


Abstract
β-arrestins are multifaceted adaptor proteins that mediate G protein-coupled receptor (GPCR) desensitization, internalization, and signaling. It is emerging that receptor-specific determinants specify these divergent functions at GPCRs, yet this remains poorly understood. Here, we set out to identify the receptor determinants responsible for β-arrestin-mediated regulation of the chemokine receptor C-X-C motif chemokine receptor 5 (CXCR5). Using bioluminescence resonance energy transfer, we show that β-arrestin1 and β-arrestin2 are dose-dependently recruited to CXCR5 by its cognate ligand C-X-C motif chemokine ligand 13 (CXCL13). The carboxy-terminal tail of CXCR5 contains several serine/threonine residues that can be divided into three discrete phospho-site clusters based on their position relative to transmembrane domain 7. Mutagenesis experiments revealed that the distal and medial phospho-site clusters, but not the proximal, are required for agonist-stimulated β-arrestin1 or β-arrestin2 recruitment to CXCR5. Consistent with this, we provide evidence that the distal and medial, but not proximal, phospho-site clusters are required for receptor desensitization. Surprisingly, the individual phospho-site clusters are not required for agonist-stimulated internalization of CXCR5. Further, we show that CXCL13-stimulated CXCR5 internalization and ERK1/2 phosphorylation, but not desensitization, remain intact in human embryonic kidney 293 cells lacking β-arrestin1 and β-arrestin2. Our study provides evidence that β-arrestins are recruited to CXCR5 and are required for desensitization but are dispensable for internalization or signaling, suggesting that discrete receptor determinants specify the divergent functions of β-arrestins.
SIGNIFICANCE STATEMENT C-X-C motif ligand 13 (CXCL13) and C-X-C motif chemokine receptor 5 (CXCR5) are important in the immune system and are linked to diseases, yet regulation of CXCR5 signaling remains poorly understood. We provide evidence that a phospho-site cluster located at the extreme distal carboxyl-terminal tail of the receptor is responsible for β-arrestin recruitment and receptor desensitization. β-arrestins are not required for CXCL13-stimulated internalization or signaling, indicating that β-arrestins perform only one of their functions at CXCR5 and that discrete receptor determinants specify the divergent functions of β-arrestins.
Introduction
G protein-coupled receptor (GPCR) signaling is tightly regulated to ensure that signals are of the appropriate magnitude and duration (Kelly et al., 2008). Dysregulated GPCR signaling is often pathogenic, and currently, a third of all FDA-approved drugs target GPCRs (Hauser et al., 2017). GPCR signaling is tightly regulated by GPCR kinases and β-arrestins (Komolov and Benovic, 2018). GPCR kinases are kinases that phosphorylate serine/threonine (Ser/Thr) residues on the cytoplasmic carboxyl-terminal tail (CT) and/or intracellular loops of GPCRs (Krupnick and Benovic, 1998). The receptor phosphates trigger the recruitment and binding of cytoplasmic proteins known as β-arrestins, which prevents G protein coupling and leads to receptor desensitization. β-arrestin binding also promotes receptor internalization by acting as an adaptor between GPCRs and the internalization machinery (Goodman et al., 1996; Laporte et al., 1999). In addition, receptor-bound β-arrestins can scaffold and activate various effector molecules, leading to β-arrestin-dependent signaling (Shenoy et al., 2006).
It is emerging that the extent to which β-arrestins can perform these three functions can be GPCR-specific or even ligand-specific, acting at the same GPCR (Maharana et al., 2022; Shukla et al., 2008). β-arrestins become activated when they interact with phosphates on the receptor, and the divergent functions may be linked to different activation states dependent upon discrete phosphorylation patterns along the CT (Nobles et al., 2011). To mediate receptor desensitization, β-arrestins may interact with phosphates on the CT, followed by inserting into a crevice created by the transmembrane bundle, or the receptor core, thereby sterically preventing G protein coupling (Shukla et al., 2014). It is also possible that β-arrestins may interact with the receptor phosphates on the CT without inserting into the receptor core, termed a “hanging” configuration, which would not impact G protein-coupling but would allow for the scaffolding function required for internalization and signaling (Cahill et al., 2017). The receptor determinants specifying the mode by which β-arrestins interact with GPCRs or specify their divergent functions remain poorly understood.
Chemokine receptors belong to a subset of GPCRs involved in the regulation of the immune system and are also involved in several diseases, such as inflammation and cancer (Kazanietz et al., 2019; Lombardi et al., 2001; Teicher and Fricker, 2010). Compared with other chemokine receptors, such as CXCR4, the molecular mechanisms regulating CXCR5 signaling remain poorly understood (Busillo and Benovic, 2007). CXCR5 and its cognate ligand CXCL13 are essential for the development of B-cell follicles (Ansel et al., 2000; Förster et al., 1996; Kazanietz et al., 2019) and have been implicated in several B-cell diseases (Dobner et al., 1992; Förster et al., 1996; Kaiser et al., 1993), cancer (Kazanietz et al., 2019) and COVID (Perreau et al., 2021). CXCR5 couples with a pertussis toxin-sensitive G protein in several cell types to activate various signaling pathways such as ERK1/2 and Akt signaling pathways (English et al., 2018). β-arrestins have been shown to be recruited to CXCL13-activated CXCR5 using various proximity-based approaches, but their function at the receptor remains unknown (Kouzeli et al., 2020; Zhuo et al., 2022).
Here, we examined the receptor determinants responsible for CXCL13-stimulated recruitment of β-arrestin1 and β-arrestin2 to CXCR5, and the impact on signaling and trafficking of CXCR5. We identified the major receptor determinants responsible for recruitment of β-arrestin1 or β-arrestin2 reside within a phospho-site cluster located at the extreme distal CT. A phospho-site cluster located medially along the CT plays a minor role in β-arrestin1 or β-arrestin2 recruitment, whereas a membrane-proximal phospho-site cluster has no role. We found that the medial and distal phospho-site clusters are required for desensitization of CXCR5 signaling, while none of the phospho-site clusters are required for agonist-stimulated internalization. Further, we found that β-arrestins are only required for CXCR5 desensitization and are dispensable for internalization or signaling. Therefore, β-arrestins mediate only one of its divergent functions at CXCR5.
Material and Methods
Cell Lines, Cell Culture, and Antibodies
Human embryonic kidney 293 (HEK293) cells were from the American Type Culture Collection (CRL-1573). Parental HEK293 cells and β-arrestin1 and β-arrestin2 double-knockout (DKO) HEK293 cells were from Dr. Asuka Inoue (Tohoku University, Japan). Cells were maintained in Dulbecco’s minimum essential medium (DMEM; Corning, 10-013-CV) supplemented with 10% fetal bovine serum (FBS; Omega Scientific, FB-11) at 37°C with 5% CO2. Cells were detached using 0.05% Trypsin-EDTA (Gibco, 25300-062). Polyethylenimine for transfections was from Polysciences (23966-1). OPTI-MEM was from ThermoFisher (31985070). Antibodies against pERK1/2 (4370S), ERK1/2 (4695S), and β-arrestin1/2 (4674) were from Cell Signaling Technologies. Anti-FLAG M2-alkaline phosphatase-conjugated antibody was from Sigma (A9469).
DNA Plasmids
Plasmids for pcDNA3 (empty vector), HA-CXCR5, FLAG-CXCR5, β-arrestin1-GFP10, β-arrestin2-GFP10, and pGloSensor-22F (pGLO) were previously described (Caballero et al., 2019; Zhuo et al., 2022; Zhuo et al., 2020). pTRE-Tight-Rluc8 was a gift from Vladislav Verkhusha (Addgene plasmid #798844; http://n2t.net/addgene:79844; RRID:Addgene_79844) (Kaberniuk et al., 2016). FLAG-tagged CXCR5 phospho-site cluster variants [Distal (Dis): T367A, S368A, T370A, T371A; Medial (Med): S358A, S359A, S361A, S363A); Proximal (Prox): S331A, S334A, T338A, T343A, S347A, S354A], di-leucine variant (L336A, L337A) and lysine variant (K328R, K339R) were made using back-to-back primers with the specific nucleotide substitutions by polymerase chain reaction using Platinum SuperFi DNA polymerase (Thermo, 12351010) and circularized using the Kinase, Ligase, and Dpn1 enzyme mix (NEB M0554S), following the manufacturer’s instructions (NEB). The FLAG-tagged CXCR5 double phospho-site variants were made from the single cluster variants. Briefly, Prox/Med was made using back-to-back primers for polymerase chain reaction amplification of the Med variant and circularizing using Kinase, Ligase, and Dpn1 enzyme mix. The Prox/Dis and Med/Dis variants were made using overlapping primers that insert the Dis substitutions to the Prox and Med variants using Gibson Assembly. HA-tagged wild-type CXCR5 or phospho-site cluster variants were tagged at the carboxyl terminus with Rluc8 by assembling a PCR amplified fragment of WT or variant in the pEYFP backbone from HA-CXCR4-EYFP (Bhandari et al., 2007) with an Rluc8 fragment containing overlapping sequence using the NEBuilder HiFi DNA assembly cloning kit (NEB, E5520S), following the manufacturer’s instructions. A complete list of new plasmids and construct assembly information described in this study are available in Supplemental Table 1.
Bioluminescence Resonance Energy Transfer (BRET) Assay for β-Arrestin Recruitment to CXCR5
Recruitment of β-arrestin1 or β-arrestin2 to CXCR5 was measured by BRET, a common approach to examine β-arrestin recruitment to ligand-activated GPCRs (Hamdan et al., 2005; Zhuo et al., 2022). HEK293 cells or HEK293 cells deleted of β-arrestin1 and β-arrestin2 (β-arrestin1/2 DKO) grown on 10 cm dishes to approximately 60–70% confluency were transiently transfected with donor plasmid HA-CXCR5-Rluc8 (100 ng) or phospho-site cluster variant receptors tagged with Rluc8 (100 ng) and 300 ng acceptor plasmid β-arrestin1-GFP10, or β-arrestin2-GFP10 or empty vector (pcDNA) using 20 μl polyethyleneimine, similar to what we have previously described (Zhuo et al., 2022). The next day, cells were detached with trypsin, counted, seeded at a density of 20,000 cells/well of a white-walled clear-bottom 96-well plate (Greiner, 655094) and grown overnight at 37°C in DMEM supplemented with 10% FBS. Cells were washed once with 100 μl of phosphate-buffered saline (PBS) and then treated with a concentration range of CXCL13 (10−10-10−5M), and 10 μM final concentration of coelenterazine-400a (Nanolight; cat. no. 340–500). White tape was applied to the bottom of the plate and BRET measurements were recorded immediately on a microplate reader (LUMIstar Omega, BMG Labtech) at room temperature with an integration time of 0.1 milliseconds every 34 seconds for 11 minutes. The BRET ratio was calculated as the emission signal at 515 +/− 30 nm divided by the emission signal at 410 +/− 80 nm. The net BRET was calculated by subtracting the BRET ratio from cells transfected with only donor plasmid. The area under the curve (AUC) was calculated for each CXCL13 concentration in GraphPad PRISM and normalized to the AUC at 10 μM CXCL13 to compare β-arrestin1 or β-arrestin2 recruitment to CXCR5 in wild-type HEK293 or β-arrestin1/2 DKO cells. To compare wild-type and variant receptors, the AUC at each CXCL13 concentration was normalized to 10 μM CXCL13 of the wild-type CXCR5. Data were analyzed using a nonlinear regression model [log[agonist] versus response (three parameters)] on GraphPad Prism 10.3.1 for macOS, GraphPad Software, San Diego, California USA, graphpad.com.
ERK1/2 Phosphorylation Assay
The β-arrestin1 and β-arrestin2 DKO HEK293 cells were transiently transfected with plasmids encoding FLAG-tagged CXCR5 (6 μg) plus β-arrestin1 and β-arrestin2 (50 ng each) or empty vector (50 ng) in 10 cm dishes with 20 μl PEI and 1 ml OPTI-MEM, similar to what we have previously described (Zhuo et al., 2022). The next day, cells were detached with trypsin+0.05% EDTA and seeded at a density of approximately 2.5 × 106 cells/well of the 6-well dish in 2 ml DMEM supplemented with 10% FBS and incubated overnight at 37°C, 5% CO2. The next day, cells were washed with 1 ml serum-free DMEM containing 20 mM HEPES, pH 7.4, and then serum starved for 3 hours in the same medium. Cells were stimulated without or with 100 nM CXCL13 for 2–45 minutes. Cells were washed with ice-cold PBS and lysed in 300 μl 2× sample buffer (37.5 mM TRIS, pH 6.5, 8% SDS, 10% glycerol, 0.7 M BME, and 0.003% (w/v) bromophenol blue). Lysates were analyzed by 10% SDS-PAGE and immunoblotting on 0.45 μm nitrocellulose membranes. Membranes were blocked with 5% (w/v) milk in Tris-buffered saline (TBS, 20 mM Tris-Cl, pH 7.5, 150 mM NaCl) containing 0.05% (v/v) Tween-20 (RPI, 9005-64-5) (TBS-T). Membranes were incubated with primary antibodies against pERK1/2 or ERK1/2 at a dilution of 1:1000 in 5%-milk/TBS-T overnight at 4°C with gentle rocking. Membranes were washed three times with TBS-T at room temperature and then incubated with horse-radish peroxidase tagged antirabbit antibody (Vector Laboratories; PI-1000) at a dilution of 1:5000 in 5% milk/TBS-T for 30 minutes at room temperature. Membranes were washed for 5–10 minutes, five times with TBS-T. Membranes were exposed to Amersham ECL Prime Western Blotting Detection Reagent (Sigma; RPM2236) solution and imaged on a Bio-Rad Chemidoc Touch Imaging System. Densitometric analysis was performed using ImageJ. Raw pERK1/2 levels were normalized to total ERK1/2 levels and further normalized to the maximum level for comparisons between time points.
Cell Surface Receptor Measurements
Receptor internalization was measured via whole-cell ELISA, similar to what we have previously described (Malik et al., 2012). Briefly, HEK293 or β-arrestin1 and β-arrestin2 DKO HEK293 cells grown on 10 cm dishes to approximately 70–80% confluency were transiently transfected with 6 μg FLAG-tagged receptor (CXCR5 or phospho-site cluster variants) or empty pcDNA3 vector as background control with 20 μl PEI as we have previously described (Caballero et al., 2019). The next day, cells were detached with trypsin+0.05% EDTA and seeded at a density of ∼150,000 cells per well of a 24-well plate that was poly-L-lysine-coated (100 μg/ml) (Sigma, P1399) and grown for an additional 24 hours. The following day, cells were rinsed twice with 500 μl warm DMEM supplemented with 20 mM HEPES, pH 7.5, and incubated with 250 μl of the same medium for 30 minutes at 37°C. Cells were treated with vehicle or CXCL13 (100 nM) for 5–60 minutes. Plates were placed immediately on ice, cells were rinsed with 500 μl ice-cold TBS, and fixed with 3.7% formaldehyde (Sigma, F1635) in TBS for 10 minutes at room temperature. Cells were washed extensively with TBS, and then incubated with TBS containing 1% (w/v) bovine serum albumin (GoldBio; A-421-250) (TBS/bovine serum albumin) for 45 minutes at room temperature while gently rocking. Cells were then incubated with the anti-FLAG M2 alkaline phosphatase-conjugated antibody at a 1:1000 dilution in TBS/bovine serum albumin for 1 hour at room temperature while gently rocking. Cells were washed three times with 500 μl TBS and incubated with 250 μl p-nitrophenyl phosphate solution (Sigma, P7998-100) for 15–45 minutes. Endpoint absorbance at 405 nm was read in a microplate reader (LUMIstar Omega, BMG Labtech). Background absorbance was subtracted, and receptor internalization was calculated as a percent loss of cell surface receptor following agonist stimulation.
cAMP Measurements
Agonist-stimulated cAMP levels were measured in real-time using the pGloSensor-22F luminescence-based cAMP biosensor (Caballero et al., 2019) or the Nluc-EPAC-VenusVenus [a BRET-based cAMP biosensor (Masuho et al., 2015)]. Receptor desensitization experiments using the pGlosensor were performed on HEK293 cells. Cells grown on 10 cm dishes to approximately 70% confluency were transfected with 3 μg FLAG-tagged receptor and 3 μg pGloSensor plasmid with 20 μl PEI. The next day, cells were seeded at a density of 20,000 cells per well of a white-walled clear-bottom 96-well plate and grown overnight at 37°C. Cells were washed once with 100 μl phenol red-free DMEM and then pretreated with either vehicle or CXCL13 (10–100 nM) for 30 minutes at 37°C in 100 μl phenol red-free DMEM containing 5 μg/ml cycloheximide (Sigma, C7698). Cells were acid-washed (1 mM ascorbic acid) once and once with PBS, before stimulation with vehicle (PBS) or 1 nM CXCL13 in the presence of 20 μM forskolin (Sigma, F3917) and 3.2 mM D-luciferin (GoldBio, LUCK). Background signal was also determined with wells containing no D-luciferin in triplicate. Total luminescence was measured every min for 20 minutes with a 0.2 integration time using a PHERAstar FSX microplate reader (BMG Labtech). Raw luminescence values were corrected by background subtraction, and the area under the curve was determined for each treatment condition using GraphPad PRISM. Values were normalized to the forskolin-treated condition.
Receptor desensitization experiments using the Nluc-EPAC-VenusVenus sensor were performed in the DKO HEK293 cell line. The Nluc-EPAC-VenusVenus sensor was kindly provided by Drs. Mikel Garcia-Marcos (Boston University) and Kirill Martemyanov (University of Florida Scripps) (Masuho et al., 2015). Cells grown on 10 cm dishes to approximately 70% confluency were transfected with 6 μg FLAG-CXCR5, 300 ng Nluc-EPAC-VV, and either 300 ng β-arrestin1 and 300 ng β-arrestin2, or 600 ng empty vector (pcDNA3) with 20 μl PEI. The next day, cells were seeded at a density of ∼20,000 cells/well onto a white-walled clear-bottom 96-well plates and grown overnight at 37°C. The next day, cells were washed once with 100 μl phenol red-free DMEM containing 20 mM HEPES and then pretreated with vehicle (PBS) or CXCL13 (10 nM) for 1 hour at 37°C. Cells were washed to remove bound chemokine by incubating with 1 mM ascorbic acid at room temperature for 5 minutes, followed by two rapid washes with phenol red-free DMEM containing 20 mM HEPES. Coelenterazine-h was added to each well (final concentration 1 μM) in phenol red-free DMEM containing 20 mM HEPES and incubated while covered at room temperature for 5 minutes. Baseline BRET measurements were made for 5 minutes before manually adding varying doses of CXCL13 (10−9.5–10−7.5 M) with 5 μM forskolin. Plates were then placed in a PHERAstar microplate reader (BMG Labtech) and BRET measurements were made for 30 minutes. The integration time was 0.08 seconds for each well per read was used. Baseline values were then averaged for each condition and then subtracted from every individual timepoint to determine ΔBRET values. The AUC of each condition was calculated using GraphPad PRISM and then normalized to the forskolin-treated condition.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 10.3.1 for macOS, GraphPad Software, www.graphpad.com. Data are represented as the mean ± S.D. from at least three independent biological replicates. A two-way ANOVA was used to compare the means between groups under different treatment conditions. A one-way ANOVA was used to compare the means of three or more groups. Student’s t test was used to compare the means of two groups. ANOVA was followed by Dunnett’s, Bonferroni’s, or Šidák’s multiple comparison test, as indicated in the figure legends. A P value of 0.05 or less was considered significant, and P values are indicated in the figure panels or legends.
Results
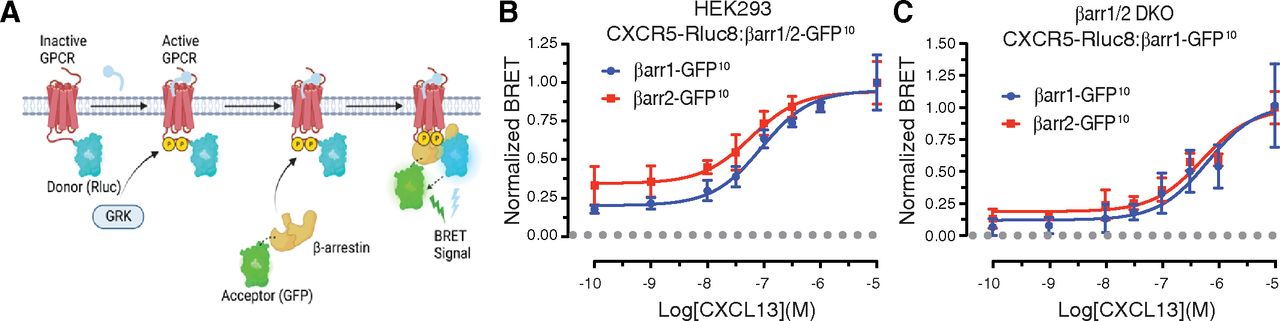
Although CXCR5 has previously been shown to interact with β-arrestins (Kouzeli et al., 2020; Zhuo et al., 2022), the molecular determinants specifying β-arrestin recruitment and the functional outcome remain unknown. To begin to address this we examined β-arrestin1 and β-arrestin2 recruitment to CXCR5 by BRET, a common approach to monitor β-arrestin recruitment to agonist-activated GPCRs (Hamdan et al., 2005; Kocan et al., 2008; Namkung et al., 2016). The donor Renilla luciferase (Rluc8) was attached in-frame to the C-terminal cytoplasmic tail (CT) of CXCR5 (CXCR5-Rluc8) and the acceptor green fluorescent protein (GFP10) was attached to the C-terminus of β-arrestin1 or β-arrestin2 (β-arrestin1/2-GFP10) (Fig. 1A). The BRET response was measured in HEK293 cells transiently expressing CXCR5-Rluc8 and either β-arrestin1-GFP10 or β-arrestin2-GFP10 and stimulated with varying concentrations of CXCL13. There was a concentration-dependent increase in the BRET response, consistent with β-arrestin1 or β-arrestin2 recruitment to ligand-activated CXCR5 (Fig. 1B). We repeated the experiment in HEK293 cells that lack β-arrestin1 and β-arrestin2 (β-arrestin1/2 double knockout, DKO) and similar to wild-type HEK293 cells, the BRET response following CXCL13 stimulation increased in a concentration-dependent manner (Fig. 1C). The potency (pEC50) values for β-arrestin1 and β-arrestin2 recruitment in wild-type (WT) or DKO HEK293 cells were similar (Table 1).

Agonist-induced recruitment of β-arrestins to CXCR5. (A) Schematic of BRET approach to measure β-arrestin recruitment to agonist-activated GPCRs. Donor Rluc8 provides energy to the acceptor (GFP) when in close proximity (∼10 nm). Net BRET is calculated by the ratio of the acceptor signal divided by the donor signal subtracted from the BRET ratio in cells expressing the donor alone. (B–C). β-arrestin1/2 recruitment to CXCR5 was measured by BRET in HEK293 cells (B) or β-arrestin1/2 double knockout HEK293 cells (DKO) (C) expressing HA-CXCR5-Rluc8 and β-arrestin1-GFP10 or β-arrestin2-GFP10. After adding coelenterazine-400a, BRET measurements were made at room temperature with an integration time of 0.1 seconds every 34 seconds for 11 minutes. The BRET response was calculated as the area under the curve at each concentration of CXCL13 and normalized to 10−5 M CXCL13 (B/C). Data represent the mean ± S.D. from three independent experiments. Data were fit by nonlinear regression [log[agonist]versus response (three parameters)] using GraphPad PRISM. The pEC50, Emax, and R2 values are reported in Table 1. The pEC50 values in panels B and C were analyzed by an unpaired t test and were not significant. The adjusted P values were greater than 0.05.
Summary of pEC50, emax, and R2 values from BRET experiments
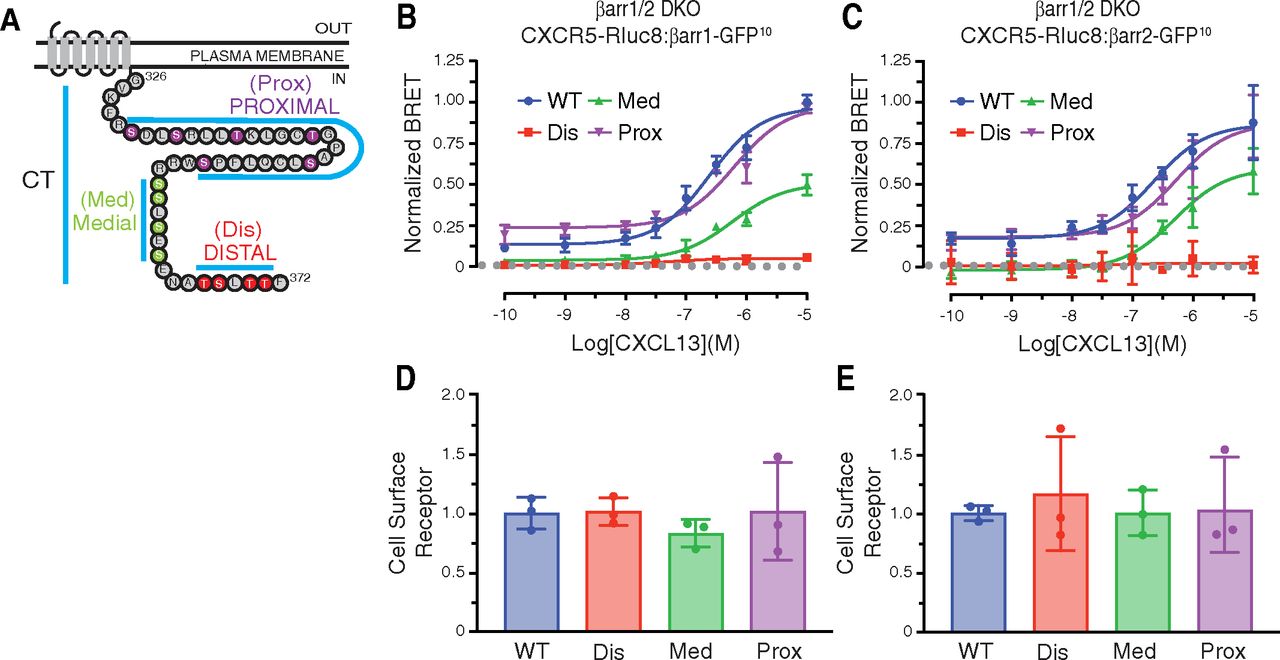
β-arrestins are recruited to GPCRs at the plasma membrane once they are phosphorylated on Ser and/or Thr residues within the cytoplasmic intracellular loops and/or the CT (Bünemann and Hosey, 1999; Luo et al., 2017). The CT of CXCR5 has 14 Ser/Thr amino acid residues that fall into three discernable clusters, which we have labeled Prox, Med, and Dis, based on their distance from predicted transmembrane domain 7 (Fig. 2A). We substituted Ser and Thr within each cluster to alanine and examined the ability of each phospho-site cluster variant to recruit β-arrestin1 or β-arrestin2 by BRET in response to CXCL13 stimulation in the β-arrestin1/2 DKO cell line. The BRET response between β-arrestin1 or β-arrestin2 and the proximal phospho-site cluster variant showed no change when compared with WT CXCR5 (Fig. 2, B and C), while the BRET response with the medial phospho-site cluster variant was only partially reduced (Fig. 2, B and C). In contrast, the BRET response between β-arrestin1 or β-arrestin2 and the distal phospho-site cluster variant receptor was effectively abolished (Fig. 2, B and C), suggesting the distal phospho-site cluster is essential for β-arrestin1 and β-arrestin2 recruitment to CXCR5. The surface level of each receptor phospho-site cluster variant in the BRET experiments was similar to the WT receptor as determined by ELISA of parallel samples (Fig. 2, D and E). Taken together, the major determinants specifying β-arrestin recruitment to CXCR5 reside within a phospho-site cluster located at the extreme distal region of the CT.

Role of phospho-site clusters in agonist-stimulated recruitment of β-arrestin1 and β-arrestin2 to CXCR5. (A) Amino acid sequence of the CXCR5 carboxyl-terminal tail (CT). Three Ser/Thr phospho-site clusters are indicated: Proximal (Prox), Medial (Med), and Distal (Dis). (B–C) BRET experiments were performed in β-arrestin1/2 DKO HEK293 cells expressing HA-CXCR5-Rluc8 variants and either β-arrestin1-GFP10 (B) or β-arrestin2-GFP10 (C) After adding coelenterazine-400a, BRET measurements were made at room temperature with an integration time of 0.1 seconds every 34 seconds for 11 minutes. The BRET response was calculated as the area under the curve at each concentration of CXCL13 and normalized to the 10−5 M CXCL13 concentration in the WT condition. Data represent the mean ± S.D. from three independent experiments. Data were fit by nonlinear regression [log[agonist]versus response (three parameters)] using GraphPad PRISM. (D–E) Parallel samples from BRET experiments were analyzed by ELISA for cell surface receptor levels of WT and variant receptors. The pEC50, Emax, and R2 values are reported in Table 1. For panels B and C, the EC50 and Emax values were analyzed by one-way ANOVA followed by Dunnett’s multiple comparison test. The adjusted P values were greater than 0.05 (P > 0.05) for all receptor variants EC50 values relative to WT receptor except for the Dis variant (P < 0.01) in panel B. The Emax for the Dis (P < 0.01) and Med (P < 0.01) variants were found to be significantly different from WT for panel B; in panel C just the Dis receptor (P < 0.01) was significantly different. For panels D and E, data were analyzed by one-way ANOVA followed by Dunnett’s multiple comparison test. The data were not significant relative to WT. The adjusted P values for all comparisons were greater than 0.05.
In general, β-arrestin binding to phosphorylated GPCRs promotes receptor internalization via clathrin-coated pits (Goodman et al., 1996; Laporte et al., 1999; Moo et al., 2021). We examined the internalization of FLAG-tagged phospho-site cluster variants in HEK293 cells by ELISA following CXCL13 stimulation from 0–60 minutes. Approximately, 70–80% WT receptor was internalized following 30–60 minutes of CXCL13 stimulation. In contrast, internalization of the distal phospho-site cluster variant (Dis) was partially reduced (∼25%) when compared with WT CXCR5, whereas internalization of the proximal (Prox) and medial (Med) phospho-site cluster variants were only slightly reduced (Fig. 3A). This was not due to differences in cell surface expression, as each receptor variant was expressed to similar levels on the cell surface compared with WT receptor (Fig. 2, D and E). Due to the possibility of phospho-site cluster redundancy, we examined agonist-stimulated internalization of double phospho-site cluster receptor variants in which the phospho-site clusters were mutated together. Cells were stimulated with CXCL13 for 30 minutes, a time when approximately 70% of the receptor has internalized (Fig. 3A). Internalization of the distal phospho-site cluster in combination with the proximal (Prox/Dis) or medial (Med/Dis) phospho-site cluster showed statistically significantly reduced internalization (∼50%) relative to WT receptor (Fig. 3B), while internalization of the proximal and medial (Prox/Med) phospho-site cluster variant remained virtually intact (Fig. 3B). These data suggest that the main determinants specifying CXCR5 internalization reside within the distal phospho-site cluster, although the proximal or medial phospho-site clusters may have a redundant role. This also reveals that while the distal phospho-site cluster is essential for β-arrestin1 and β-arrestin2 recruitment (Fig. 2, B and C), it is only partially required for internalization, suggesting that β-arrestins are not essential for CXCL13-stimulated internalization of CXCR5.

Agonist-stimulated internalization of CXCR5 and phospho-site cluster variants. (A–B) HEK293 cells expressing FLAG-tagged single (A) or combined (B) phospho-site cluster receptor variants were stimulated with 100 nM CXCL13 for 0–60 minutes (A) or 30 minutes (B). Cell surface receptor was determined by ELISA using an antibody against the N-terminally exposed FLAG epitope. Receptor internalization for each independent experiment was calculated as a decrease in cell surface receptor level in the CXCL13-treated condition relative to the vehicle-treated condition. Data represent the mean ± S.D. from three independent experiments. Data for panel A were analyzed by two-way ANOVA, followed by Dunnett’s multiple comparisons test. WT and Dis significantly differed at 30 minutes (P = 0.04) and 45 minutes (P = 0.03). Data for panel B were analyzed by one-way ANOVA, followed by Dunnett’s multiple comparisons test, and the adjusted P values are shown.
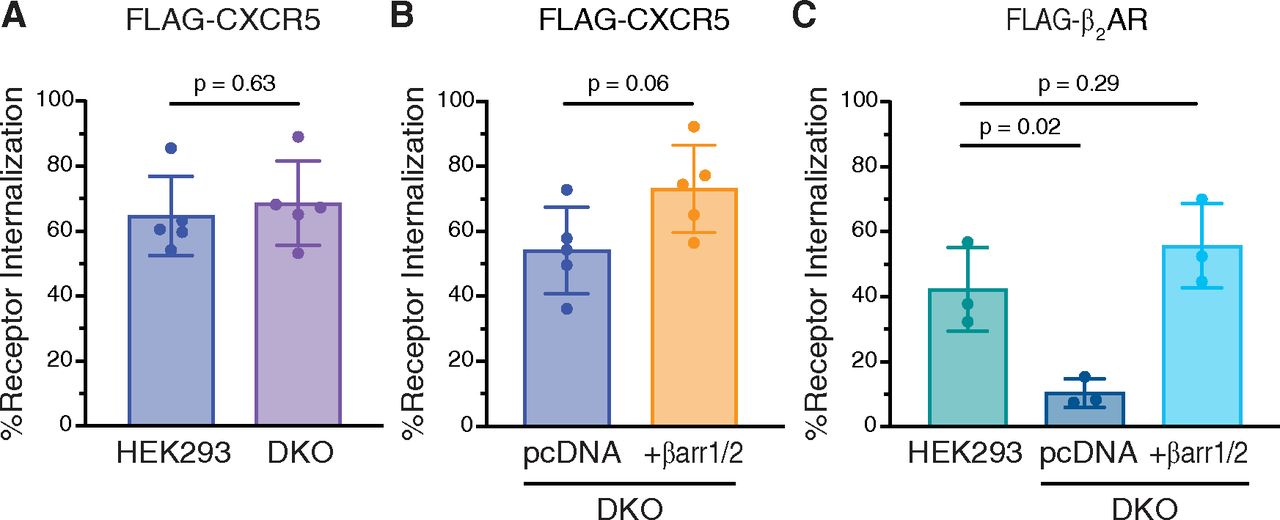
There is emerging evidence that certain GPCRs undergo internalization in a β-arrestin-independent manner, although they are still able to recruit β-arrestins following agonist activation (Moo et al., 2021). With this in mind, we examined CXCL13-stimulated internalization of FLAG-tagged CXCR5 in the β-arrestin1/2 DKO HEK293 cell line by ELISA. CXCL13-stimulated CXCR5 internalization was similar in wild-type HEK293 or β-arrestin1/2 DKO cells (Fig. 4A), suggesting that β-arrestin1 and β-arrestin2 are not essential for CXCR5 internalization. When β-arrestin1 and β-arrestin2 were re-expressed in the β-arrestin1/2 DKO cell line, CXCR5 internalization partially increased (∼25%) compared with the control empty vector (pcDNA) suggesting that while not essential, overexpression of β-arrestins may be sufficient for CXCR5 internalization (Fig. 4B). This is in contrast to agonist-stimulated internalization of the β2-adrenergic receptor (β2AR), which was nearly abolished in the β-arrestin1/2 DKO cell line compared with WT HEK293 cells and was rescued by re-expression of β-arrestin1 and β-arrestin2 (Fig. 4C), and which is consistent with this GPCR internalizing via the β-arrestin-dependent mode (Goodman et al., 1996; Laporte et al., 1999). Further, agonist-stimulated internalization of the three CXCR5 phospho-site cluster receptor variants was similar to the WT receptor in the β-arrestin1/2 DKO cell line (Fig. 5A). Together, these data suggest that β-arrestins are not essential for CXCL13-stimulated CXCR5 internalization.

Role of β-arrestins in agonist-stimulated internalization of CXCR5. (A–B) Agonist-stimulated internalization of FLAG-CXCR5 in either WT HEK293 or β-arrestin1/2 DKO HEK293 cells (A) and DKO cells with empty vector (pcDNA) or β-arrestin1 and β-arrestin2 (+βarr1/2) re-expression (B). Cells were stimulated with 100 nM CXCL13 for 30 minutes at 37°C. Cell surface receptor levels were determined by ELISA using an antibody against the N-terminally exposed FLAG epitope. Receptor internalization for each independent experiment was calculated as a decrease in the cell surface receptor level in the CXCL13-treated condition relative to the vehicle-treated condition. Data represent the mean ± S.D. from five independent experiments. (C) Agonist-stimulated internalization of FLAG-β2AR in WT HEK293 cells or DKO cells with (+βarr1/2) or without (pcDNA) β-arrestin1/2 re-expression. Cells were stimulated with 10 μM isoproterenol for 30 minutes at 37°C. Receptor internalization for each independent experiment was calculated as a decrease in the cell surface receptor level in the isoproterenol-treated condition relative to the vehicle-treated condition. Data represent the mean ± S.D. from three independent experiments. Data for panels A and B were analyzed by Student’s t test. Data for panel C were analyzed by one-way ANOVA, followed by Dunnett’s multiple comparisons test to WT HEK293. Adjusted P values are shown.

Agonist stimulated internalization of CXCR5 receptor variants in β-arrestin1/2 DKO cells. (A) β-arrestin1/2 DKO HEK293 cells expressing FLAG-tagged CXCR5 WT or single phospho-site cluster receptor variants were stimulated with 100 nM CXCL13 for 0–60 minutes. (B–C) WT HEK293 cells (B) or β-arrestin1/2 DKO HEK293 cells (C) expressing the dileucine (2L/A) variant or putative ubiquitin-deficient variant (2K/R) were stimulated with 100 nM CXCL13 for 30 minutes at 37°C. Cell surface receptor levels were determined by ELISA, and receptor internalization for each independent experiment was calculated as a decrease in cell surface receptor level in the CXCL13-treated condition relative to the vehicle-treated condition. Data represent the mean ± S.D. from six (A) or three (B/C) independent experiments. Data were analyzed by two-way ANOVA (A) or one-way ANOVA (B/C), followed by multiple comparisons to WT control with Dunnett’s multiple comparison test. Adjusted P values are shown.
Several GPCRs have been shown to follow a β-arrestin-independent mode of internalization that is dependent upon a dileucine motif located within the CT. For example, dileucine motifs in chemokine receptors CXCR2 and CXCR4 are required for their internalization following agonist stimulation (Fan et al., 2001; Orsini et al., 1999). CXCR5 has a putative dileucine motif located within the CT (332DLSRLL337), but its role in CXCR5 internalization remains unknown. To examine this, we substituted leucine residues 336 and 337 for alanine (L336/337A; 2L/A) and examined CXCL13-stimulated internalization in WT HEK293 cells or the β-arrestin1/2 DKO cells by ELISA. Internalization of the 2L/A variant was similar to the WT receptor in HEK293 cells (Fig. 5B). However, in the β-arrestin1/2 DKO cell line, there was a ∼25% decrease in internalization relative to the WT receptor (Fig. 5C). These data suggest that while the dileucine mode of internalization is not essential for CXCR5 internalization, it may be partly redundant with the β-arrestin mode. However, since internalization was only decreased by ∼25% in the β-arrestin1/2 DKO cells, CXCR5 internalization occurs mainly via a yet-to-be-determined mechanism.
Ubiquitination may also mediate GPCR internalization, although mainly in Saccharomyces cerevisiae, whereby ubiquitin moieties attached to lysine residues on the receptor interact with ubiquitin-binding endocytic adaptor proteins that link receptors to the internalization machinery (Kennedy and Marchese, 2015; Patwardhan et al., 2021). CXCR5 has two lysine residues within the CT (K328, K339) that could serve as potential ubiquitination sites. To examine their role in CXCR5 internalization, we substituted both lysine residues for arginine (K328/339R; 2K/R) and examined CXCL13-stimulated internalization in WT HEK293 cells and the β-arrestin1/2 DKO cell line by ELISA. Internalization of the 2K/R variant was similar to WT receptor in HEK293 or β-arrestin1/2 DKO cells (Fig. 5, B and C). Although it remains to be determined whether these sites are indeed modified by ubiquitin, these data suggest that ubiquitination is not essential for CXCR5 internalization, nor is it redundant with the β-arrestin mode of internalization.
β-arrestins also mediate GPCR signaling by scaffolding effector molecules, such as mitogen-activated protein kinases and extracellular regulated kinases 1 and 2 (ERK1/2) (Gurevich and Gurevich, 2006). We examined CXCL13-stimulated ERK1/2 phosphorylation in the β-arrestin1/2 DKO cell line after we restored β-arrestin1/2 expression by transient transfection. There was no observable difference in time-dependent phosphorylation of ERK1/2 in β-arrestin1/2 DKO cells transfected with or without β-arrestin1 and β-arrestin2 (Fig. 6A and quantified in Fig. 6B). Cell surface receptor levels were similar in both cell lines (Fig. 6C). These data provide evidence that β-arrestins are not required for agonist-stimulated CXCR5-mediated phosphorylation of ERK1/2, at least in HEK293 cells.

β-arrestin1 and β-arrestin2 are not essential for CXCR5-mediated ERK1/2 activation. (A) CXCL13-stimulated ERK1/2 phosphorylation was examined in β-arrestin1/2 DKO cells expressing FLAG-tagged CXCR5 with (+βarr1/2) or without (pcDNA) β-arrestin1 and β-arrestin2 re-expression. Cells were stimulated with 100 nM CXCL13 for 0–45 minutes at 37°C, and whole-cell lysates were analyzed by immunoblotting for phosphorylated (pERK1/2) and total ERK1/2 (ERK1/2). Representative immunoblots probed with the indicated antibodies are shown. (B) ERK1/2 phosphorylation was quantified by densitometry, normalized to ERK1/2 levels, and then compared with the 2 minutes condition transfected with pcDNA. Data represent the mean ± S.D. from three independent experiments. (C) Parallel samples from each experiment were analyzed by ELISA for cell surface receptor levels of CXCR5. Data were normalized to pcDNA condition and represent the mean ± S.D. For panel B, data were analyzed by two-way ANOVA followed by Šídák’s multiple comparison test for pcDNA versus βarr1/2 at each time point. Data were not significant with adjusted P values greater than 0.05 for all comparisons (B). For panel C, data were analyzed by a student’s t test and were not significant with an adjusted P value greater than 0.05.
In addition to their role in internalization and signaling, the classical function of β-arrestins is to mediate GPCR desensitization (Kelly et al., 2008). CXCR5 couples to a pertussis-toxin-sensitive G-protein (English et al., 2018), which inhibits adenylyl cyclase (AC) mediated cAMP production, and is best quantified in the presence of the AC activator forskolin (FSK) (Tsvetanova and von Zastrow, 2014). We measured cAMP levels in live cells using the pGloSensor, which luminesces when bound to cAMP (Tsvetanova and von Zastrow, 2014). HEK293 cells transiently expressing the pGloSensor and WT receptor or the phospho-site cluster variants were pretreated with vehicle or CXCL13 (10, 30, and 100 nM) for 30 minutes at 37°C. Cells were acid-washed to facilitate the removal of bound chemokine and rechallenged with vehicle or CXCL13 (1 nM) in the presence of FSK. In cells expressing WT receptor or the proximal phospho-site cluster variant, there was a loss in the ability of the CXCL13 challenge to inhibit cAMP production in cells concentration-dependently pretreated with the chemokine, consistent with receptor desensitization (Fig. 7). In contrast, the distal and medial phospho-site cluster variants, which are defective in the recruitment of β-arrestin1 or β-arrestin2 (Fig. 2, B and C), showed a similar reduction of FSK-stimulated cAMP levels when challenged with CXCL13 in cells pretreated with the chemokine, suggesting these receptor variants do not desensitize (Fig. 7).

Agonist-induced desensitization of CXCR5 and phospho-site cluster variants. Desensitization of CXCL13-promoted inhibition of FSK-stimulated intracellular cAMP production. HEK293 cells expressing WT CXCR5 or phospho-site cluster variants and the pGloSensor, which luminesces in proportion to cAMP levels, were pretreated with vehicle or increasing concentrations of CXCL13 (10, 30, and 100 nM) for 30 minutes at 37°C. Cells were washed once with ascorbic acid to remove bound chemokine, followed by washing them with PBS. Cells were immediately stimulated with 1 nM CXCL13 or vehicle in phenol red-free DMEM containing 10% FBS, 20 μM forskolin, and 3.2 mM luciferin, and luminescence was immediately measured in a microplate reader at room temperature every minute for 20 minutes. After background subtraction, luminescence values were normalized to the forskolin-treated control condition within each transfection condition. Data represent the mean ± S.D. from three independent experiments. The starting levels of cAMP were virtually identical (within ∼1–2% of maximum luminescence observed with FSK alone) without or with all CXCL13 concentrations within each transfection condition. Data within each transfection condition were analyzed by two-way ANOVA, followed by Dunnett’s multiple comparison test. The adjusted P values are shown.
These data are consistent with the classical paradigm of receptor desensitization, in which β-arrestin binding promotes G protein uncoupling and reduces cellular responsiveness to subsequent stimulation. To explore this further, we examined CXCR5 desensitization in the β-arrestin1/2 DKO HEK293 cell line. For this, we used the BRET-based Nluc-EPAC (exchange protein activated by cAMP)-VenusVenus cAMP sensor (Masuho et al., 2015). The BRET signal decreases when the sensor binds to cAMP, as previously described (Masuho et al., 2015). Cells expressing CXCR5, Nluc-EPAC-VV, and either empty vector (pcDNA) or β-arrestin1 and β-arrestin2 were pretreated with either vehicle or 10 nM CXCL13 for 1 hour at 37°C and, after acid washing, were challenged with varying concentrations of CXCL13 in the presence of FSK (Fig. 8, A and B). In pcDNA transfected cells, concentration-dependent inhibition of cAMP production by CXCL13 was similar between cells pretreated with CXCL13 and vehicle, suggesting that CXCR5 does not desensitize in β-arrestin DKO cells (Fig. 8A). In contrast, CXCR5 desensitization was restored when β-arrestins were added back to the DKO cells (Fig. 8B). This is discernable as a significant decrease in the EC50 after CXCL13 pretreatment in cells re-expressing β-arrestins (Fig. 8C). Surface expression of CXCR5 was similar in all experiments (Fig. 8D) as was re-expression of β-arrestins (Fig. 8E). These data suggest that β-arrestins are essential for CXCR5 desensitization.

β-arrestins are essential for CXCR5 desensitization. (A–B) β-arrestin1/2 knockout HEK293 (DKO) cells were transfected with WT FLAG-tagged CXCR5, BRET Nluc-EPAC-VV cAMP biosensor, and empty vector (pcDNA) (A) or β-arrestin1/2 (B). Cells were pretreated with vehicle or CXCL13 (10 nM) for 1 hour at 37°C before challenging with varying doses of CXCL13 in the presence of forskolin (FSK). Data represent the mean ± S.D. from 4 or 2 (-8 and -7 doses) independent experiments. Curves were fit using nonlinear regression (log(agonist) versus response (three parameters) in GraphPad PRISM. (C) The data from (A) and (B) are shown as the mean ± 95% CI of pEC50 values. Data were analyzed by one-way ANOVA and Bonferroni’s multiple comparison tests between vehicle (“−”) and CXCL13 (“+”) pretreatments within each transfection condition. The adjusted P values are shown. (D) Parallel samples from each experiment were analyzed by ELISA for cell surface receptor levels. Data were normalized to pcDNA condition and represent the mean ± S.D. Data were analyzed by a student’s t test. The adjusted P value is shown. (E) Representative immunoblots probed with the indicated antibodies are shown.
Discussion
β-arrestins are considered to be master regulators of GPCR signaling by mediating desensitization, internalization, and signaling (Wess et al., 2023). However, it is emerging that β-arrestins may not mediate each of these functions at a given GPCR or when the same GPCR is activated by a different ligand. However, the reason for this remains poorly understood. Here, we provide evidence that β-arrestins are required for desensitization of the chemokine receptor CXCR5 but are dispensable for internalization or signaling. Although we have yet to characterize the mode of CXCR5 internalization, phosphorylation of the receptor is likely required. Therefore, our study suggests that the divergent function of β-arrestins may be linked to the location of phospho-sites on GPCRs and reveals another GPCR that uses a β-arrestin-independent mode of endocytosis.
We mapped the major determinants specifying β-arrestin recruitment to CXCR5 to a phospho-site cluster located at the extreme distal CT of the receptor (Fig. 2). The are multiple phosphor-site clusters along the CT, and it remains unclear why one phospho-site cluster would favor β-arrestin recruitment over others, although it could be related to the relative affinity of β-arrestins for these clusters. There is evidence that β-arrestins have different affinities for different phosphorylation sites (Latorraca et al., 2020; Zhou et al., 2017). An additional feature that may be relevant is that the location of the phospho-cluster is at the extreme CT of CXCR5, which is similarly positioned when compared with other GPCRs (Oakley et al., 2001). The distal phospho-site cluster along the CT of the PTH1 receptor has more influence on the recruitment and binding of β-arrestin1 and β-arrestin2 than a more proximal cluster (Haider et al., 2022). Similarly, the distal phospho-site cluster along the CT of CXCR4 also has a greater influence on β-arrestin1 and β-arrestin2 recruitment when compared with more membrane-proximal phospho-sites (Busillo et al., 2010). We note that in our experiments, binding between receptor and β-arrestin was measured by BRET, which is dependent upon the close proximity of the donor and acceptor molecules but can also be influenced by their orientation (Hamdan et al., 2006). We cannot rule out the possibility that BRET is somehow impacted by the orientation of the donor and acceptor molecules and not receptor binding per se. Further, we have yet to examine whether any Ser or Thr residues on the CT of CXCR5 are indeed phosphorylated.
A major finding from our study is that β-arrestins mediate only one of its three canonical functions at CXCR5. The reason for this remains unknown, but it may be linked to the two distinct modes by which β-arrestins are believed to interact with phosphorylated GPCRs (Cahill et al., 2017; Thomsen et al., 2016). In one binding mode, β-arrestins are fully engaged with the phosphorylated receptor via additional interactions within the cytoplasmic core formed by the transmembrane domains. The fully engaged β-arrestin likely interferes with G protein coupling and thus mediates desensitization. In a second binding mode, known as the “hanging” configuration, β-arrestins bind only to the phosphorylated CT without engaging with the cytoplasmic core, which would allow for the scaffolding function required for receptor internalization and signaling, but would not interfere with G protein coupling (Cahill et al., 2017; Kumari et al., 2017). It is possible that β-arrestin binding to extreme distal phospho-sites on the CT of CXCR5 mainly adopts a fully engaged configuration that facilitates desensitization but not receptor internalization or signaling. The binding mode of β-arrestin to CXCR5 and the role of receptor phosphorylation requires further investigation.
Our study adds to the growing list of GPCRs for which agonist-stimulated internalization is independent of β-arrestins (Moo et al., 2021). However, the adaptors remain largely unknown, although direct interactions with clathrin adaptors such as AP2 have been reported to mediate β-arrestin-independent internalizations of some GPCRs (Patwardhan et al., 2021). Di-leucine motifs, which are recognized by AP2 on the CT of several chemokine receptors, have been linked to agonist-stimulated internalization (Fan et al., 2001; Kraft et al., 2001; Orsini et al., 1999). For CXCR5, the di-leucine motif variant (2L/A) showed a small reduction in internalization (∼25%) in the β-arrestin1/2 DKO cell line (Fig. 5C) but not in the WT HEK293 cell line (Fig. 5 A), suggesting these two modes of internalization may be partially redundant, but that CXCR5 mainly follows a di-leucine and β-arrestin independent mode of internalization. We also examined the role of ubiquitination on CXCR5 internalization, although mammalian GPCR internalization generally does not require receptor ubiquitination (Patwardhan et al., 2021). Mutation of lysine residues within the C-tail of CXCR5 did not impact agonist-stimulated internalization of CXCR5 (Fig. 5C), although we have yet to confirm whether CXCR5 is indeed ubiquitinated. Nevertheless, further work is required to identify the mode of CXCR5 internalization.
An interesting feature of this β-arrestin-independent mode of CXCR5 internalization is that it likely also requires receptor phosphorylation. Our data suggest that multiple phospho-sites along the CT are required for agonist-stimulated internalization of CXCR5. GPCRs are mainly phosphorylated by second-messenger-dependent protein kinases or GPCR kinases, although their role in CXCR5 internalization remains to be investigated (Marchese et al., 2003). A potentially important aspect of the distal phospho-site cluster that is essential for β-arrestin phospho-sites (370TTF372; Fig. 2A) are part of a type-1 PDZ-ligand consensus sequence (S/T-X-ϕ, where X = any amino acid and ϕ = large hydrophobic amino acid) (Valgardson et al., 2019). PDZ-ligands are recognized by PDZ (PSD-95/Glg1/ZO-1)-domain containing proteins involved in various cellular processes, including signaling and membrane trafficking. Several chemokine receptors have a PDZ-ligand at the extreme CT, but it seems to be required for postinternalization sorting or signal transduction and not internalization from the plasma membrane (Baugher and Richmond, 2008; Delhaye et al., 2007; Romero et al., 2011; Valgardson et al., 2019). It is of interest to determine whether PDZ proteins indeed regulate CXCR5 signaling or trafficking and how they might integrate with β-arrestin binding and function.
β-arrestins are known to mediate GPCR signaling after they bind to a GPCR. This is best exemplified by GPCRs such as angiotensin II (AT1aR) and V2 vasopressin (V2R) receptors, which require β-arrestins for phosphorylation of ERK1/2 (Charest et al., 2007; Wei et al., 2003). Distal phospho-site clusters on the CT of these GPCRs, but not more membrane-proximal phospho-clusters, are necessary for high affinity β-arrestin binding and ERK1/2 phosphorylation from endosomes (Gurevich and Gurevich, 2006; Oakley et al., 2000). In contrast, we provide evidence that β-arrestins are not required for downstream signaling to this pathway upon binding to CXCR5 (Fig. 6). This was not completely unexpected since a previously published study showed that CXCL13-mediated ERK1/2 phosphorylation occurs via a pertussis toxin-sensitive G protein in HeLa cells or B cells (English et al., 2018). This is also consistent with other GPCRs, such as CXCR4, CXCR2, and β2AR, which do not require β-arrestin1/2 for ERK1/2 phosphorylation (Grundmann et al., 2018; Malik et al., 2012; Zhao et al., 2004). However, this does not necessarily rule out a role for β-arrestin1 and/or β-arrestin2 in CXCR5 signaling in other cell types. In addition, β-arrestins can scaffold other signaling molecules (Alekhina and Marchese, 2016), and it is possible that β-arrestins are required for different signaling pathways upon binding to CXCR5 (Zhuo et al., 2022).
CXCR5 is primarily expressed in B cells and is involved in the organization of B-cells within lymphoid follicles, which is required for humoral immunity (Ansel et al., 2000; Förster et al., 1996). Several GPCR agonists acting on their cognate receptors expressed on B cells serve as guidance cues in a highly regulated manner with spatial and temporal overlap within lymphoid follicles (Cyster, 2010). While our study is restricted to HEK293 cells and the mode of CXCR5 internalization in B cells remains to be determined, it is possible that a β-arrestin-independent mode of internalization could be physiologically advantageous if other simultaneously activated GPCRs are regulated by β-arrestins. This could reduce competition between β-arrestins and endocytic adaptors for GPCRs, and ensure the fidelity of GPCR signaling in a complex physiological setting (Gurevich and Gurevich, 2006; Klein et al., 2001; Puthenveedu et al., 2010; Wolfe and Trejo, 2007).
In summary, our study shows that β-arrestin1 and β-arrestin2 are recruited to agonist-activated CXCR5 by a phospho-site cluster located at the extreme distal CT. The binding of β-arrestins to CXCR5 is required only for receptor desensitization but not internalization or signaling. Therefore, the role of β-arrestins at CXCR5 has evolved to mediate only one of its canonical functions, which makes CXCR5 a potentially useful model GPCR to study the molecular mechanisms that drive the divergent functions of β-arrestins.
Acknowledgments
The author thank Asaka Inoue (Tohoku University, Sendai, Japan) for generously sharing knockout cell lines and Mikel Garcia-Marcos (Boston University) and Kirill Martemyanov (University of Florida Scripps) for kindly sharing the Nluc-EPAC-VV plasmid.
Data Availability
The authors declare that all the data supporting the findings of this study are contained within the paper and Supplemental Material.
Authorship Contributions
Participated in research design: Crecelius, Marchese.
Conducted experiments: Crecelius, Manz, Benzow.
Contributed new reagents or analytic tools: Crecelius, Marchese.
Performed data analysis: Crecelius, Manz, Benzow, Marchese.
Wrote or contributed to the writing of the manuscript: Crecelius, Marchese.
Footnotes
- Received May 6, 2024.
- Accepted October 7, 2024.
This work was supported by National Institutes of Health National Institute of General Medical Sciences [Grants R01GM143808, R01GM106727-09S1, and R35GM149253] (to A.M.).
The authors declare no competing interests.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AUC
- area under the curve
- BRET
- bioluminescence resonance energy transfer
- CT
- carboxyl-terminal tail
- CXCL13
- C-X-C motif chemokine ligand 13
- CXCR5
- C-X-C motif chemokine receptor 5
- DMEM
- Dulbecco’s minimum essential medium
- Dis
- distal
- FBS
- fetal bovine serum
- FSK
- forskolin
- GPCR
- G protein-coupled receptor
- GRK
- GPCR kinase
- HEK293
- human embryonic kidney cells 293
- Med
- medial
- PBS
- phosphate-buffered saline
- Prox
- proximal
- Ser
- serine
- TBS
- Tris-buffered saline
- TBS-T
- Tris-buffered saline with Tween
- Thr
- threonine
- WT
- wild-type
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}