrinsic activity. Differences between more than two groups were subject to analysis of variance (ANOVA). Significant ANOVA results were followed by Dunnett's post hoc analysis. The analysis was performed using InStat 3.05 software (GraphPad).
Results
D2 Receptor Agonists Bind to hD2L Receptor Stably Expressed in CHO Cells. The dopamine hD2L receptor was expressed stably in a CHO cell line at approximately 4.6 pmol receptor/mg membrane protein (data not shown). Before characterizing the functional profile of the test compounds, their binding characteristics to the hD2L receptor in CHO cells were assessed. Each of the drugs competed for hD2L sites labeled with [3H]spiperone. The D2 ligands had a rank order of affinity of RNPA ≫ DNX > DNS = SNPA > DHX > QP. RNPA had a K0.5 around 1 nM, whereas DNX, DNS, and SNPA had K0.5 values in the tens of nanomolars range, similar to the endogenous ligand dopamine (data not shown), and DHX and QP had K0.5 values in the 100 to 200 nM range (Table 1).
Binding affinities and functional potencies of ligands for hD2L receptors in CHO cells
Binding data represent the mean ± S.E.M. from two independent experiments preformed in triplicate. Functional data represent the means ± S.E.M. from two to four independent experiments. See Materials and Methods for details.
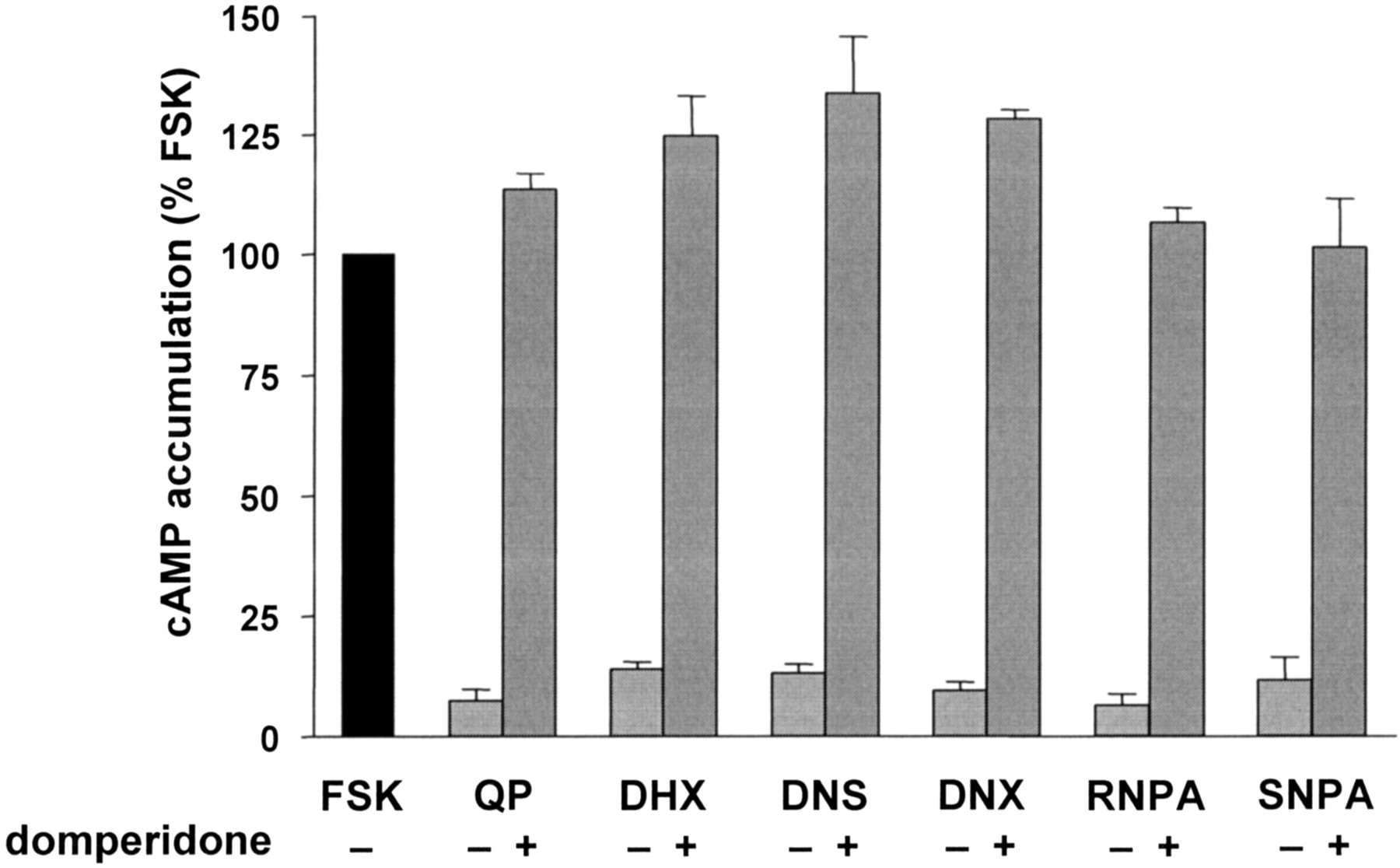
D2 Receptor Agonists Inhibit FSK-Stimulated cAMP Synthesis in CHO hD2L Cells. Activation of adenylate cyclase was stimulated using 10 μM FSK, and then the ability of each agonist to inhibit FSK-stimulated cAMP accumulation was measured (Fig. 1). The mean activities of cAMP accumulation were the following: basal, 30 ± 8 fmol cAMP/well/min; FSK-stimulated, 1100 ± 200 fmol cAMP/well/min; and QP inhibition of FSK-stimulated, 20 ± 3 fmol cAMP/well/min. The rank order of potency for the compounds was RNPA ≫ QP = DNX > SNPA > DHX = DNS. The addition of domperidone completely blocked the ability of each ligand to inhibit FSK-stimulated cAMP synthesis (Fig. 2), indicating that the adenylate cyclase inhibition was D2L receptor-mediated. In some cases, the presence of domperidone increased cAMP accumulation to a level greater than that elicited by FSK alone, suggesting that there may be some constitutive hD2L activity in this particular overexpression system.

Inhibition of FSK-stimulated cAMP synthesis in whole cells. Cells were incubated with FSK (10 μM) and/or drug for 15 min at 37°C. All D2 receptor agonists maximally inhibited FSK-stimulated cAMP accumulation with a rank order of potency RNPA ≫ QP = DNX > SNPA > DHX = DNS. Data are expressed as a percentage of FSK-stimulated cAMP accumulation relative to the maximal inhibition of QP. Each value represents the mean ± S.E.M. of two to four independent experiments conducted in triplicate.

Antagonist blockade of D2 receptor agonist inhibition of FSK-stimulated cAMP synthesis. Domperidone (10 μM) was able to block inhibitory effects of each test compound (1 μM; 100 nM RNPA) on FSK-stimulated cAMP accumulation. Data are expressed as a percentage of FSK-stimulated cAMP accumulation. Each value represents the mean ± S.E.M. of two to four independent experiments conducted in triplicate.
D2 Receptor Agonists Stimulate MAP Kinase Phosphorylation in CHO hD2L Cells. Each of the dopamine receptor ligands had full intrinsic activity for phosphorylation of ERK1/2, with the exception of SNPA, which had an intrinsic activity (relative to quinpirole) of only ∼75% (Fig. 4). QP stimulation over basal had a mean fold change of 1.94 ± 0.24. The agonists had a rank order of potency of RNPA ≫ QP = DNX > DHX = DNS > SNPA (Fig. 3). As with the adenylate cyclase assays, the addition of domperidone completely blocked the ability of each compound to stimulate phosphorylation of ERK1/2 (Fig. 4), again showing hD2L receptor mediation. Table 1 shows the EC50 values (in nanomolars) for both inhibition of adenylate cyclase and phosphorylation of MAP kinase. QP, DNS, and DNX have similar potencies for the two functional endpoints. It is interesting that DHX has a higher potency for phosphorylation of ERK1/2 than for inhibition of cAMP accumulation, whereas both enantiomers of NPA have a significantly higher potency for inhibition of cAMP synthesis than for activation of MAP kinase. Table 1 also shows the relative affinity/potency ratios for both adenylate cyclase and MAP kinase activities. In addition, a Furchgott analysis (Kenakin, 1997) was performed on the functional data for adenylate cyclase and MAP kinase. As can be seen clearly in Fig. 7, there are significant differences in the rank order of potency in one function versus another, consistent with the prediction from the functional selectivity hypothesis.

Antagonist blockade of D2 receptor agonist-stimulated phosphorylation of MAP kinase. Domperidone (10 μM) was able to block the stimulatory action of the test compounds (1 μM; 100 nM RNPA) for phosphorylation of p44/p42 MAP kinase. Data are expressed as a percentage of the maximal stimulation of quinpirole over basal. All ligands had similar intrinsic activity compared with QP, except for SNPA (ANOVA, Dunnett post hoc, ★, p < 0.05). Each value represents the mean ± S.E.M. of three to four independent experiments conducted in quadruplicate.
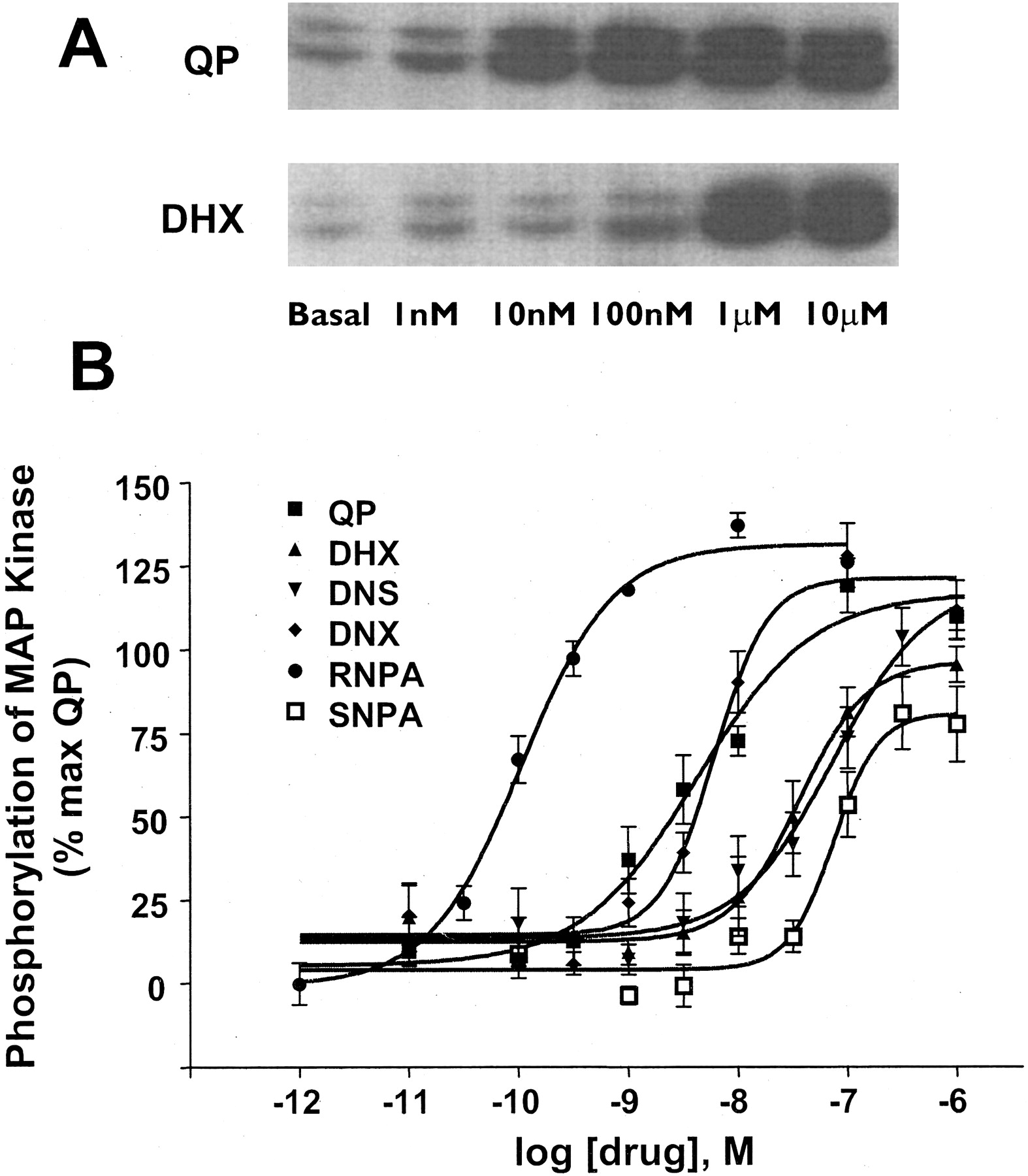
Phosphorylation of MAP kinase. The ability of test ligands to stimulate phosphorylation of p44/p42 MAP kinase was measured in CHO hD2L cells. A, representative Western blot indicating the ability of QP and DHX to stimulate phosphorylation of p44/p42 MAP kinase over basal in a dose-dependent fashion. B, quantification of p44/p42 MAP-kinase phosphorylation using an enzyme-linked immunosorbent assay. Cells were incubated with varying concentrations of test compounds for 10 min at room temperature (20°C). DHX, DNS, DNX, and RNPA stimulated phosphorylation of MAP kinase with intrinsic activity similar to that of QP, whereas SNPA was a partial agonist (Fig. 4). The rank order of potency was RNPA ≫ QP = DNX > DHX > DNS = SNPA. Data are expressed as a percentage of the maximal stimulation of quinpirole over basal phosphorylation. Each value represents the mean ± S.E.M. of three to four independent experiments conducted in quadruplicate.
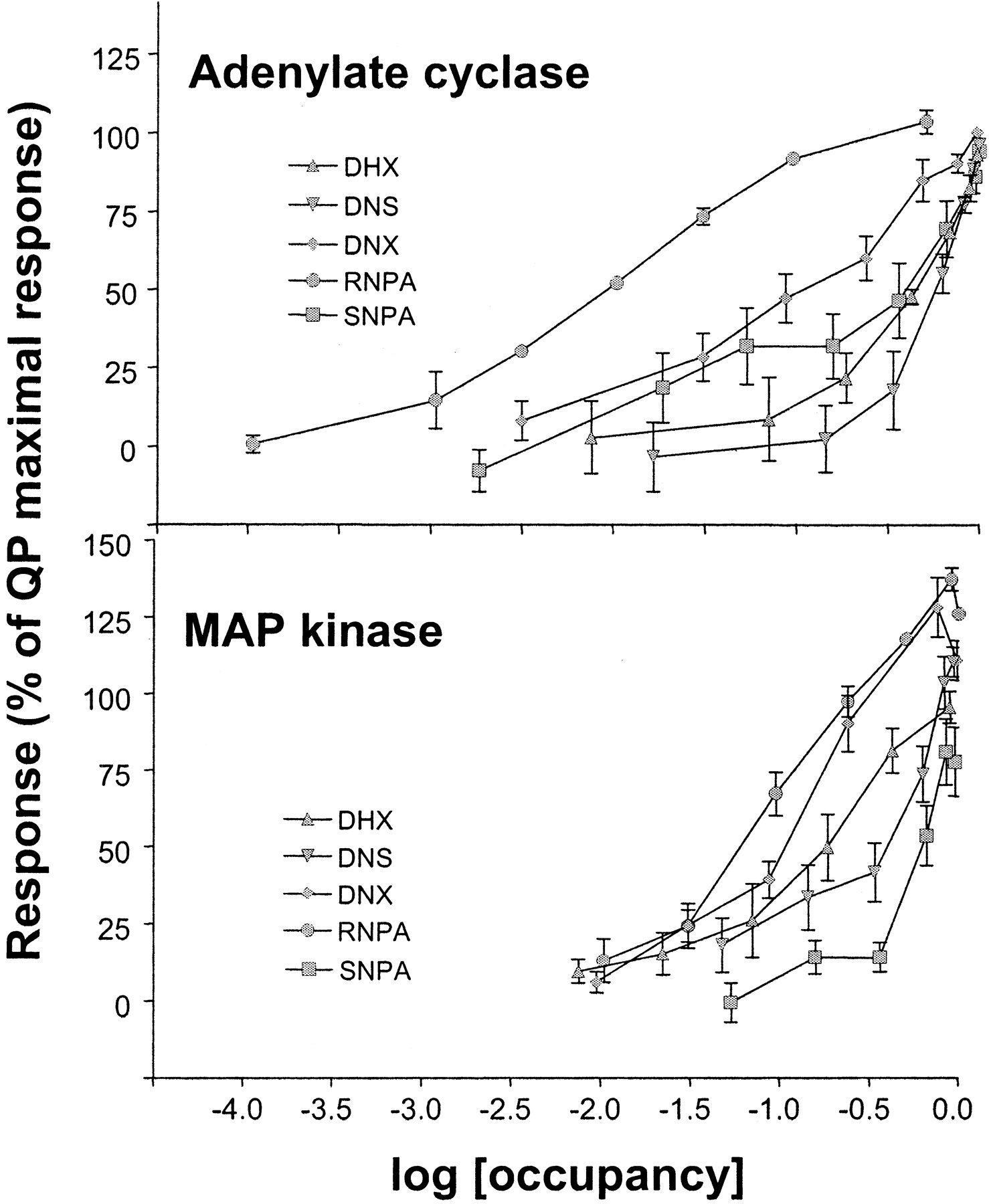
Furchgott analysis of adenylate cyclase and MAP kinase functional data. The relative intrinsic activity of each ligand to inhibit cAMP synthesis or activate MAP kinase phosphorylation was compared by plotting an occupancy-response curve. The lateral displacement between the curves represents the relative intrinsic activities of the ligands for either adenylate cyclase inhibition or MAP kinase phosphorylation. Notice that the relative intrinsic activities of the ligands vary between the two functional endpoints (e.g., RNPA versus DHX). In addition, the rank order of adjusted potency is dependent on the functional assays (e.g., SNPA versus DNS).
Selected D2 Receptor Agonists Activate GIRK Channels in CHO hD2L Cells. Dopamine hD2L receptors also couple to the activation of GIRK channels that have been transiently transfected into CHO cells. The ability of each test ligand to activate GIRK channels compared with quinpirole was measured in voltage-clamp experiments (Fig. 5). Although full dose-response curves were not generated in this experiment, multiple concentrations of each agonist were tested, including concentrations causing ≥95% receptor occupancy. Figure 5, B and D, demonstrate that the GIRK channels do not desensitize over multiple applications of ligand, because QP produces the same maximal activation after a second application of ligand. DNX was the only agonist that had intrinsic activity for activation of K+ current similar to quinpirole (Fig. 6). Both DHX and DNS were partial agonists, activating approximately 50% of the current elicited by quinpirole. We were surprised to find that the binding of neither RNPA nor SNPA caused hD2L coupling to activation of GIRK channels in this cell line.
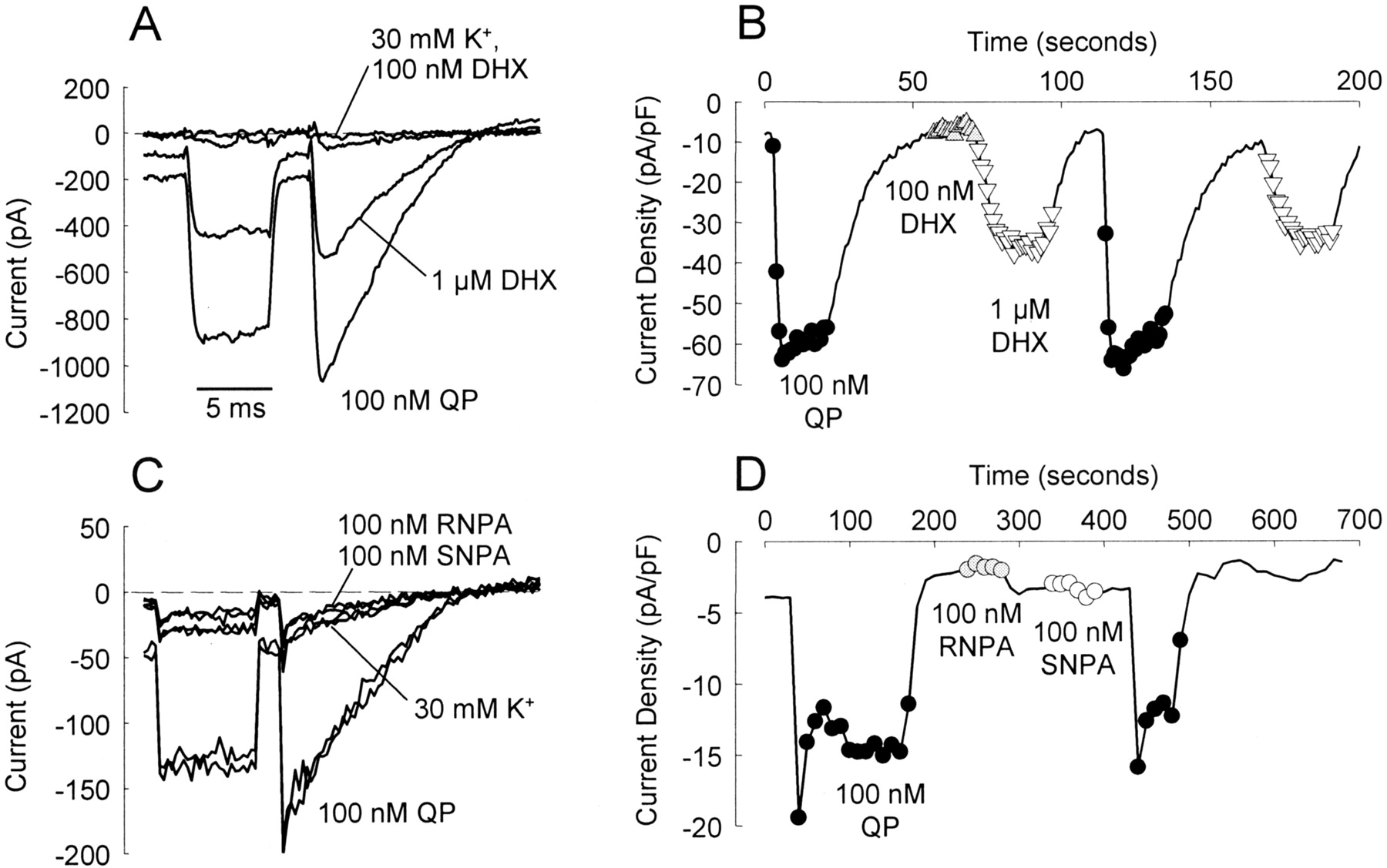
Comparison of ability of various D2 ligands to activate GIRK currents. A and C, representative K+ current records induced during a first hyperpolarizing step (-100 mV) followed by a voltage ramp from -120 to +40 mV from two CHO hD2L cells transiently transfected with hKir3.2. In each case, currents were elicited in normal external K+ (5 mM), elevated external K+ (30 mM), and elevated K+ containing different concentrations of each test compound as indicated. B, time course of current change at -100 mV for the cell in A. Solid line represents data in elevated K+ alone, whereas symbols are recordings in 100 nM quinpirole (•), 100 nM DHX (gray triangles), or 1 μM DHX (▿). D, time course of current change at -100 mV for cells in C. Solid line represents elevated K+ condition, whereas symbols represent application of 100 nM or quinpirole (•), RNPA (gray circles), or SNPA (○). Data for Fig. 6 were collected by following similar protocols.

Activation of GIRK currents. The ability of test ligands to activate GIRK channel currents was measured in CHO hD2L cells transiently transfected with hKir3.2. The ability of DHX and DNS to activate GIRK channels is substantially decreased compared with quinpirole. RNPA and SNPA produced no appreciable activation of GIRK currents, whereas DNX activates with maximal intrinsic activity similar to that of QP. Currents were elicited by a hyperpolarizing step followed by a ramp as shown in Fig. 5. The bar graph represents the means ± S.E.M. of agonist-activated currents measured during the hyperpolarizing step in a whole-cell, patch-clamp configuration. Multiple recordings from a single cell were averaged for a solitary value; n, the number of cells from which recordings were taken, including recordings made on the same day and across multiple days. Data were expressed as a percentage of QP activation in the same cell and subsequently averaged for all cells exposed to a particular ligand.
Discussion
Functional selectivity/agonist trafficking of receptor signaling is characterized by the ability of ligands to activate differentially particular functions linked to a single receptor. Certain atypical dopamine agonists studied here have been found previously to activate selectively various D2-mediated functions in brain, pituitary lactotrophs, and the MN9D D2L cell line (Kilts et al., 2002; Mottola et al., 2002). The current study examined selective signaling at the D2L receptor, with a wider range of agonists and functional endpoints in a second clonal cell line in which transduction of D2L-mediated signals may involve different effector mechanisms. In addition, this study is the first to characterize two novel rigid dopamine receptor agonists (DNS and DNX) at the hD2L receptor, as well as measure the responses elicited by several of the agonists at the downstream effectors MAP kinase and GIRK channels.
First, the affinity of the test ligands was determined in CHO cells stably expressing the hD2L receptor. RNPA had the highest hD2L affinity (K0.5 = 1 nM); DNX, DNS, and SNPA were intermediate (K0.5 = 30-50 nM); whereas QP and DHX had the lowest affinity (K0.5 = 100-200 nM). Next, these D2 receptor agonists were characterized at three distinct D2L-mediated functional endpoints. Each of the agonists examined had full intrinsic activity for inhibition of FSK-stimulated adenylate cyclase activity. This is the first demonstration that DNS has maximal intrinsic activity for inhibition of FSK-stimulated cAMP synthesis through the dopamine hD2L receptor. The ability of SNPA to act as a full agonist for the inhibition of cAMP accumulation in the CHO hD2L cell line is in contrast to its partial inhibition of adenylate cyclase in MN9D D2L cells (Kilts et al., 2002). The fact that SNPA seems to be a full, rather than a partial, agonist for inhibition of cAMP synthesis is most probably caused by the high degree of overexpression of the hD2L receptor in these CHO cells. Overexpression of receptors can result in markedly increased receptor reserve, and it is well known that partial agonists may seem to have full intrinsic activity in this condition (Adham et al., 1993; Watts et al., 1995; Pohjanoksa et al., 1997). When high receptor reserve is present, it is useful to also consider the relative potency of a drug for a particular functional endpoint and not simply for its maximal intrinsic activity.
Over the past 7 years, the ability of several GPCRs to regulate MAP kinase pathways has been investigated (Sugden and Clerk, 1997; Lopez-Ilasaca, 1998). Recently the first demonstration that dopamine D2 receptors can activate the MAP kinase pathway was shown in both C6 and CHO cells transfected with either the dopamine D2S or D2L receptor (Luo et al., 1998; Choi et al., 1999). The pertussis toxin sensitivity of the D2-mediated stimulation of ERK1/2 phosphorylation indicates that this pathway is, at least initially, mediated through the activation of G proteins (Choi et al., 1999). Each of the dopamine receptor agonists tested was able to stimulate the phosphorylation of ERK1/2. All of the test ligands had maximal intrinsic activity compared with quinpirole with the exception of SNPA, which was a partial agonist.
Experimental observations of the type we describe (i.e., in which a drug activates one effector more readily than another through a single GPCR) do not prove that the ligand in question is functionally selective. If a GPCR couples more efficiently to one signaling pathway over another, then it might be expected that a drug could have higher intrinsic activity and/or potency for that particular effector. From this perspective, at a single GPCR, all compounds should cause the same relative effects across different functional endpoints, even if the quantization of the effects varies. In the current data, this is clearly not the case because the relative potencies for inhibition of adenylate cyclase and phosphorylation of MAP kinase were significantly different (Table 1 and Figs. 7 and 8). When we compare the relative potencies for the test ligands to inhibit adenylate cyclase and phosphorylate MAP kinase, we observe that DNS, DNX, and QP couple similarly to the two effectors, whereas DHX has a higher potency for activation of MAP kinase than for inhibition of adenylate cyclase. On the other hand, RNPA and SNPA demonstrate the opposite coupling preference, showing a higher potency for inhibition of adenylate cyclase than for activation of MAP kinase (Fig. 8). These data indicate that the hD2L receptor does not by itself preferentially couple to inhibition of adenylate cyclase or stimulation of MAP kinase; rather, the specific dopamine D2 receptor agonist drives the receptor to couple more efficiently to one pathway or the other. The abilities of the test compounds to inhibit cAMP synthesis or to activate MAP kinase are qualitatively different, indicating that these drugs are functionally selective at the hD2L receptor.

Comparison of the ability of D2 agonists to activate specific functional effectors. A, relative intrinsic activity of D2 agonists compared with the prototypical D2 agonist quinpirole for inhibition of cAMP synthesis, phosphorylation of MAP kinase, and activation of GIRK channels. B, relative potency of D2 agonists for inhibition of cAMP synthesis and phosphorylation of MAP kinase. Ligands with similar potency for both cAMP and MAP kinase have a log(fold change) of nearly zero (QP, DNS and DNX), whereas ligands with decreased potency for MAP kinase compared with cAMP inhibition (RNPA and SNPA) have a negative change, and ligands with an increased potency for MAP kinase compared with cAMP inhibition (DHX) display a positive change. Data for each ligand are expressed as the log of the EC50 (in nanomolars) for each function divided by their respective EC50 value for cAMP inhibition.
Dopamine hD2L receptors also couple to the activation of hKir3.2 channels transiently transfected into CHO cells, presumably through Gβγ. The ability of each of the test ligands to activate GIRK currents varied markedly. DNX was the only agonist to activate this pathway with the same maximal intrinsic activity as quinpirole. DHX and DNS were partial agonists, whereas RNPA and SNPA elicited no response. The discovery that DNX is maximally active whereas DHX and DNS are partial agonists is especially intriguing considering the very similar chemical structure of the compounds. DNX and DNS differ in structure by a single replacement of an ether (DNX) for a methylene bridge (DNS) at a site in the molecule not believed to be critical for D2 receptor binding (Ghosh et al., 1996; Grubbs et al., 2004). The fact that these two compounds are structurally so similar and yet have distinctly different abilities to activate GIRK currents may make them useful tools in elucidating the mechanisms behind ligand-induced receptor active states.
Another interesting point is the observation that RNPA and SNPA did not activate GIRK channels, which was at first glance surprising considering published in vivo data showing that both compounds inhibit substantia nigra cell firing (Cox et al., 1988) and inhibit dopamine release in MN9D cells transfected with the D2L receptor (Kilts et al., 2002). These findings underscore the importance of accounting for both the functional endpoint being measured and the experimental system in which ligands are defined. These results are not without precedence, however. For example, in the CCL1.3 cell line, the D2L receptor was found to couple to Gαi2 and Gαi3 but not to Gαi1 or GαoA to inhibit cAMP synthesis, whereas in the MN9D cell line, D2L receptors couple to Gαi2 but not to GαoA (O'Hara et al., 1996). In NS20Y cells, D2L receptors couple to inhibition of cAMP synthesis through Gαo but not Gαi1, Gαi2, or Gαi3 (Watts et al., 1998). Different ligands were used as the reference point in these studies (dopamine in CCL1.3 and MN9D lines versus QP in the NS20Y line), suggesting that these agonists may create different receptor active states and recruit different G protein α subunits. On the other hand, the complement of signaling proteins available in each cell line is likely to be different (i.e., adenylate cyclase isoforms, regulators of G protein signaling, or activators of G protein signaling, etc.) and may consequently influence coupling.
Regardless of the molecular mechanisms that are responsible for such effects, it seems clear that traditional classification of ligands as “full agonist”, “partial agonist”, “antagonists”, or “inverse agonist” can no longer explain the types of data reported and discussed here. Our findings, and those with other GPCRs, suggest that it will be the rule rather than the exception that GPCR ligands can create active states of a receptor that favor differential activation of particular functional responses (e.g., cAMP inhibition or GIRK channel activation in the present study). A corollary is that the complement of interacting proteins available to a receptor becomes critical both physiologically and in comparing experimental systems (vide supra). Therefore, the apparent contradiction of how the NPA isomers failed to activate GIRK channels in the CHO cell line yet robustly inhibit substantia nigra dopamine neuron firing in situ may reflect the functionally selective characteristics of these drugs, as well as the differences in interacting proteins in CHO cells versus nigral neurons. In particular, both NPA isomers may induce a receptor conformation that efficiently couples to only particular Gαβγ combinations (vide infra) or promotes interactions with particular regulator of G protein signaling proteins. If these particular proteins are not present in CHO cells, NPA isomers will be unable to activate GIRK channels. On the contrary, if the correct interacting proteins are present in nigra dopamine neurons, then NPA will be able to inhibit cell firing through GIRK channel activation. In a recent review, Kenakin (2002) makes just this point, that each ligand/receptor/G protein complex must be thought of as a distinct protein species capable of a unique conformational identity and pattern of signaling.
Also intriguing is the commonly held idea that Gβγ dimers indiscriminately activate GIRK channels (Wickman et al., 1994). Thus, any ligand that activated inhibition of cAMP synthesis through activation of a Gαi/o protein (as each of the test compounds did) might also be expected to activate GIRK channels through release of Gβγ. Studies investigating the ability of different Gβγ dimers to inhibit Ca2+ channels in human embryonic kidney 293 cells indicate that although each of the β subunits could couple to channel inhibition, coupling was dependent on the type of γ subunit expressed (Zhou et al., 2000). In contrast, Leaney et al. (2000) showed that the α subunit determines the specificity of coupling between several Gαi/o-coupled receptors and K+ channels in human embryonic kidney 293 cells. These studies suggest that the specificity of GPCR coupling to GIRK and Ca2+ channels may be dependent on either Gα or Gβγ expression and may be regulated differently, depending on the experimental system. Other factors, such as compartmentalization or G protein expression levels, may also play a role in determining the ability of certain agonists to activate GIRK channels.
Finally, the observation that some of the test ligands were able to activate GIRK channels whereas others either were partial agonists or were unable to elicit a response suggests that these drugs are functionally selective for GIRK channel activation. The possibility that dopamine D2L receptors couple less efficiently to GIRK channel activation might suggest that the phenomenon of strength of signaling may underlie the current results (Kenakin, 1995). However, considering the opposite functional profile of the agonists for inhibition of adenylate cyclase and MAP kinase phosphorylation, it seems more probable that these drugs are activating GIRK currents through distinct ligand-specific receptor states. The overexpression of hD2L receptors may be masking partial agonism for inhibition of cAMP or phosphorylation of MAP kinase, thereby complicating the comparison of the maximal intrinsic activities of each drug to inhibit adenylate cyclase or stimulate MAP kinase phosphorylation versus activate GIRK channels.
The data clearly demonstrate that DHX, DNS, RNPA, and SNPA exhibit differential activation for particular effectors linked to the dopamine hD2L receptor. The ability of these agonists to induce/stabilize specific receptor active states that couple the receptor more or less efficiently to each function may be a key molecular mechanism underlying this functional selectivity. Indeed, there is direct evidence for ligand-specific receptor states with the β2-adrenergic receptor (Ghanouni et al., 2001). Indirect evidence for the existence of multiple D2 receptor active states comes from a study by Wiens et al. (1998) in which point mutations were made in several serine residues believed to be important in ligand binding. Their results indicated that the ability of different D2 receptor agonists (including DHX) to bind the mutant receptors, inhibit cAMP accumulation, activate GIRK channels, and stimulate guanosine 5′-O-(3-[35S]thio)triphosphate binding was affected differentially by the serine mutations. It is further hypothesized that distinct ligand-active states may couple preferentially to different G proteins, thereby translating specific receptor-active conformations into selective activation of individual functional responses. It was demonstrated recently that several dopamine receptor agonists have varying abilities to stimulate specific Gαi/o proteins in Sf9 cells expressing hD2L receptors (Gazi et al., 2003).
The current data provide clear evidence for functional selectivity/agonist trafficking of hD2L receptor signaling in CHO cells in which ligands have discrete and distinct functional profiles. These data also have relevance to the general question of whether such findings of agonist trafficking of receptor signaling have physiological relevance. Previous studies including not only in vitro experiments but also in situ and in vivo studies indicate that D2 functional selectivity is of clear physiological importance (Smith et al., 1997; Kilts et al., 2002; Mottola et al., 2002). Indeed, unlike quinpirole, neither dihydrexidine nor dinapsoline has been shown to cause emesis in susceptible animal species (unpublished data), which is also consistent with their functionally selective D2 properties. It also has been demonstrated recently that functional selectivity is a critical mechanism in the actions of the novel antipsychotic drug aripiprazole (Lawler et al., 1999; Shapiro et al., 2003). Thus, the characterization of compounds that can cause differential activation of functional pathways through a single GPCR is of immense potential usefulness both for understanding neuropharmacological mechanisms and for discovering compounds with improved clinical use. We also believe that if this is as universal as we believe (see Introduction), it is timely for pharmacology as a discipline to settle on a single terminology and introduce the concept into basic teaching
Acknowledgments
We thank Dr. David Siderovski and Dr. Terry Kenakin for their helpful discussions of the data.
Footnotes
-
This work was supported by National Institutes of Health research grants MH53356 (to R.B.M.), MH40537 (to R.B.M.), NS18788 (to G.S.O.), MH42705 (to D.E.N.), and training grant NS07431.
-
R.B.M. and D.E.N. have a significant financial interest in DarPharma, Inc., the company currently holding license rights to dihydrexidine, dinapsoline, and dinoxyline. All opinions are those of the authors and do not represent opinions of either the universities or DarPharma, Inc.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; DHX, dihydrexidine, trans-10,11-dihydroxy-5,6,6a,7,8,12b-hexahydrobenzo[a]phenanthridine; DNS, dinapsoline, 8,9-dihydroxy-2,3,7,11b-tetrahydro-1H-naph[1,2,3-de]isoquinoline; DNX, dinoxyline, 8,9-dihydro-1,2,3,11b-tetrahydrochromeno[4,3,2,-de]isoquinoline; ERK, extracellular signal-regulated kinase; GIRK, G protein-coupled inwardly rectifying potassium; K0.5, concentration-corrected IC50 when nH ≠ 1.0.; MAP, mitogen-activated protein; QP, (-)-quinpirole; LY171555, trans-(-)-4,4a,5,6,7,8,8a, 9-octahydro-5-propyl-1H-pyrazolo[3,4-g]quinoline; RNPA, R(-)-propylnorapomorphine, R(-)-10,11-dihydroxy-N-n-propylnoraporphine; SNPA, S(+)-propylnorapomorphine, S(+)-10,11-dihydroxy-N-n-propylnoraporphine; FSK, forskolin; CHO, Chinese hamster ovary; PMSF, phenylmethylsulfonyl fluoride; HRP, horseradish peroxidase; ANOVA, analysis of variance; NPA, propylnorapomorphine.
- Received October 10, 2003.
- Accepted March 24, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}