


Abstract
Phosphorylation of G protein-coupled receptors (GPCRs) is a key event for cell signaling and regulation of receptor function. Previously, using tandem mass spectrometry, we identified two phosphorylation sites at the distal C-terminal tail of the chemokine receptor CXCR4, but were unable to determine which specific residues were phosphorylated. Here, we demonstrate that serines (Ser) 346 and/or 347 (Ser-346/7) of CXCR4 are phosphorylated upon stimulation with the agonist CXCL12 as well as a CXCR4 pepducin, ATI-2341. ATI-2341, a Gαiβγ heterotrimer–biased CXCR4 agonist, induced more robust phosphorylation of Ser-346/7 compared with CXCL12. Knockdown of G protein-coupled receptor kinase (GRK) 2, GRK3, or GRK6 reduced CXCL12-induced phosphorylation of Ser-346/7 with GRK3 knockdown having the strongest effect, while inhibition of the conventional protein kinase C (PKC) isoforms, particularly PKCα, reduced phosphorylation of Ser-346/7 induced by either CXCL12 or ATI-2341. The loss of GRK3- or PKC-mediated phosphorylation of Ser-346/7 impaired the recruitment of β-arrestin to CXCR4. We also found that a pseudo-substrate peptide inhibitor for PKCζ effectively inhibited CXCR4 phosphorylation and signaling, most likely by functioning as a nonspecific CXCR4 antagonist. Together, these studies demonstrate the role Ser-346/7 plays in arrestin recruitment and initiation of receptor desensitization and provide insight into the dysregulation of CXCR4 observed in patients with various forms of WHIM syndrome.
Introduction
The chemokine receptor CXCR4 is critical for embryonic germ cell development, hematopoiesis, and directed chemotaxis (Nagasawa et al., 1996; Doitsidou et al., 2002; Ara et al., 2003) and is involved in the progression of a number of diseases. For example, CXCR4 acts as a coreceptor for T-cell tropic isolates of human immunodeficiency virus (Feng et al., 1996); mutations that either truncate or mutate the C-terminal tail of CXCR4 lead to a rare immunodeficiency characterized by warts, hypogammaglobulinemia, recurrent bacterial infection, and myelokathexis known as WHIM syndrome (Diaz and Gulino, 2005; Liu et al., 2012) and dysregulation of CXCR4 can lead to cancer progression and metastases (Müller et al., 2001; Busillo and Benovic, 2007).
Upon activation, G protein-coupled receptors (GPCRs) are rapidly phosphorylated, typically by members of the GPCR kinase (GRK) family (Krupnick and Benovic, 1998; Pitcher et al., 1998). This triggers the recruitment of arrestins, which prevents further activation of their cognate G protein and leads to receptor desensitization, internalization, and initiation of arrestin-dependent signaling (DeWire et al., 2007; Moore et al., 2007). Recent studies have shown that GPCRs can be phosphorylated by multiple kinases and that individual kinases are often able to phosphorylate multiple sites (Tobin et al., 2008; Busillo et al., 2010; Doll et al., 2011; Nobles et al., 2011). The C-terminal tail of CXCR4 contains 18 potential phosphorylation sites; 15 serines and three threonines. The agonist CXCL12 was previously shown to promote phosphorylation of serine (Ser)-339 in CXCR4 while phorbol esters and epidermal growth factor stimulation also led to phosphorylation of Ser-339, suggesting that it was a substrate for protein kinase C (PKC) (Woerner et al., 2005). Using a combination of liquid chromatography–tandem mass spectrometry and phospho-specific antibodies, we identified seven serine residues that were phosphorylated following CXCL12 stimulation: Ser-321, Ser-324, Ser-325, Ser-330, Ser-339, a residue between Ser-346 and Ser-348, and either Ser-351 or Ser-352 (Busillo et al., 2010). Moreover, we showed that Ser-330 and Ser-339 were phosphorylated by GRK6, while serines 324 and/or 325 (Ser-324/5) were primarily phosphorylated by PKC and GRK6 (Busillo et al., 2010). Recently, Ser346 and/or 347 (Ser-346/7) was shown to be rapidly phosphorylated by GRK2 and GRK3 following CXCL12 stimulation (Mueller et al., 2013). Moreover, phosphorylation of Ser-346/7 may be required for phosphorylation of Ser-324/5 and Ser-330 (Mueller et al., 2013). In addition to the studies identifying specific phosphorylation sites, several studies have implicated GRK2 (Orsini et al., 1999; Jiménez-Sainz et al., 2006; Busillo et al., 2010), GRK3 (Diaz and Gulino, 2005; Balabanian et al., 2008; Busillo et al., 2010; Liu et al., 2012; Tarrant et al., 2013), GRK6 (Fong et al., 2002; McCormick et al., 2009), and PKC (Signoret et al., 1997; Orsini et al., 1999) as being involved in regulating CXCR4 activity and signaling.
Multiple kinases play a critical role in phosphorylating and/or regulating CXCR4 following activation. While there are some data that suggest specific roles for GRKs as mentioned previously, the PKC isoforms that are involved in the phosphorylation and regulation of CXCR4 are still unclear. Furthermore, how site-specific phosphorylation is linked with the various downstream signaling pathways and cellular functions of CXCR4 still needs to be better defined. Agonist-promoted phosphorylation of GPCRs initiates the recruitment and high-affinity binding of arrestins (Shenoy and Lefkowitz, 2005). In this regard, deletion of GRK6 and β-arrestin2 enhances CXCR4 function in mice (Fong et al., 2002), while knockdown of GRK6 and β-arrestin2 significantly enhances calcium mobilization following CXCR4 activation (Busillo et al., 2010). It has also been shown that there is impaired recruitment of GRK6 and β-arrestin2 to a mutant CXCR4 that is associated with WHIM syndrome (McCormick et al., 2009), while overexpression of β-arrestin2 was unable to rescue internalization of CXCR4 lacking the last 15 amino acids (Balabanian et al., 2008). On the other hand, loss of GRK3, GRK6, or β-arrestin1 reduces extracellular signal-regulated kinase activation (Busillo et al., 2010), suggesting that receptor phosphorylation can also play a positive role in signal transduction following activation of CXCR4.
Together, these data suggest that GRK- and PKC-mediated phosphorylation of CXCR4 results in the differential recruitment of arrestins to promote desensitization and G protein-independent signaling (Fong et al., 2002; Busillo et al., 2010; Doll et al., 2011). Previously, we were unable to determine the kinase(s) that mediated phosphorylation of the distal tail of CXCR4 (Busillo et al., 2010). In this study, we sought to determine the kinase(s) responsible for phosphorylation of Ser-346 to Ser-348 and how site-specific phosphorylation of these residues affects the recruitment of β-arrestin. We show that GRK3- and PKC-mediated phosphorylation of Ser-346/7 contributes to the recruitment of β-arrestin2.
Materials and Methods
Materials.
HEK293 cells were obtained from Microbix Biosystems, Inc. (Toronto) and Lipofectamine 2000 and Opti-MEM were obtained from Invitrogen (Carlsbad, CA). Anti-GRK3 polyclonal rabbit antibodies and anti-PKCα and PKCβ monoclonal mouse antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-CXCR4 monoclonal antibodies and anti-β-arrestin monoclonal antibodies were obtained from BD Biosciences Pharmingen (San Diego, CA). Anti-GRK4-6 and anti-GRK2/3 monoclonal antibodies were obtained from Millipore (Billerica, MA). GRK2 monoclonal and anti-CXCR4 phospho-serine 324/5 (pS324/5) antibodies were produced in our laboratory (Busillo et al., 2010), while anti-CXCR4 pS346/7 polyclonal antibody was previously reported and validated using a CXCR4-S346-348A mutant (Mueller et al., 2013). Anti-flag-tagged monoclonal and polyclonal antibodies and anti-α-tubulin monoclonal antibodies were obtained from Sigma-Aldrich (St. Louis, MO). Anti-PKCζ polyclonal rabbit antibodies were obtained from Cell Signaling Technology (Danvers, MA). The CXCR4 pepducin ATI-2341 was obtained from Anchor Therapeutics (Boston, MA) or was synthesized by Peptide 2.0 Inc. (Chantilly, VA). On-TARGETplus short interfering RNA (siRNA) reagents were obtained from GE Dharmacon (Lafayette, CO).
Cell Culture and Transfection.
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 25 mM HEPES, pH 7.2, and 0.1 mM nonessential amino acids in a 5% CO2 incubator at 37°C. Cells stably expressing human CXCR4 were selected and maintained in complete DMEM supplemented with 0.8 mg/ml G418 and penicillin/streptomycin. For transfection of DNA or siRNAs, HEK293 cells were grown to ∼70% confluency in 100-mm dishes and transfected with the indicated amount of DNA or 600 pmol of ON-TARGETplus siRNA using Lipofectamine 2000 in Opti-MEM, and 24 hours later cells were split into 6-well plates for assay the next day.
Detection of CXCR4 Phosphorylation in HEK293 Cells Stably Expressing Flag-Tagged CXCR4 by Immunoblotting.
Cells were plated into 12-well plates and cultured for 24 hours. Cells were then starved with serum-free DMEM for 4 hours prior to stimulation. Cells were then washed twice with ice-cold phosphate-buffered saline (PBS) on ice to stop the reaction, and lysed with buffer (20 mM HEPES, pH 7.2, 10 mM EDTA, 150 mM NaCl, 1% Triton X-100, one tablet each of mini-complete protease inhibitor and PhosSTOP phosphatase inhibitor (Roche, Indianapolis, IN) per 10 ml) at 4°C on a rocker for 30 minutes. The lysates were cleared by centrifugation at 14,000 rpm in an Eppendorf centrifuge for 20 minutes at 4°C. For assays with kinase inhibitors, cells were pretreated with the indicated inhibitor or vehicle for 20 minutes prior to stimulation. Total protein in cell lysates was measured with Bio-Rad Protein Assay (Hercules, CA). An equal amount of total protein was electrophoresed on a 10% SDS polyacrylamide gel, transferred to nitrocellulose, and immunoblotted with the following primary antibodies overnight at 4°C: monoclonal anti-CXCR4 (1:1000), polyclonal anti-pS346/7-CXCR4 (1:500), polyclonal anti-pS324/5-CXCR4 (1:1000), polyclonal anti-GRK3 (1:500), monoclonal anti-GRK4-6 (1:3000), monoclonal anti-PKCα (1:1000), monoclonal anti-PKCβ2 (1:200), polyclonal anti-PKCζ (1:1000), or monoclonal anti-β-arrestin1 (1:1000, detects both β-arrestin1 and 2). Blots were washed extensively with Tris-buffered saline plus 0.05% Tween 20, incubated with a horseradish peroxidase–labeled secondary antibody, washed with Tris-buffered saline plus 0.05% Tween-20, and then proteins were detected using chemiluminescence. The blots were then stripped and reprobed using an antitubulin (1:7500) monoclonal antibody. Western blots were visualized and quantified using a C-DiGit Blot Scanner and Image Studio Software (LI-COR, Lincoln, NE).
Bioluminescence Resonance Energy Transfer (BRET) Assay.
For the BRET studies, 24 hours before transfection the HEK293 cells were split into 6-well plates. Transient transfections were performed using Lipofectamine 2000. CXCR4-RLucII (10 ng) was cotransfected with GFP10-β-arrestin2 (100 ng). The total amount of DNA transfected in each well was adjusted to 2 μg with pcDNA3. After overnight incubation, transfected cells were detached and seeded into a 96-well white plate (50,000 cells/well) that was pretreated with poly-L-ornithine hydrobromide (Sigma-Aldrich). The cells were then incubated with phenol red–free complete DMEM for an additional 24 hours before being tested. For the BRET measurements, cells were washed once with 200 μl of PBS plus glucose (Life Technologies, Grand Island, NY) and Deep Blue C (coelenterazine 400A) was added (final concentration of 2.5 μM in PBS) 5 minutes before reading. Readings were collected using an Infinite F500 multidetector plate reader (Tecan, Mannedorf, Switzerland). The BRET signal was determined by calculating the ratio of the light intensity emitted by the GFP10-β-arrestin2 over the light intensity emitted by CXCR4-RLucII. The values were corrected by subtracting the background BRET signal detected when the CXCR4-RLucII construct was expressed alone. CXCL12 was added just before reading the BRET signal.
Measurement of Intracellular Calcium Mobilization.
Calcium mobilization was performed as previously described (Iwata et al., 2005) with slight modification. In brief, HEK293 cells that stably express CXCR4 were harvested, washed twice with PBS, and resuspended at 5 × 106 cells/ml in Hanks’ balanced salt solution (140 mM NaCl, 5 mM KCl, 10 mM HEPES, pH 7.4, 1 mM CaCl2, 1 mM MgCl2, and 1 mg/ml glucose) (Invitrogen) containing 0.025% Hanks’ bovine serum albumin. The cells were then loaded with 2 μM Fura-2 acetoxymethyl ester derivative (Fura-2/AM; Invitrogen) for 30 minutes at 37°C. The cells were washed once in Hanks’ solution, resuspended in Hanks’ bovine serum albumin, incubated at room temperature for 15 minutes, and washed twice in Hanks’ solution. The cells were then resuspended in Hanks’ solution at a concentration of 3 × 107 cells/ml. To measure calcium mobilization, 1.5 × 106 cells/1.6 ml in a quartz cuvette was preincubated with the designated concentrations of PKCζ inhibitor for 1 minute and then stimulated with the indicated agonists. Calcium mobilization was measured using excitation at 340 and 380 nm and emission at 510 nm in a fluorescence spectrometer (LS55; PerkinElmer Life and Analytical Sciences, Waltham, MA). Calibration was performed using 0.1% Triton X-100 for total fluorophore release and 15 mM EGTA to chelate free calcium. Intracellular calcium concentrations were calculated using a fluorescence spectrometer measurement program.
Statistics.
Western blots were quantified on a LI-COR C-DiGit scanner. Data are shown as the mean ± S.E.M. with P values determined by comparing the data from experimental versus control from at least three independent experiments using a two-tailed paired t test in Excel. Where noted, n equals the number of independent experiments that were performed.
Results
Phosphorylation of Ser-346/7 Following CXCL12 Stimulation.
Phosphorylation of GPCRs is a key regulatory event following agonist activation. The C-terminal tail of CXCR4 (Fig. 1A) contains 15 serine and three threonine residues, and at least seven residues are phosphorylated following receptor activation (Busillo et al., 2010). Truncation and alanine scanning mutagenesis indicated that a number of potential phosphorylation sites are important for receptor desensitization, internalization, and degradation (Orsini et al., 1999; Marchese and Benovic, 2001). Our previous mass spectrometry data suggested that one serine residue between Ser-346 and Ser-348 was phosphorylated following CXCL12 stimulation (Busillo et al., 2010), while recent data suggested that Ser-346/7 was rapidly phosphorylated following CXCL12 stimulation (Mueller et al., 2013). Therefore, we sought to further characterize the CXCL12-promoted phosphorylation of CXCR4 in this cluster of serine residues (Ser-346 to Ser-348) in cells that stably express FLAG-tagged CXCR4. HEK293 cells stably expressing CXCR4 were stimulated with 50 nM CXCL12 for various times up to 60 minutes, and cell lysates were probed with pS346/7 antibodies to assess the kinetics of phosphorylation (Fig. 1B). Phosphorylation of Ser-346/7 was detected as early as 2 minutes, peaked by 5 minutes, and was returning to baseline levels by 60 minutes (Fig. 1B). These data demonstrate that Ser-346/7 is rapidly phosphorylated following receptor activation by CXCL12, and suggest that phosphorylation of these residues likely has an important function in receptor regulation.

GRKs and PKC mediate phosphorylation of CXCR4 at Ser-346/7 following stimulation with CXCL12. (A) Sequence of CXCR4 C-terminus with Ser-324/5 and Ser-346/7 highlighted in red. (B) HEK293 cells stably expressing CXCR4 were incubated in serum-free medium for 4 hours, and then stimulated with 50 nM CXCL12 for the indicated times. Cells were washed twice with cold PBS and lysed. Phosphorylation of CXCR4 Ser-346/7 was detected by western blot using a phospho-specific antibody (pS346/7). Shown is a representative western blot using pS346/7 and tubulin antibodies from at least three independent experiments. (C) Shown is a representative western blot from at least three experiments where endogenous GRKs were knocked down using ON-TARGETplus GRK siRNAs in HEK293 cells stably expressing CXCR4 for 48 hours. (D) HEK293 cells stably expressing CXCR4 were stimulated with CXCL12 (5 nM) following knockdown of endogenous GRK2, GRK3, or GRK6 and the phosphorylation of CXCR4 at Ser-346/7 was detected by western blot. Shown is a representative blot (upper panel) and summary of the 5-minute time point (lower panel) from three independent experiments (**P < 0.01 compared with control siRNA); arbitrary units (AU). (E) HEK293 cells stably expressing CXCR4 were incubated in serum-free medium for 4 hours. Bis I (2.5 μM) was added to the cells 30 minutes prior to being stimulated with CXCL12. The phosphorylation of CXCR4 Ser-346/7 was detected by western blot. Shown is a representative blot (upper panel) and summary (lower panel) from three independent experiments (*P < 0.05; **P < 0.01 compared with control); AU. (F) GRK3 was knocked down in HEK293 cells stably expressing CXCR4 while Bis I (2.5 μM) was added 30 minutes prior to stimulation with CXCL12 (50 nM). Shown is a representative blot (upper panel) and summary from three independent experiments (**P < 0.01 compared with Bis I alone, ***P < 0.001 compared with control, +P < 0.01 compared with siGRK3 alone).
Serine 346/7 Is Phosphorylated by GRKs and PKC.
The distal tail of CXCR4 has been shown to be critical for arrestin recruitment following CXCR4 activation (Balabanian et al., 2008; McCormick et al., 2009; Busillo et al., 2010). Our previous results suggested that either GRK2 or GRK3 could be responsible for phosphorylation of the distal tail of CXCR4 (Busillo et al., 2010). Indeed, a recent report suggested that Ser-346/7 is phosphorylated by GRK2/3, although siRNA-mediated knockdown of GRK2/3 did not completely eliminate the phosphorylation (Mueller et al., 2013). Therefore, we wanted to determine the contribution of other kinases in Ser-346/7 phosphorylation following CXCL12 stimulation. Endogenous GRK2, GRK3, and GRK6 in HEK293 cells stably transfected with FLAG-CXCR4 were knocked down using GRK-selective siRNAs (Fig. 1C), and Ser-346/7 phosphorylation triggered by the agonist CXCL12 was detected using a site-specific phospho-antibody. To enhance our ability to inhibit agonist-activated phosphorylation, cells were stimulated with a relatively low concentration of CXCL12 (5 nM). Phosphorylation of Ser-346/7 was reduced by ∼50% at 5 minutes following knockdown of GRK3 compared with control siRNA (P < 0.01), while knockdown of GRK2 or GRK6 had lesser effects (Fig. 1D). Knocking down GRK2 and GRK3 together did not further decrease phosphorylation of Ser-346/7 (Fig. 1D). In contrast, phosphorylation of Ser-346/7 following knockdown of GRK2, GRK3, or GRK6 was not significantly reduced at 15 minutes (Fig. 1D). These results suggest that while GRK2, GRK3, and GRK6 may be involved in the phosphorylation of Ser-346/7 following stimulation with CXCL12, GRK3 is the primary GRK involved in HEK293 cells.
The phosphorylation of GPCRs is often mediated by multiple kinases (Tobin et al., 2008). Indeed, our data indicate that several GRKs are involved in the phosphorylation of Ser-346/7. However, agonist-promoted phosphorylation is still evident following GRK knockdown, suggesting that other kinases might also be involved (Fig. 1D). PKCs are ubiquitously expressed in mammalian cells and phosphorylate many proteins, including GPCRs (Newton, 1995; Orsini et al., 1999). Using a broad spectrum PKC inhibitor [bisindolylmaleimide I (Bis I)], we previously found that multiple PKCs contributed to phosphorylation of Ser-324/5 (Busillo et al., 2010). To determine if PKC isoforms play a role in the phosphorylation of Ser-346/7, HEK293 cells stably expressing FLAG-CXCR4 were pretreated with Bis I (2.5 µM) for 30 minutes prior to stimulation with CXCL12. Inhibition of PKC significantly reduced the basal phosphorylation of Ser-346/7 as well as CXCL12-promoted phosphorylation at 5 and 30 minutes (Fig. 1E). Thus, our results suggest that both GRK3 (Fig. 1D) and PKC (Fig. 1E) contribute to phosphorylation of Ser-346/7. We next evaluated the combined effect of GRK3 knockdown and PKC inhibition on Ser-346/7 phosphorylation. GRK3 knockdown or inhibition of PKC each reduced phosphorylation of Ser-346/7 by ∼60% at 5 minutes, while combined knockdown of GRK3 and PKC inhibition reduced CXCL12-promoted phosphorylation of Ser-346/7 by >80% at 5 minutes (Fig. 1F). These data demonstrate that both GRK3 and PKC contribute to Ser-346/7 phosphorylation following stimulation with CXCL12.
The Pepducin ATI-2341 Promotes Phosphorylation of Ser346/7.
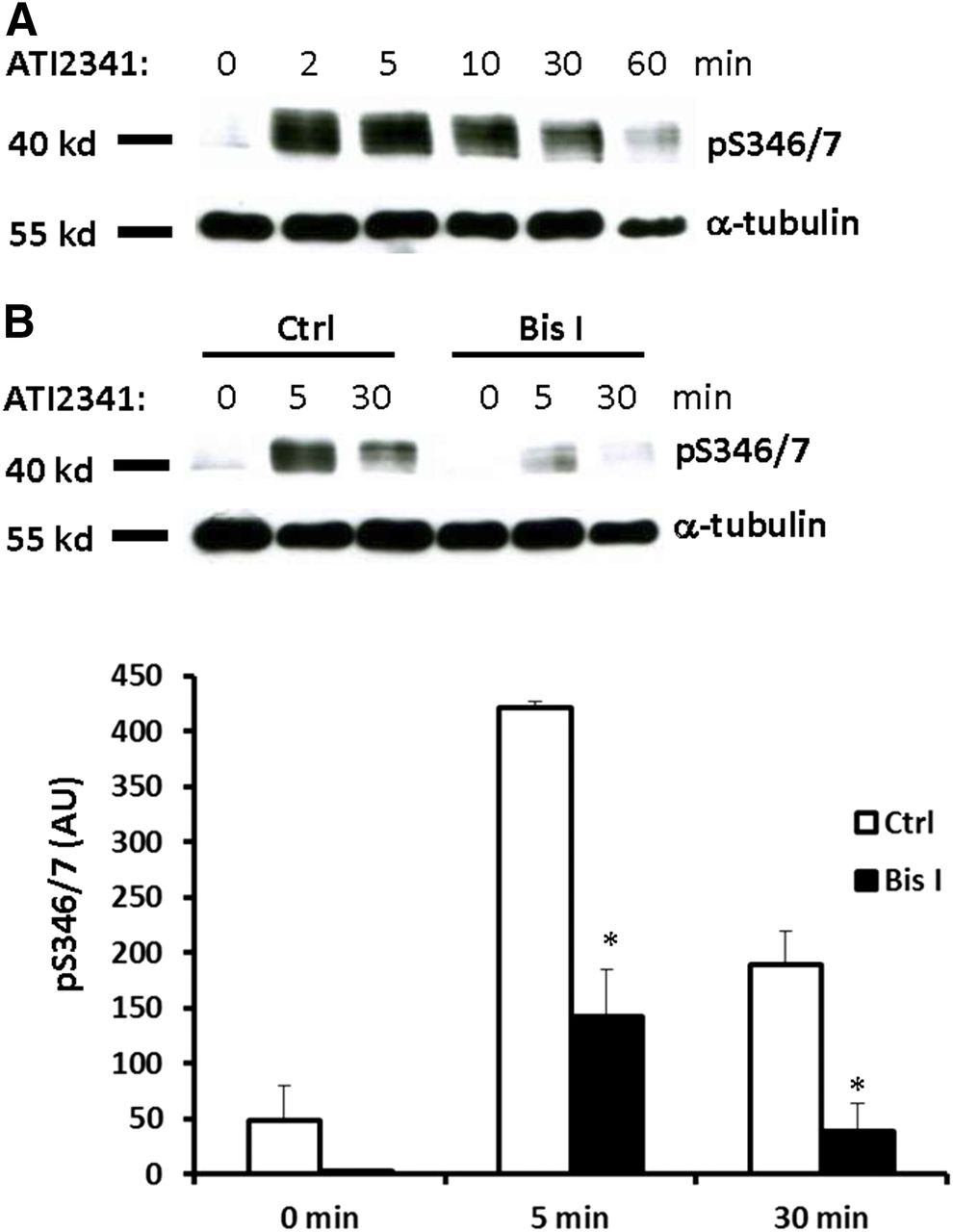
Pepducins are a class of molecules that are derived from the intracellular loops of GPCRs (Carr and Benovic, 2016). ATI-2341 is a pepducin derived from intracellular loop 1 of CXCR4 that functions as a Gαiβγ heterotrimer–biased agonist to promote G protein-dependent signaling downstream from CXCR4 (Tchernychev et al., 2010; Quoyer et al., 2013). Furthermore, ATI-2341 has been shown to promote the phosphorylation of Ser-324/5, which is dependent on PKC but not GRK6 (Quoyer et al., 2013), and also induce CXCR4 internalization (Tchernychev et al., 2010). To determine if ATI-2341 can also promote the phosphorylation of Ser-346/7, HEK293 cells stably expressing CXCR4 were stimulated with ATI-2341 (3 µM) and phosphorylation of Ser-346/7 was detected. Surprisingly, ATI-2341 induced more robust and faster phosphorylation of Ser-346/7 than CXCL12 (compare Figs. 2A and 1B). PKC inhibition reduced ATI-2341-induced phosphorylation of Ser-346/7 by >60% (Fig. 2B). This provides further evidence that PKC plays a central role in the phosphorylation of Ser-346/7 at the distal portion of the CXCR4 C-terminal tail. Although Ser-346/7 does not fall within a putative consensus sequence for PKC, our data indicate that PKC plays an important role in CXCR4 phosphorylation at this site.
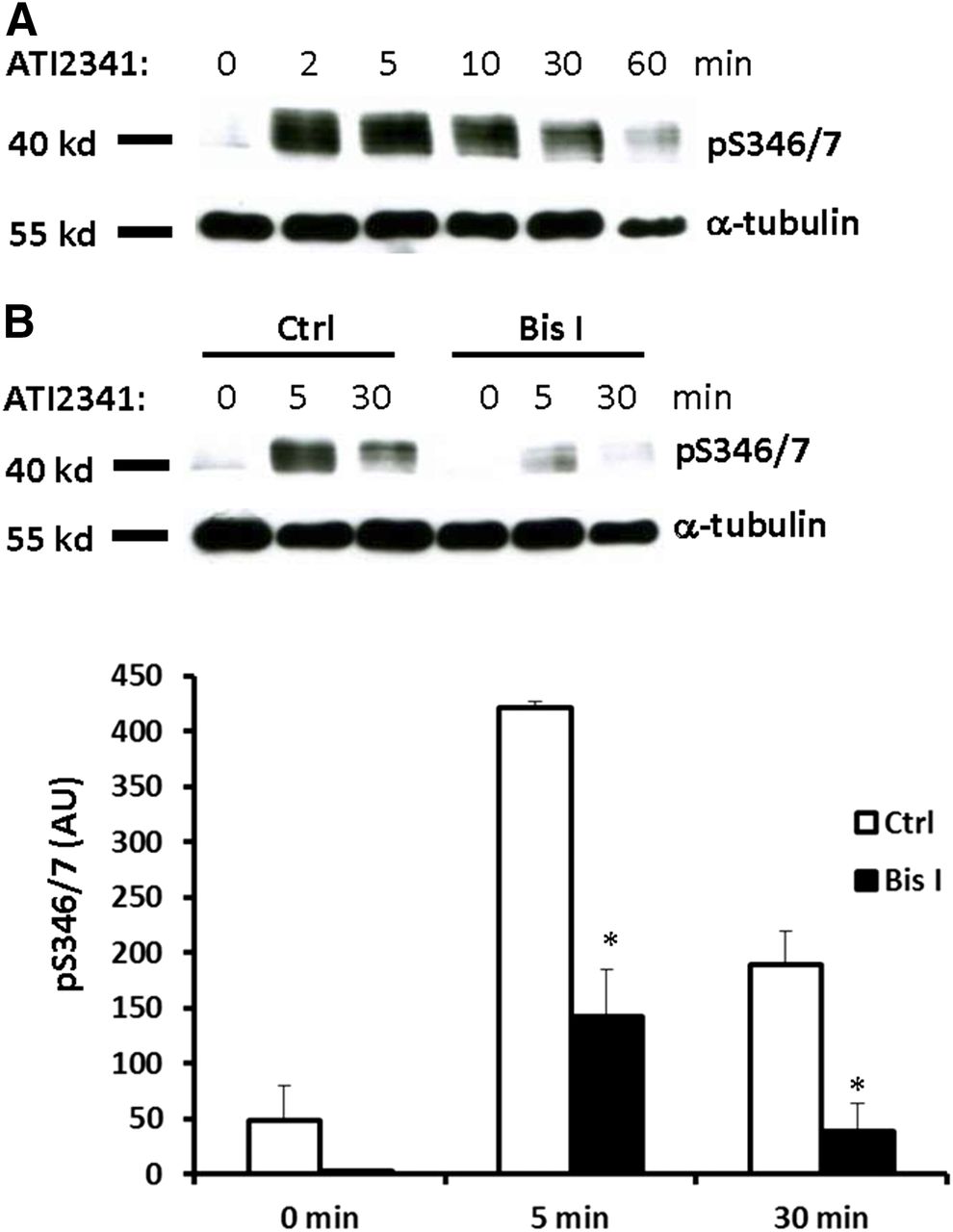
The Pepducin ATI-2341 induces phosphorylation of CXCR4 at Ser-346/7 in a PKC-dependent manner. (A) HEK293 cells stably expressing CXCR4 were starved with serum-free medium for 4 hours, and then stimulated with 3 μM ATI-2341 for the indicated times. Whole cell lysates were probed with the pS346/7 antibody. Shown is a representative blot from three independent experiments. (B) HEK293 cells stably expressing CXCR4 were pretreated with Bis I (2.5 μM) for 30 minutes, and then stimulated with ATI-2341 (3 μM). Phosphorylation of CXCR4 was detected with the pS346/7 antibody. Shown is a representative blot (upper panel) and summary (lower panel) from four independent experiments (*P < 0.05 compared with control); arbitrary units (AU).
Ser-346/7 Is Not Phosphorylated by Casein Kinase (CK) 1, CK2, or Glycogen Synthase Kinase 3 (GSK3).
Ser-346/7 falls within a putative consensus site for phosphorylation by CK1, CK2, and GSK3 (Pinna and Ruzzene, 1996; Tobin, 2002). Therefore, we also evaluated if inhibition of CK1, CK2, and GSK3 affected phosphorylation of Ser-346/7. HEK293 cells stably expressing CXCR4 were preincubated with the kinase inhibitors Bis I (PKC inhibitor; 2.5 µM), D4476 (CK1 inhibitor; 20 µM), 4,5,6,7-tetrabromobenzotriazole (CK2 inhibitor; 20 µM), or CT99021 (GSK3 inhibitor; 10 µM) for 20 minutes, and then either directly lysed (for basal phosphorylation) or stimulated with 50 nM CXCL12 for 5 minutes. We previously found that Ser-324/5 is phosphorylated by PKC (Busillo et al., 2010), which falls within a PKC consensus site; therefore, blots were also probed for pS324/5. Inhibition of PKC effectively reduced basal and CXCL12-promoted phosphorylation of both Ser-346/7 and Ser-324/5 (Fig. 3, A, B, D–G). In contrast, inhibition of CK1, CK2, or GSK3 had no effect on basal phosphorylation (Fig. 3, A, D, and E) or CXCL12-promoted phosphorylation (Fig. 3, B, F, and G) at either Ser-346/7 or Ser-324/5. While these results suggest that CK1, CK2, and GSK3 do not contribute to CXCR4 phosphorylation of Ser-324/5 or Ser-346/7, we did not demonstrate that the inhibitor treatments effectively inhibited the specific kinases. Thus, while we used inhibitor concentrations that have proven effective in previous studies (Pinna and Ruzzene, 1996; Tobin, 2002), we cannot completely rule out a potential role for these kinases in CXCR4 phosphorylation.

Phosphorylation of CXCR4 at Ser-346/7 and Ser-324/5 is regulated by PKC and not CK1, CK2, or GSK3β. HEK293 cells stably expressing CXCR4 were preincubated with Bis I (pan-PKC inhibitor; 2.5 μM), D4476 (CK1 inhibitor; 20 μM), 4,5,6,7-tetrabromobenzotriazole (TBB) (CK2 inhibitor; 20 μM), or CT99021 (GSK3 inhibitor; 10 μM) for 20 minutes. Cells were either untreated (basal phosphorylation) or stimulated with CXCL12 (50 nM) for 5 minutes or PMA (200 nM) for 10 minutes. (A) Shown are representative blots from two independent experiments for basal and (B) three experiments for CXCL12-promoted phosphorylation of CXCR4. (C) Shown is a representative blot from three independent experiments for PMA-promoted phosphorylation of CXCR4. (D–I) Shown are summaries from two to three independent experiments; arbitrary units (AU).
To further characterize the role PKC plays in phosphorylating Ser-346/7, we determined if directly activating PKC with phorbol 12-myristate 13-acetate (PMA) resulted in phosphorylation of Ser-346/7. As shown in Fig. 3C, PMA resulted in robust phosphorylation of both Ser-346/7 and Ser-324/5 at 10 minutes. As expected, inhibition of PKC with Bis I effectively blocked PMA-promoted phosphorylation of Ser346/7 and Ser-324/5, while inhibition of CK1, CK2, or GSK3 had no effect (Fig. 3, C, H, and I). Together, these data provide further evidence for a role for PKC in phosphorylation of Ser-346/7 following activation of CXCR4.
Identification of PKC Isoforms that Phosphorylate Ser-346/7.
The PKC family contains 10 isoforms that are classified as conventional, novel, and atypical according to their regulatory domains (Reyland, 2009). Therefore, we wanted to differentiate the PKC isoforms responsible for phosphorylation of Ser-346/7. Since Bis I broadly inhibits the conventional (PKCα/β/γ) and novel (PKCδ/ε/θ/η) PKC isoforms (Wu-Zhang and Newton, 2013), we attempted to narrow down the isoform responsible for phosphorylation of Ser-346/7 by pretreating cells with Gö 6976 (to inhibit the conventional isoforms PKCα and β), CGX1037 (to inhibit the novel isoform PKCδ), and myristolated PKCζ pseudosubstrate peptide (to inhibit the atypical isoform PKCζ). We also assessed the effect of phosphatidylinositol 3-kinase inhibition with wortmannin. Gö 6976 reduced the basal phosphorylation of Ser-346/7 by 60% and Ser-324/5 by 70% and was comparable to the effects seen with Bis I (Fig. 4, A–C). In contrast, inhibition of PKCδ, PKCζ, or phosphatidylinositol 3-kinase had no effect on basal phosphorylation of either Ser-346/7 or Ser-324/5 (Fig. 4, A–C).

Identification of the PKC isoform that regulates basal phosphorylation of CXCR4 Ser-346/7 and Ser-324/5. HEK293 cells stably expressing CXCR4 were pretreated with Bis I (pan-PKC inhibitor; 2.5 μM), Gö 6976 (PKCα and PKCβ inhibitor; 1 μM), CGX1037 (PKCδ inhibitor; 2.5 μM), PKCζ pseudo-substrate (PKCζ inhibitor; 10 μM), or wortmannin (phosphatidylinositol 3-kinase inhibitor; 100 nM) for 20 minutes. Cells were then lysed and whole cell lysates were probed with either pS346/7 or pS324/5 antibodies. (A) Shown is a representative blot from three individual experiments. Quantification of pS346/7 (B) and pS324/5 (C) from three independent experiments (**P < 0.01 compared with medium alone); arbitrary units (AU).
Next, we examined the effect of these inhibitors on CXCL12- and ATI-2341-induced phosphorylation of Ser-346/7 and Ser-324/5. Inhibition of PKCα/β (Gö 6976) reduced CXCL12-promoted phosphorylation of Ser-346/7 (Fig. 5, A and C) and Ser-324/5 (Fig. 5, A and E) at 5 minutes; however, it was not as effective as Bis I (Fig. 5, A, C, and E). Interestingly, whereas inhibition of PKCζ did not affect the basal phosphorylation of CXCR4 (Fig. 4), CXCL12-promoted phosphorylation of Ser-346/7 and Ser-324/5 was significantly reduced by inhibition of PKCζ (Fig. 5, A, C, and E). Moreover, the inhibition of PKCζ reduced phosphorylation of Ser-324/5 to a similar extent as Bis I (Fig. 5, A and E). Similar results were obtained when ATI-2341-promoted phosphorylation of CXCR4 at 5 minutes was examined (Fig. 5, B, D, and F), although inhibition of PKCζ was more effective at reducing phosphorylation of Ser-346/7 following treatment with ATI-2341 (Fig. 5, B and D). Inhibition of PKCδ resulted in ∼20% reduction of ATI-2341-induced phosphorylation of Ser346/7 but had no effect on phosphorylation of Ser324/5 (Fig. 5). The effective reduction in CXCR4 phosphorylation following stimulation with ATI-2341 is in line with the observation that ATI-2341 triggers PKC-dependent phosphorylation of CXCR4 (Quoyer et al., 2013).

Phosphorylation of Ser346/7 and Ser324/5 is mediated by PKC following stimulation with either CXCL12 or ATI-2341. HEK293 cells stably expressing CXCR4 were preincubated with Bis I (2.5 μM), Gö 6976 (1 μM), CGX1037 (2.5 μM), PKCζ pseudo-substrate (10 μM), or wortmannin (100 nM) for 20 minutes. Cells were then stimulated with 50 nM CXCL12 (A) or 3 μM ATI-2341 (B) for 5 minutes and whole cell lysates were probed for either pS346/7 or pS324/5 by western blot. Quantification of pS346/7 following either CXCL12 (C) (n = 4) or ATI-2341 (D) (n = 5) stimulation (*P < 0.05 and **P < 0.01 compared with vehicle); arbitrary units (AU). Quantification of pS324/5 following either CXCL12 (E) (n = 4) or ATI-2341 (F) (n = 5) stimulation (*P < 0.05, **P < 0.01, and ***P < 0.001 compared with vehicle); arbitrary units (AU). The measurement of CXCR4 phosphorylation at Ser-346/7 involved detection of all bands while Ser-324/5 involved detection of just the upper band.
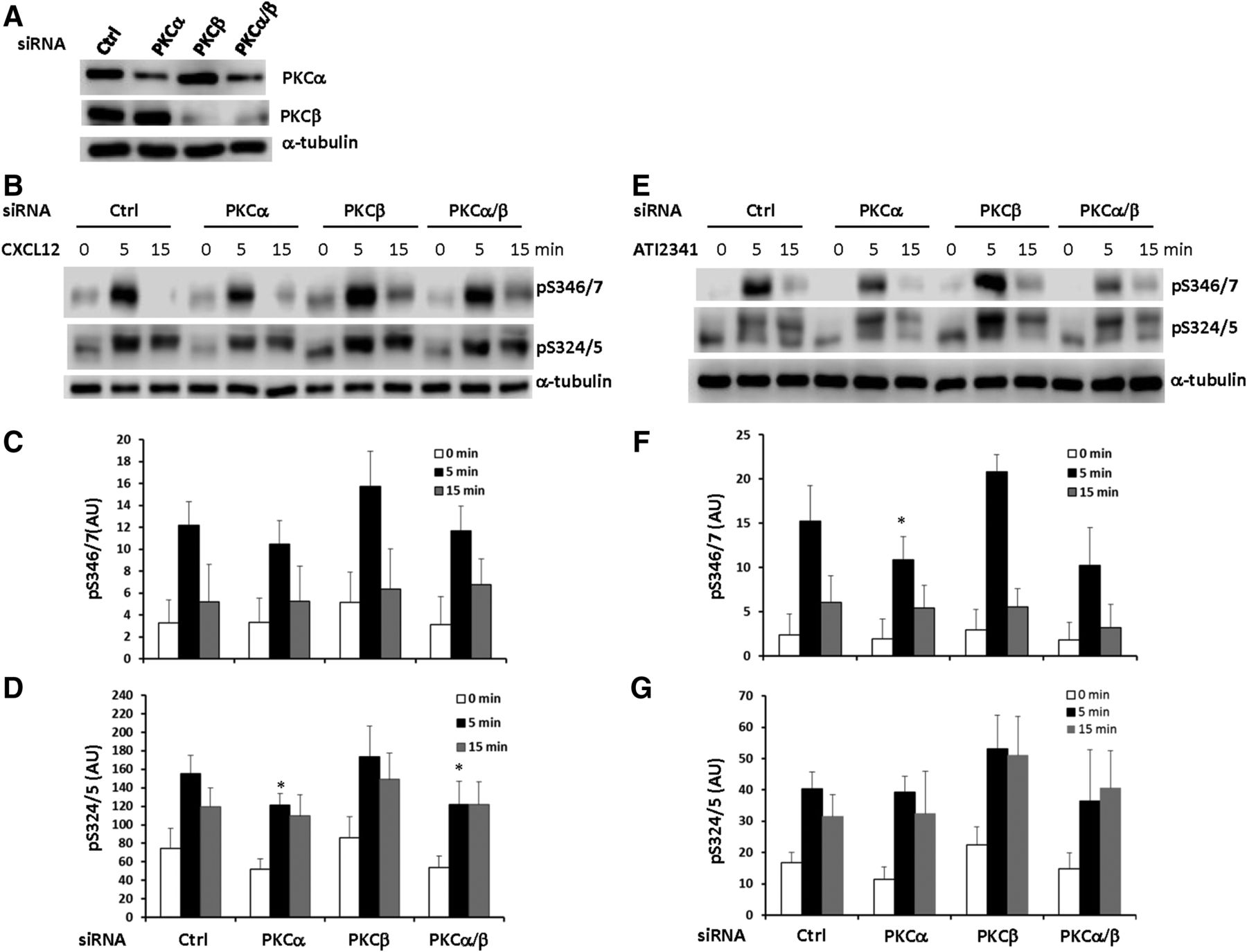
Since the PKCα/β-specific inhibitor Gö 6976 was fairly effective in inhibiting CXCR4 phosphorylation (Fig. 5), we tried to further define which PKC isoform is involved in Ser-346/7 and Ser-324/5 phosphorylation using a knockdown approach. Endogenous PKCα and PKCβ in HEK293 cells stably expressing CXCR4 were knocked down using ON-TARGETplus siRNAs (Fig. 6A). PKCα was reduced ∼70% while PKCβ was decreased >90%. While there was no significant effect of PKCβ knockdown, there was some reduction of CXCL12- and ATI-2341-promoted phosphorylation of Ser-346/7 and Ser324/5 at 5 minutes in cells with PKCα knockdown, albeit only ∼20% (Fig. 6, B–G). The lack of a larger effect may reflect the incomplete knockdown of PKCα expression as well as potential contributions from other PKC isoforms. Thus, our data suggest that PKCα plays a role in agonist-promoted phosphorylation of CXCR4.

Role of conventional PKC in the phosphorylation of Ser-346/7 and Ser-324/5. (A) HEK293 cells stably expressing CXCR4 were transfected with ON-TARGETplus PKCα, PKCβ, or PKCα/β siRNA for 48 hours and endogenous PKCα and PKCβ expression was examined by western blotting (shown is a representative blot from four independent experiments). Following knockdown of PKCα and/or PKCβ, HEK293 cells were incubated with or without 50 nM CXCL12 (B) or 3 μM ATI-2341(E) for 5–15 minutes and whole cell lysates were probed for pS346/7 and pS324/5. Shown are representative western blots from four individual experiments. Quantification of relative pS346/7 and pS324/5 levels following stimulation with CXCL12 (C and D) or ATI-2341 (F and G) are summarized (*P < 0.05 compared with control); arbitrary units (AU).
Phosphorylation of CXCR4 by GRK3 or PKC Facilitates Recruitment of β-Arrestin2.
The recruitment of arrestins to agonist-activated and phosphorylated GPCRs is critical for receptor desensitization as well as to initiate arrestin-dependent signaling (DeWire et al., 2007; Moore et al., 2007). Previous results suggested that β-arrestin2 was involved in receptor desensitization, whereas β-arrestin1 was involved in signaling following CXCR4 activation (Busillo et al., 2010). CXCL12 stimulation results in the recruitment of β-arrestin2 (Quoyer et al., 2013), which is lost when the C-terminal tail is truncated (McCormick et al., 2009). Given the critical role the distal C-terminal tail plays in desensitization and internalization of CXCR4 (Orsini et al., 1999; Balabanian et al., 2008; McCormick et al., 2009), we hypothesized that the GRK- and PKC-mediated phosphorylation of Ser-346/7 might be involved in the recruitment of β-arrestin2. Therefore, we used BRET to investigate the interaction between CXCR4 and β-arrestin2 (Quoyer et al., 2013). The distal C-terminal tail of CXCR4 contains five serine residues (Ser-346/7/8 and Ser-351/2). Our mass spectrometry data suggested that there were two potential phosphorylation sites in these two serine clusters (Busillo et al., 2010); therefore, we mutated all five residues to alanine (CXCR4-5AT). HEK293 cells were cotransfected with GFP10-β-arrestin2 and either wild-type or 5AT mutant CXCR4-RLucII to investigate the influence of PKCζ inhibitor on the recruitment of β-arrestin2 to CXCR4 using BRET (Fig. 7A). In the absence of CXCL12, there was no change in the BRET signal for either wild-type or CXCR4-5AT and β-arrestin2 (Fig. 7B). Stimulation with CXCL12 induced the interaction between wild-type CXCR4 and β-arrestin2, which peaked at ∼12 minutes after the addition of CXCL12 (Fig. 7B). While CXCL12 promoted a change in the BRET signal between CXCR4-5AT and β-arrestin2, the BRET signal was ∼70% lower and the peak was slightly delayed compared with wild-type CXCR4 (Fig. 7B). Next, we knocked down GRK3 and determined the BRET signal between wild-type CXCR4 and β-arrestin2. Knockdown of GRK3 reduced the BRET signal by ∼20% (Fig. 7C). In addition, broadly inhibiting PKC with Bis I reduced the BRET signal by ∼15% (Fig. 7D). The combination of GRK3 knockdown and PKC inhibition did not have an additional effect (data not shown). Together, these data indicate that both GRK3- and PKC-mediated phosphorylation of Ser-346/7 are involved in β-arrestin2 recruitment. However, since the association between CXCR4 and β-arrestin2 was not completely lost when this cluster of serines was mutated to alanine, phosphorylation at other sites may also be involved in β-arrestin recruitment.

Phosphorylation of CXCR4 by GRK3 or PKC regulates the interaction of CXCR4 with β-arrestin2. (A) Schematic of β-arrestin recruitment by CXCR4 in a BRET assay. Upon agonist stimulation, GFP10-β-arrestin2 is recruited to CXCR4-RLucII and BRET signal is increased. (B) HEK293 cells were cotransfected with either wild-type or 5AT mutant CXCR4-RLucII with β-arrestin2-GFP10. The interaction between CXCR4 and β-arrestin2 was detected with or without stimulation of 100 nM CXCL12 in the BRET assay after 48-hour cotransfection. Data show a summary of seven individual experiments. (C) HEK293 cells were transfected with ON-TARGETplus GRK3 siRNA; 24 hours later cells were cotransfected with CXCR4-RLucII and β-arrestin2-GFP10. Cells were stimulated with 100 nM CXCL12 after 48-hour cotransfection and interaction between CXCR4 and β-arrestin2 was detected in the BRET assay. Data show a summary of six individual experiments. (D) HEK293 cells transiently cotransfected with CXCR4-RLucII and β-arrestin2-GFP10 were treated with or without 2.5 μM Bis I for 20 minutes. Cells were then stimulated with 100 nM CXCL12 and interaction between CXCR4 and β-arrestin2 was detected. Data show a summary of three individual experiments.
Given the significant effect inhibition of PKCζ had on CXCR4 phosphorylation (Fig. 5), we also wanted to explore if PKCζ-promoted phosphorylation of Ser-346/7 affected the recruitment of β-arrestin2. Interestingly, the PKCζ inhibitor reduced the BRET signal in a dose-dependent manner and completely blocked β-arrestin2 recruitment at the highest dose (10 μM) tested (Fig. 8A). In fact, inhibition of PKCζ was more efficient at blocking β-arrestin2 recruitment than blocking the conventional and novel PKC isoforms with Bis I (compare Figs. 8A and 7D). To determine if this was a nonspecific effect of the pseudo-substrate peptide inhibitor for PKCζ (Wu-Zhang et al., 2012), we also looked at the recruitment of β-arrestin2 to the β2-adrenergic receptor, which is GRK dependent (Violin et al., 2006). While inhibition of PKCζ blocked the association between CXCR4 and β-arrestin2, it had no effect on the recruitment of β-arrestin2 to the β2-adrenergic receptor (Fig. 8B). In addition, different concentrations of PKCζ inhibitor were also tested for inhibition of calcium flux activated by CXCL12 or ATI-2341. We found that 50 nM CXCL12 activated a modest calcium flux in CXCR4 stably expressing HEK293 cells, and preincubation with the PKCζ inhibitor effectively blocked calcium mobilization with an IC50 value of ∼2 μM (Fig. 8C). In comparison, 3 μM ATI-2341 induced a more robust calcium flux, and pretreatment of the cells with the PKCζ inhibitor effectively inhibited calcium signaling with an IC50 value of ∼0.5 μM (Fig. 8D). In contrast, pretreatment of HEK293 cells with the PKCζ inhibitor at concentrations as high as 5 μM had no effect on calcium flux activated by 100 μM carbachol through endogenous M3 muscarinic acetylcholine receptors (Fig. 8E). These data demonstrate that the PKCζ inhibitor can effectively and selectively block the function of CXCR4.
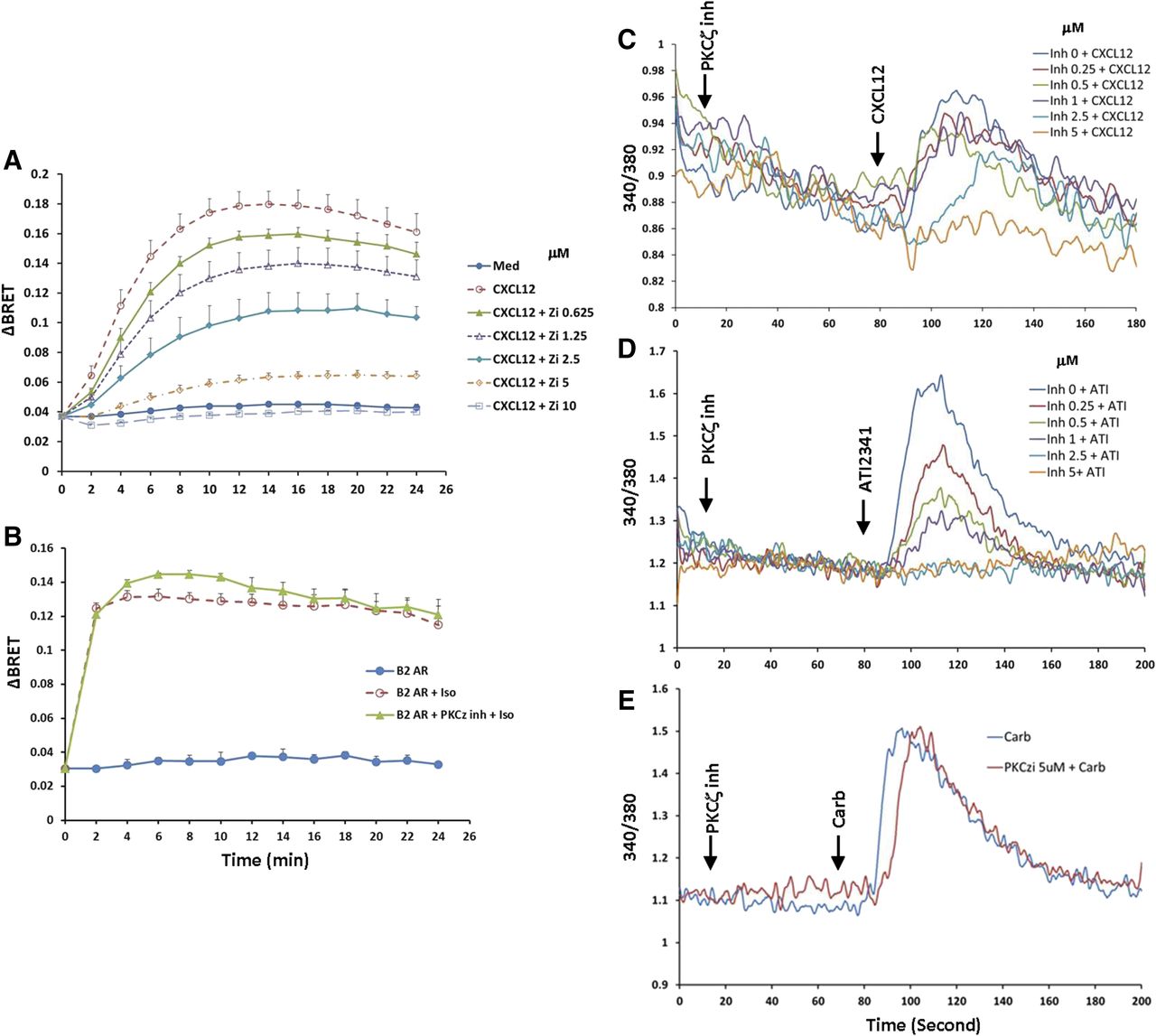
Inhibition of CXCR4 function by PKCζ myristoylated pseudosubstrate peptide inhibitor. (A) HEK293 cells transiently cotransfected with CXCR4-RLucII and β-arrestin2 GFP10 were treated with different concentrations of PKCζ inhibitor for 20 minutes, and then cells were incubated with 100 nM CXCL12 and BRET signal was detected. Data show a summary of five individual experiments. (B) HEK293 cells transiently cotransfected with β2-adrenergic receptor (β2AR)-RLucII and β-arrestin2-GFP10 were treated with or without 10 μM PKCζ inhibitor for 20 minutes. Cells were stimulated with 10 μM isoproterenol and the interaction between β2AR and β-arrestin2 was detected. Data show a summary of four individual assays. (C and D) Inhibition of calcium mobilization through CXCR4 induced by PKCζ inhibitor. Stable CXCR4 HEK293 cells loaded with 2 μM Fura2-AM were incubated with different concentrations of PKCζ inhibitor for 1 minute at 37°C, and then stimulated with either 50 nM CXCL12 (C) or 3 μM ATI-2341 (D), and calcium flux was detected. Thess data are representative of at least three individual experiments. (E) Effect of PKCζ inhibitor on calcium mobilization through the M3 muscarinic acetylcholine receptor. For comparison, a representative of at least three individual experiments using the stable CXCR4 HEK293 cells stimulated with 100 μM carbachol with or without preincubation with PKCζ inhibitor is shown.
Next, we used a siRNA approach to knock down PKCζ in HEK293 cells that stably express CXCR4 (Fig. 9A). In contrast to the PKCζ inhibitor, knockdown of endogenous PKCζ actually enhanced phosphorylation at Ser-346/7 and Ser-324/5, particularly at 15 minutes (Fig. 9B). PKCζ knockdown also led to modestly enhanced recruitment of β-arrestin2 following stimulation with 20 nM CXCL12 (Fig. 9C). Thus, it appears that PKCζ is not involved in CXCR4 phosphorylation and that the effects of the PKCζ inhibitor on CXCR4 function might involve a direct effect on the receptor. Together, these data show that the distal C-terminal tail of CXCR4 plays a critical role in β-arrestin2 recruitment following activation. Furthermore, the data suggest that GRK- and PKC-mediated phosphorylation of this region leads to the recruitment of β-arrestin2 to CXCR4 following activation.

Knockdown of endogenous PKCζ enhances phosphorylation and β-arrestin2 recruitment to CXCR4. (A) HEK293 cells stably expressing CXCR4 were transfected with PKCζ ON-TARGETplus siRNA or scrambled control for 48 hours and the expression of endogenous PKCζ was probed using an anti-PKCζ antibody. Shown is a representative western blot from three independent experiments. (B) Following knockdown of PKCζ, HEK293 cells stably expressing CXCR4 were incubated with or without CXCL12 (50 nM) for up to 15 minutes and whole cell lysates were probed for pS346/7 and pS324/5. Shown are representative western blots from three individual experiments. (C) Following knockdown of PKCζ, HEK293 cells were cotransfected with CXCR4-RLucII and β-arrestin2-GFP10. Cells were stimulated with 20 nM CXCL12 and the relative interaction between CXCR4 and β-arrestin2 was determined by BRET assay. Data shown are a summary of six independent experiments.
Discussion
GPCRs play a very important role in many biologic functions in various cells and tissues. Activated GPCRs are primarily regulated by three mechanisms: desensitization, internalization, and degradation. Receptor desensitization is often initiated by phosphorylation and subsequent high-affinity binding of arrestin, which uncouples the receptor from its cognate G protein. Classically, it was thought that the GRKs were primarily responsible for phosphorylating activated GPCRs (Krupnick and Benovic, 1998); however, it is becoming evident that the process of receptor phosphorylation is very complex and involves multiple kinase families (Premont and Gainetdinov, 2007; Tobin et al., 2008). The complexity of receptor phosphorylation is further complicated by the fact that multiple kinases can phosphorylate a particular site and some kinases can phosphorylate multiple sites (Tobin et al., 2008). For example, within the C-terminal tail of CXCR4, we have shown that Ser-324/5 can be phosphorylated by both GRK6 and PKC and that GRK6 phosphorylates two additional sites (Ser-330 and Ser-339) (Busillo et al., 2010). In the present report, we extend our previous work characterizing site-specific phosphorylation of CXCR4 and provide evidence that phosphorylation of Ser-346/7 is mediated by GRK3 and PKC, in particular PKCα, and is involved in the recruitment of β-arrestin2, initiating the process of receptor desensitization.
The phosphorylation of CXCR4 mainly occurs in the serine/threonine-rich C-terminal tail. To date, at least seven phosphorylation sites have been identified through a variety of different techniques. We recently identified and confirmed several phosphorylation sites within the C-terminal tail of CXCR4 using a combination of mass spectrometry and phospho-specific antibodies. In addition, with the help of RNA interference and various kinase inhibitors, we identified the protein kinases that were responsible for site-specific phosphorylation of Ser-324/5, Ser-330, and Ser-339 (Busillo et al., 2010). Two of the phosphorylation sites detected by mass spectrometry were located between amino acids 346 and 352 of CXCR4, which contain two clusters of serine residues (Ser-346-348 and Ser-351/352). While we were unable to identify the exact residues phosphorylated or the kinases involved, we hypothesized that GRK2 and/or GRK3 could be responsible (Busillo et al., 2010). Recently, it was demonstrated that Ser-346/7 is phosphorylated in response to CXCL12 and PMA. Moreover, CXCL12-induced phosphorylation of Ser-346/7 was mediated by GRK2/3, which appears to impact additional phosphorylation events within the C-terminal tail of CXCR4 (Mueller et al., 2013). In the present study, we demonstrated that activation of CXCR4, with either CXCL12 or the pepducin ATI-2341 (Quoyer et al., 2013), results in rapid phosphorylation of Ser-346/7. Consistent with previous results (Mueller et al., 2013), we found that phosphorylation at Ser-346/7 peaked slightly earlier than Ser-324/5 (Fig. 1B) (Busillo et al., 2010). Furthermore, we also found that knockdown of GRK2, GRK3, or GRK6 reduced phosphorylation of Ser346/7 with GRK3 playing the major role (Fig. 1D). Interestingly, the majority of WHIM mutations described to date involve truncations of the last 10–15 amino acids of CXCR4 or have a selective loss of GRK3 (Diaz and Gulino, 2005; Balabanian et al., 2008). These data suggest that the GRK3-mediated phosphorylation of Ser-346/7 is critical for regulating the desensitization of CXCR4. In agreement with this, knockdown of GRK3 (Fig. 7C) and mutation of the last five serine residues of CXCR4 (Ser346/7/8, Ser-351, and Ser-352) (Fig. 7B) significantly reduced the association between CXCR4 and β-arrestin2. However, since mutation of the last five serine residues did not completely abolish the recruitment of β-arrestin2, phosphorylation of other residues is likely involved in the process.
As indicated previously, knockdown of GRK3 reduced phosphorylation of Ser-346/7 but did not eliminate it completely. Since PMA also induced phosphorylation of these residues (Fig. 3C) (Mueller et al., 2013), we investigated the role of PKC. We provide evidence for a role of PKC in phosphorylating Ser-346/7 (Fig. 1E) and recruiting β-arrestin2 (Fig. 7D). Following CXCR4 activation by CXCL12, both PKC and GRK3 are needed for robust phosphorylation of Ser346/7 (Fig. 1F). Additionally, ATI-2341, which allosterically activates CXCR4, results in the PKC-dependent phosphorylation of Ser-346/7 (Fig. 2, A and B). This is consistent with ATI-2341 selectively activating the Gαiβγ heterotrimer, resulting in the PKC-dependent phosphorylation of Ser-324/5 (Quoyer et al., 2013). In an attempt to narrow down the PKC isoforms involved in phosphorylation of Ser-346/7, we found that the conventional and/or novel isoforms of PKC are likely involved. Specifically, we showed that two PKC inhibitors, Bis I (conventional and novel PKC isoforms) and Gö 6976 (conventional PKC isoforms), reduced the basal phosphorylation of Ser-346/7 (Fig. 4A), whereas inhibition of PKCδ or PKCζ had no effect. In contrast, phosphorylation of Ser-346/7 following stimulation with either CXCL12 (Fig. 5, A and C) or ATI-2341 (Fig. 5, B and D) was reduced by inhibition of the conventional PKC isoforms (Bis I and Gö 6976) and PKCζ. This, in turn, reduced the recruitment of β-arrestin2 (Figs. 7D and 8A). The PKCζ inhibitor (a PKCζ pseudo-substrate) was much more effective at blocking arrestin recruitment to CXCR4 (Fig. 8A), while it had no effect on the recruitment of β-arrestin2 to the β2-adrenergic receptor (Fig. 8B). Together, these data suggest that the conventional PKC isoforms regulate the basal phosphorylation state of CXCR4, whereas PKCζ might be involved in phosphorylating the activated receptor and initiating arrestin recruitment. However, when we knocked down PKCζ (Fig. 9A), the phosphorylation of both Ser-324/5 and Ser-346/7 was modestly increased (Fig. 9B), resulting in enhanced recruitment of β-arrestin2 (Fig. 9C). Given the nonspecific actions of the PKC pseudo-substrates (Smith et al., 1990; Wu-Zhang and Newton, 2013) and the low affinity for PKCζ in cells (Wu-Zhang et al., 2012), it is difficult to interpret the data using the PKCζ inhibitor. However, it is worth noting that the PKCζ pseudo-substrate is a highly basic peptide, and might be directly binding to and inhibiting CXCR4. Indeed, many of the known inhibitors of CXCR4, including AMD3100, IT1t, CVX15, and ALX-40C, all possess a positive charge and inhibit CXCR4 by interacting with the CXCL12 binding pocket (Wu et al., 2010). In line with this hypothesis, we observed a dose-dependent inhibition of calcium mobilization downstream from CXCR4 activation but not downstream from the M3 muscarinic acetylcholine receptor (Fig. 8, C–E) (Luo et al., 2008). We also cannot rule out the possibility that the PKCζ pseudo-substrate is inhibiting a number of different kinases (Ling et al., 2002; Wu-Zhang et al., 2012; Wu-Zhang and Newton, 2013), including the conventional PKC isoforms (Bogard and Tavalin, 2015).
Since the amino acid sequence around Ser-346/7 (TESESSSFHSS) does not contain a linear PKC consensus sequence for phosphorylation, which requires a basic residue at −2 or +2, we searched for other kinases having consensus sequences that would better fit and we identified three kinases: CK1, CK2, and GSK3β. These kinases are abundantly expressed and have previously been shown to phosphorylate GPCRs (Kramer et al., 2000; Lee et al., 2001; Torrecilla et al., 2007; Luo et al., 2008). We showed that only inhibition of PKC significantly reduced both the basal and agonist-promoted phosphorylation of Ser-346/7 (Fig. 3, A–C), ruling out CK1, CK2, and GSK3β. Furthermore, we found that both PMA and ATI-2341 promoted phosphorylation of Ser-346/7. These data suggest that PKC is one of the main kinases responsible for the regulation of CXCR4 activity. While this region of CXCR4 does not contain a consensus site for PKC phosphorylation, one possibility is that the activation of CXCR4 causes the C-terminal tail to adopt a conformation that creates a structural PKC consensus motif, which has recently been demonstrated for both PKC and protein kinase A phosphorylation of various substrates (Duarte et al., 2014). Another possibility is that PKC-mediated phosphorylation of other residues within the C-terminal tail of CXCR4 allows for subsequent phosphorylation of Ser-346/7. Moreover, we cannot exclude the possibility that additional protein kinases downstream from PKC may be involved in the phosphorylation of CXCR4.
Although the GRK family has historically been considered the main kinase family to phosphorylate GPCRs, there is strong evidence to support a role for other protein kinases in mediating agonist-dependent phosphorylation of activated GPCRs. Advances in technology, such as tandem mass spectrometry and phospho-specific antibodies, have helped in identifying phosphorylation sites in many GPCRs (Tobin et al., 1997; Blaukat et al., 2001; Trester-Zedlitz et al., 2005; Busillo et al., 2010; Nobles et al., 2011). Given that most receptors are phosphorylated on multiple residues, it is difficult to link a functional outcome with a specific phosphorylation site. This is further complicated by the fact that the pattern of phosphorylation (the “phosphorylation barcode”) in one tissue may differ from the pattern elicited in another tissue. However, we have shown that there is overlap in kinase specificity for phosphorylating specific residues within CXCR4. This suggests that specific residues are critical for regulating the activity and function of CXCR4, and that redundancy exists between kinases to ensure that proper regulation is maintained in various tissues. This is of particular interest for CXCR4, given that the receptor plays a critical role in diverse physiologic processes and is one of the most commonly expressed receptors found on tumor cells (Busillo and Benovic, 2007). As we begin to better understand the kinetic and functional significance of site-specific phosphorylation, we will be able to better understand how this contributes to the dysregulation of CXCR4.
Acknowledgments
The authors thank Dr. Michel Bouvier for the CXCR4-RLucII and GFP10-β-arrestin2 BRET constructs. DNA sequence analysis was performed in the Genomics Shared Resource at the Sidney Kimmel Cancer Center, Thomas Jefferson University.
Authorship Contributions
Participated in research design: Luo, Busillo, Benovic.
Conducted experiments: Luo.
Contributed new reagents or analytic tools: Stumm.
Performed data analysis: Luo, Benovic.
Wrote or contributed to the writing of the manuscript: Luo, Busillo, Stumm, Benovic.
Footnotes
- Received August 10, 2016.
- Accepted March 17, 2017.
↵1 Current affiliation: Merck & Co., Inc., Upper Gwynedd, Pennsylvania.
Research reported in this publication was supported in part by the National Institutes of Health [Grants R01GM044944, R01CA129626, and P30CA056036], by a grant from the Pennsylvania Department of Health, and by Deutsche Forschungsgemein-Schaft [Grant STU295/7-1 to R.S.].
Abbreviations
- Bis I
- bisindolylmaleimide I
- BRET
- bioluminescence resonance energy transfer
- CK
- casein kinase
- DMEM
- Dulbecco’s modified Eagle’s medium
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- GSK3
- glycogen synthase kinase 3
- PBS
- phosphate-buffered saline
- PKC
- protein kinase C
- PMA
- phorbol 12-myristate 13-acetate
- Ser
- serine
- siRNA
- short interfering RNA
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}