


Abstract
An Asp-Arg-Tyr triad occurs in a majority of rhodopsin-like G protein-coupled receptors. The fully conserved Arg is critical for G protein activation, but the function of the flanking residues is not well understood. We expressed in COS-7 cells m1 muscarinic receptors that were mutated at Asp122 and Tyr124. Most mutations at either position strongly attenuated or prevented the expression of binding sites for the antagonist [3H]N-methylscopolamine. However, sites that were expressed displayed unaltered affinity for the antagonist. Receptor protein, visualized with a carboxyl-terminally directed antibody, was reduced but never completely abolished. The effects of these mutations were partially reversed by the deletion of 129 amino acids from the third intracellular loop of the receptor. In several cases, comparison of immunocytochemistry with binding measurements suggested the presence of substantial amounts of inactive, presumably misfolded, receptor protein. Some of the variants that bound [3H]N-methylscopolamine underwent small changes in their affinities for acetylcholine. All retained nearly normal abilities to mediate an acetylcholine-induced phosphoinositide response. We propose that Asp122 and Tyr124 make intramolecular contacts whose integrity is important for efficient receptor folding but that they do not participate directly in signaling. The role of these residues is completely distinct from that of Arg123, whose mutation abolishes signaling without diminishing receptor expression.
The Asp-Arg-Tyr sequence that is found at the carboxyl terminus of TM domain 3 of the mAChRs is a characteristic signature of the rhodopsin-like family of GPCRs (1, 2). The arginine [Arg123 in the m1 mAChR; residue 3.50 in the nomenclature of van Rhee and Jacobson (3)] is completely conserved. Substitution of this amino acid does not affect receptor expression and has only limited effects on ligand binding, but it strongly disrupts G protein binding and activation (3-6). Maintenance of a positive charge at this position is a critical requirement for function (6).
The flanking residues, corresponding to Asp122 and Tyr124 in the m1 mAChR, are also very highly conserved. An aspartic acid or a glutamic acid is found in 99% and a tyrosine is found in 75% of the rhodopsin-like GPCRs (2). However, some variations occur. For example, position 3.49 is asparagine in the platelet-activating factor receptor and histidine in the P2 purinergic receptors. Position 3.51 shows somewhat more diversity, particularly in the neuropeptide and endocrine hormone receptor subfamily, in which tryptophan, phenylalanine, cysteine, alanine, histidine, and serine can all be found at this locus (7).
The functional role of these residues is unclear. The majority of investigations of residue 3.49 have focused on the assumption that the negatively charged side chain usually found in this position is important for receptor function and examined the effect of altering the carboxylate to an amide group (8-11). In all except one instance, that of the β2-adrenoceptor (8), this did not lead to complete abolition of signaling; indeed, in rhodopsin, it promoted constitutive activation (12), and it has been argued that the protonation of residue 3.49 is integral to the photoreceptor activation mechanism (13). In the luteinizing hormone receptor, its alteration affected receptor processing rather than signaling; the mutant receptor failed to insert into the cell-surface membrane but retained its ability to activate adenylyl cyclase (14).
Position 3.51 has not been investigated so extensively. In studies of the m1 mAChR (5) and rhodopsin (15), single-point mutations had inhibitory effects on signaling and possibly on receptor processing. In the gonadotropin-releasing hormone receptor, uniquely, a serine occurs in this position, but reversion to tyrosine did not affect signaling capability, and gonadotropin-releasing hormone affinity and receptor internalization were enhanced (16).
During investigations of receptor-response coupling by the m1 mAChR, we found, unexpectedly, that the side-chain deletion mutant D122A failed to give binding activity when expressed in COS-7 cells. This prompted investigation of a more extensive series of mutations of Asp122 and Tyr124. The outcome suggests that neither of these residues is critical for signaling via the phosphoinositide pathway. Instead, both of them may form intramolecular contacts whose integrity is important for receptor folding. Sequence variation at these two positions may be particularly restricted in receptors, such as the m1 mAChR, which possess a long third intracellular loop.
Experimental Procedures
Site-directed mutagenesis and expression of muscarinic receptors.
Mutations were made by applying the polymerase chain reaction (17) or the Amersham (Arlington Heights, IL) phosphorothioate method to a 488-bp SacI/Xma1 fragment containing TM 2–5 of the rat m1 mAChR, as previously described (6). After verification by dideoxy sequencing, the mutated sequence was religated into the rm1 sequence contained in the pCD vector (6).
Wild-type and mutant receptors were expressed in COS-7 cells by transfection using a BioRad (Hercules, CA) Gene Pulser at 180 V and 960 μF with 10 μg of DNA (unless otherwise specified)/0.4-cm cuvette (4 × 107 cells; 0.8 ml). Cells were grown in α-minimal essential medium supplemented with 10% newborn calf serum, antibiotics, and 2 mm glutamine for 72 hr, with the medium changed at 24 hr after transfection. Levels of expression of wild-type receptors were typically ∼1 pmol/mg of protein.
Ligand binding assays.
After 72 hr, the transfected COS-7 cells were washed with phosphate-buffered saline; harvested; homogenized in 20 mm Na-HEPES and 10 mm EDTA, pH 7.5, at 0°; pelleted; resuspended in 20 mm Na-HEPES and 1 mm EDTA, pH 7.5; snap frozen; and stored at −80°. For binding assays, membranes were thawed, pelleted, and rehomogenized in binding buffer (containing 20 mm Na-HEPES, 100 mm NaCl, 1 mm MgCl2, pH 7.5) to a protein concentration of ∼15 μg/ml. Aliquots of 1 ml were incubated with the appropriate concentration of [3H]NMS (supplemented with unlabeled ligands as required) for 60 min at 30°, which was shown to be sufficient to allow the attainment of equilibrium. We used 10−11 to 3 × 10−9 m [3H]NMS in the saturation binding assays and 3 × 10−10 m[3H]NMS in competition binding assays. Nonspecific binding was determined with 10−6 m(−)-3-quinuclidinyl benzilate. The filters underwent three ice-cold washes within ∼15 sec. Radioactivity was determined by liquid scintillation counting. All binding assays were performed in quadruplicate. Protein concentrations were determined according to the Lowry method after trichloroacetic acid precipitation of the membrane protein.
PI turnover assays.
PI turnover assays were as previously described (6). Transfected cells were seeded onto 12-well plates, and after 24 hr, they were labeled with 1 μCi/mlmyo-d-[3H]inositol. At 72 hr after transfection, the cells were washed in warm Krebs-bicarbonate solution containing 10 mm LiCl. After a 30-min incubation, the agonists were added for an additional 30 min. This was shown to be within the linear period of the assay. After removal of the medium, the cells were extracted with 0.5 ml of cold 5% perchloric acid, and after neutralization, the [3H]inositol phosphates were isolated by chromatography on Dowex AG 1X8 columns and quantified by liquid scintillation counting. Assays were carried out in triplicate.
Data analysis.
Binding curves were fitted to the Hill equation or to one-site (18), two-site (19), or ternary complex (20) models of binding using Sigma Plot 5.0 (Jandel) as described previously (6, 21). The wild-type ACh binding curve could be resolved into a major low affinity component (∼62% of the total) and a minor component with a ∼10-fold higher affinity. The estimates of apparent affinity constants were equivalent, within the error, for the two-site and ternary complex methods of analysis. Where necessary, affinity constants were corrected for the Cheng-Prusoff shift (21). PI dose-response curves were fitted to a four-parameter logistic function, yielding an EC50 value and a slope factor that was characteristically near 1.0.
Efficacy values for different mutant receptors were estimated by exploiting a simplified version of the ternary complex model
By rearrangement of eq. 13 of Leff and Harper (22), the following equation may be obtained:
The functional response was assumed to be proportional to [ARG]/[Gt], where [Gt] is the total concentration of receptor-accessible G protein and [Rt] is the total concentration of receptor binding sites. A value of 20 was taken for the ratio [Rt]/[Gt] for the wild-type receptors; this was sufficient to account for the shift in the ACh dose-response curve caused by PrBCM blockade. The relative values of [Rt] and K were estimated from the binding experiments. K was taken to be equal to the low affinity binding constant for ACh. This parameter is insensitive to GTP (6), which is consistent with its representing binding to uncoupled receptors. Values of the composite parameter [K∗G][Gt] were then calculated to reproduce the measured EC50 values of the PI responses generated by the different mutant receptors. This parameter is a measure of the tendency of a given agonist/receptor complex to bind G protein. It is governed by both the affinity constant of the G protein for the agonist/receptor complex and the total concentration of receptor-accessible G protein, and it gives a guide to the efficacies of the mutant receptors relative to the wild-type.
Immunocytochemistry.
Transfected cells were plated onto coverslips previously exposed to 25 μg/ml fibronectin. After 3 days, the cells were washed three times with ice-cold phosphate-buffered saline and fixed with ice-cold 4% paraformaldehyde in phosphate-buffered saline containing 0.05% Triton X-100/Nonidet P-40 (1:1 v/v) for 5 min. After washing, the cells were blocked with 5% nonfat dry milk for 60 min at room temperature, washed, and incubated for 60 min at 37° with a 1:100 dilution of an immunoaffinity-purified rabbit antiserum raised against the carboxyl-terminal 13 amino acids of the m1 mAChR, which are identical in the rat and human sequences. After washing, the cells were incubated with a 1:5000 dilution of a goat anti-rabbit IgG/alkaline phosphatase conjugate. After further washing with Tris-buffered saline, color was developed by incubation with nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate. Reaction product was quantified with a Molecular Dynamics (Sunnyvale, CA) laser scanning densitometer and Image-Quant software.
Materials.
Pfu polymerase was from Stratagene (La Jolla, CA). Restriction endonucleases, T4 ligase, and calf intestinal phosphatase were from Promega (Madison, WI) or Boehringer-Mannheim (Indianapolis, IN). [3H]NMS (85 Ci mmol−1),myo-d-[3H]inositol (80 Ci mmol−1), Sequenase, and the phosphorothioate mutagenesis kit were from Amersham. Alkaline phosphatase-conjugated goat anti-rabbit IgG was from Promega. Other reagents were from Sigma Chemical (St. Louis, MO) or Research Biochemicals (Natick, MA). PrBCM was cyclized to the aziridinium ion in 10 mm sodium phosphate, pH 7.5, for 60 min at room temperature before use.
Results
Expression of antagonist binding sites.
Mutations of Asp122 strongly decreased the expression of m1 mAChRs in COS-7 cells, as measured by saturation binding assays with the antagonist [3H]NMS (Table 1). The mutation of Asp122 to histidine was tolerated best, yielding 30% of the wild-type level, whereas the mutations of Asp122 to glutamic acid and asparagine gave only 10% expression. Other mutations, either to polar residues (serine or lysine) or to neutral or nonpolar amino acids (cysteine, alanine, or leucine) gave little or no binding activity. The affinity constants of the His122, Asn122, and Glu122 mutants showed no significant change from the wild-type value (Kd = 10−10 m). Virtually identical levels were measured with the membrane-permeable antagonist [3H]quinuclidinyl benzilate (data not shown).
Binding of [3H]NMS to m1 mAChRs mutated at Asp122 or Tyr124
Mutations of Tyr124 also diminished the expression of [3H]NMS binding sites (Table 1). Conservative replacements by the aromatic residues phenylalanine or tryptophan were reasonably well tolerated, but the other substitutions gave little (e.g., histidine, methionine, cysteine; 5% of wild-type) or no (e.g., isoleucine, arginine, glutamic acid) binding. Again, changes occurred in the numbers rather than in the affinities of the sites.
Immunocytochemical measurements of expression.
Receptor protein was visualized in fixed, permeabilized COS-7 cells using an antibody directed against the carboxyl-terminal sequence of the m1 mAChR. After transfection with wild-type DNA, clones of ∼10 cells (on average) were stained. These were well separated when small amounts of DNA were used (0.1–0.3 μg; Fig. 1a) but often merged into extensive areas of staining with larger amounts (10 μg; Fig.1b). No immunoreactivity could be detected in untransfected controls or in cells transfected with a receptor in which the carboxyl-terminal epitope had been substituted.
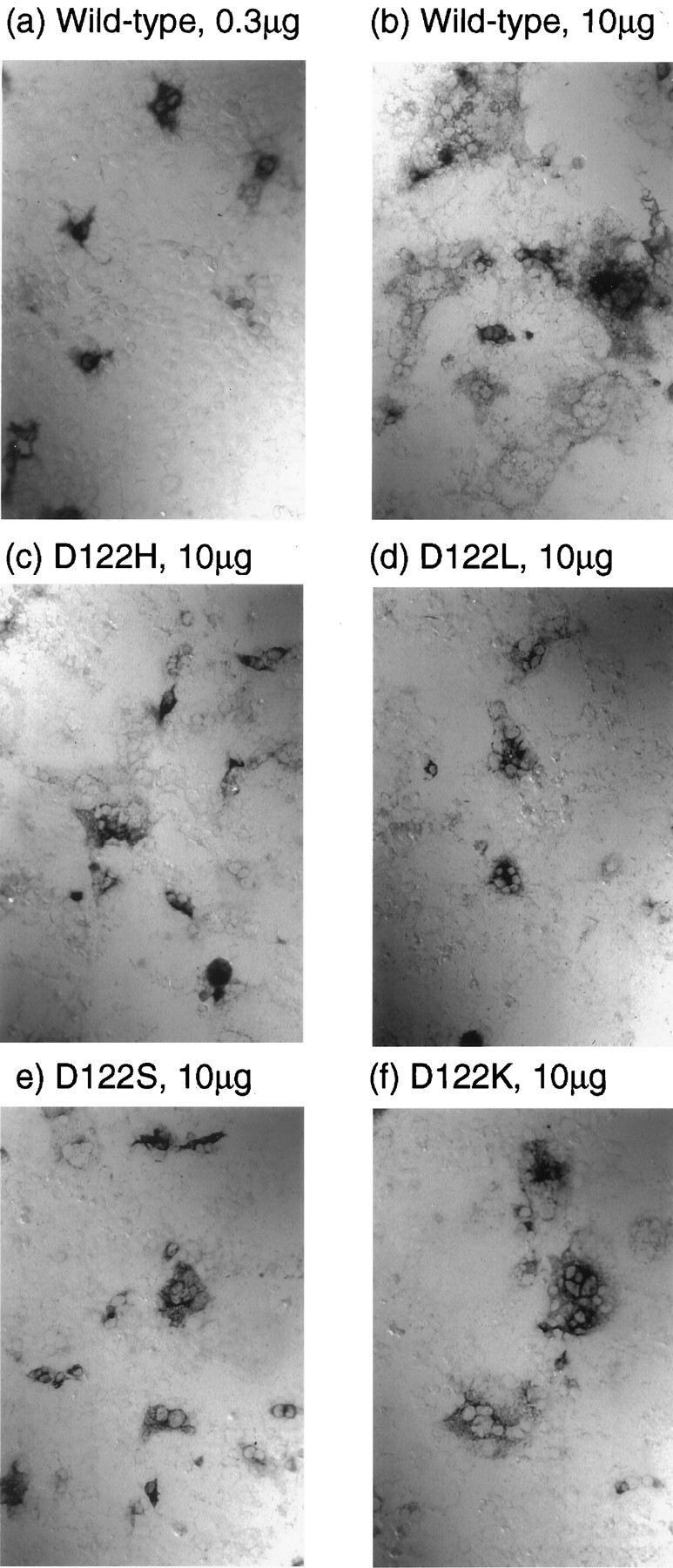
Immunocytochemical visualization of wild-type and mutant m1 mAChRs in transfected COS-7 cells using an antibody directed against a carboxyl-terminal epitope. Methods are described in Experimental Procedures. Magnification, 200×. a, Wild-type, 0.3 μg of DNA. b, Wild-type, 10 μg of DNA. c, D122H, 10 μg of DNA. d, D122L, 10 μg of DNA. e, D122S, 10 μg of DNA. f, D122K, 10 μg of DNA.
All of the mutant sequences that showed diminished levels of [3H]NMS binding (e.g., D122H; Fig. 1c) gave detectably lower levels of immunostaining than the wild-type receptor. In most cases, reductions in the size and intensity of staining of the immunoreactive clones appeared to outweigh any decrease in their frequency. In the case of the Asp122 mutants, the nonpolar substitutions, particularly D122L (Fig. 1d), yielded the lowest levels of staining. However, polar substitutions such as D122S (Fig. 1e) or D122L (Fig. 1f) gave significant immunoreactivity, despite their failure to generate measurable levels of radioligand binding. The Asn122, Glu122, and His122 (Fig. 1c) mutants gave a paler version of the wild-type pattern of staining. Large variations were also seen with the Tyr124 mutants. For example, Y124F generated a wild-type pattern, whereas Y124M yielded a lower level of staining.
Careful examination at higher magnification showed that for both wild-type and mutant receptors, staining was apparently concentrated around the periphery of the cells, presumably in the cell-surface membrane. However, the immunocytochemical methods used in the current study were not sufficiently resolving to provide fine details of intracellular localization. For the above mutants, gross densitometric measurements of reaction product showed the following expression levels relative to wild-type: His122, 50%; Lys122, 70%; Asn122, 60%; Glu122, 30%; Ser122, 30%; Cys122, 20%; Ala122, 10%; Leu122, <10%; Phe124, 90%; and Met124, 45%.
ACh binding.
In agreement with previous experiments, ACh binding to wild-type m1 mAChRs expressed in COS-7 cells was characterized by a slightly flat binding curve (Hill coefficient = 0.80), which can be described parametrically by a major fraction (∼60%) of sites with a Kd value of 20 μm and a minor fraction (∼40%) of sites with a Kd value of 1.6 μm (6, 23). Mutation of Asp122 to asparagine or glutamic acid caused a 4-fold increase in ACh affinity, whereas mutation to histidine caused a slight (2-fold) decrease (Table2). The slope of the binding curve was not altered, so low and high affinity binding constants seemed to be equally affected. In contrast, mutation of Tyr124 had little effect on ACh binding, with the exception of an apparent 6-fold increase in the high afffinity binding constant for the His124 mutant (Table 2). In none of these cases was there any systematic change in the relative fraction of high affinity sites from the previously determined wild-type value of 0.38 (6), which also represents ACh binding to m2 mAChRs expressed in COS cells (23). This parameter was therefore fixed in calculation of the binding constants in Table 2.
Binding of acetylcholine to m1 mAChRs mutated at Asp122 or Tyr124
PI response.
All of the mutant sequences that gave measurable levels of expression of [3H]NMS binding were able to mediate a PI response to ACh. The maxima were similar to that evoked by the wild-type receptor (Table 3). ACh potency at the D122H mutant was 10% of the wild-type value, but the responses from the Glu122 and Asn122 variants were quantitatively similar to the wild-type, despite their low levels of expression. Even the Y124C mutant, which had levels of binding equivalent to only 4% of the wild-type, supported a maximum PI response, with an apparent potency reduced to one-20th of the wild-type value. The mutations had no effect on the basal PI response.
Phosphoinositide response to acetylcholine of m1 mAChRs mutated at Asp122 or Tyr124
To control for differences in the expression levels, transfected cells were pretreated with various concentrations of the irreversible muscarinic blocking agent PrBCM aziridinium ion before assay of the PI response to ACh. Fig. 2A shows that the wild-type dose-response curve was shifted to 10-fold higher ACh concentrations by blockade of 90% of the cell-surface receptors (measured by subsequent binding of [3H]NMS to membrane preparations from cells treated in parallel) without any reduction in the maximum response. There was an approximately linear relationship between the EC50 value and the fraction of unalkylated receptors (Fig.2A, inset). This suggests a large spare receptor ratio (>10). In contrast, blockade of a similar fraction of the Asn122 receptors caused a significant reduction in the maximum response (Fig.2B), suggesting a smaller receptor reserve, which is consistent with the lower expression level of this construct. An intermediate result was obtained with the His122 mutant (data not shown).

Effect of different methods of altering the level of functional m1 mAChRs in COS-7 cells on the ACh-induced PI response. Details are given in Experimental Procedures. Points, mean ± standard error of triplicate measurements. A, COS-7 cells transfected with 10 μg of wild-type DNA and pretreated with different concentrations of PrBCM aziridinium ion before measurement of the PI response. Inset, effect of the pretreatment on the concentration of binding sites for [3H]NMS, measured subsequently in a membrane preparation. B, COS-7 cells transfected with 10 μg of D122N DNA and pretreated with different concentrations of PrBCM aziridinium ion; the extent of blockade was similar to that in A. C, COS-7 cells were transfected with different amounts of wild-type m1 mAChR DNA before measurement of the PI response to ACh. The results shown are representative of three experiments.
It was notable that although it resulted in a similar change in the global levels of receptor binding, transfection with different amounts of plasmid DNA led to a proportional alteration in the maximum PI response but did not affect the EC50 value (Fig. 2C). This is in accord with the observation (Fig. 1, a and b) that it is the proportion of cells transfected rather than the level of receptor expression in any given transfected cell that is altered by this maneuver.
Enhancement of expression by a deletion in the third intracellular loop.
We found that deletion of 129 amino acids from the third intracellular loop of the m1 mAChR (residues 225–353; dLoop) resulted in a significant increase (66%) in the level of receptor binding expressed in COS-7 cells (Table 4) and a consistent increase in expression of the receptor protein as measured by immunocytochemistry (60% by densitometry). An equivalent deletion caused an approximately proportional increase in the expression of the D122E, D122N, and D122H constructs (Table 4) and led to the reappearance of small amounts of binding activity in several mutants (D122S, D122A, and D122L) that were devoid of activity in the context of the wild-type sequence. There were even larger (∼10-fold) increases in three of the four Tyr124 mutants examined in this way (i.e., Y124H, Y124M, and Y124C).
Effect of deletion of amino acids 225–353 of the third intracellular loop of the m1 mAChR on expression of Asp122 and Tyr124 mutants
The loop deletion mutation did not affect the [3H]NMS or ACh affinity of the Asp122 receptor or that of the Asp122 or Tyr124 mutants previously characterized in the context of the wild-type sequence. It enabled measurements to be made of NMS and ACh binding constants for the D122A, D122S, and D122L mutants, which had previously been inaccessible because of their failure to express at measurable levels. The NMS affinities of the serine, alanine, and leucine mutants were identical to the wild-type values, whereas the ACh affinity of the serine variant was increased ∼4-fold with respect to the wild-type receptor, as was observed for the asparagine and glutamic acid mutants (Table 2).
The loop-deletion construct mediated a robust PI response to ACh, with a maximum close to that given by the wild-type receptor but a significantly decreased potency (5-fold; Table 5). The potencies of the loop-deleted variants of the D122E, D122N, and D122H mutants (whose expression increased ∼2-fold) were also reduced, but the EC50 values for the Y124H, Y124M, and Y124C variants (whose expression increased ∼10-fold) remained similar to the values observed in the wild-type context. The D122A, D122S, and D122L mutants developed the ability to elicit a PI response when expressed in the loop-deleted sequence, although their maximum responses were attenuated compared with the wild-type values (Table 5).
Effect of deletion of amino acids 225–353 of the third intracellular loop of the m1 mAChR on the phosphoinositide response to acetylcholine mediated by the Asp122 and Tyr124 mutants
Comparison of immunocytochemistry and binding may reveal the presence of receptor protein that is inactive.
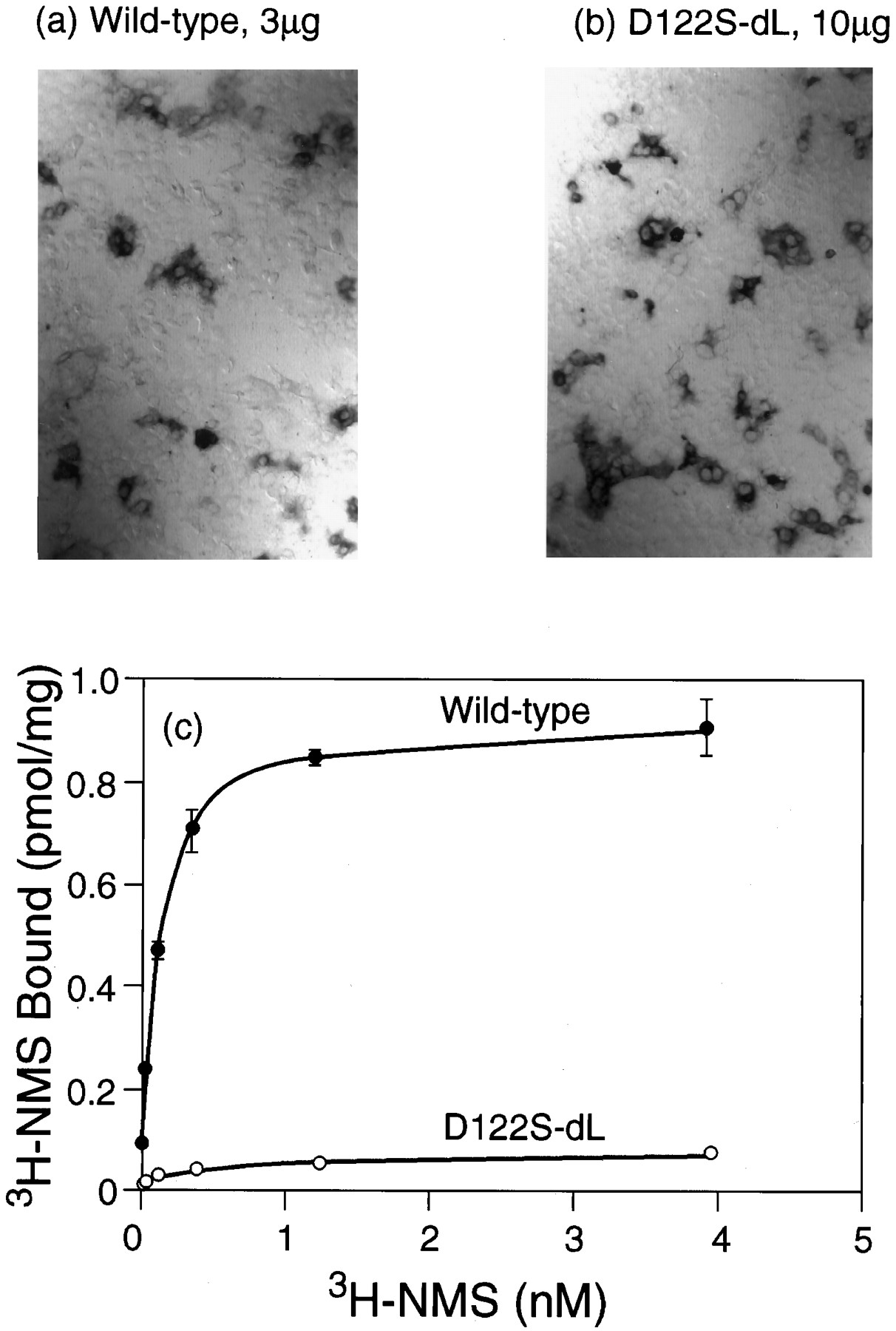
As noted above, immunocytochemical visualization frequently suggested the presence of larger amounts of receptor protein than might be expected on the basis of the corresponding binding assays. These discrepancies became more acute in the context of the loop-deletion construct, which led to large increases in the expression of immunocytochemically detectable protein from all of the previously poorly expressed mutants (e.g., 3-fold for Ser122 by densitometry). These increases were not always accompanied by the appearance of equivalent amounts of binding activity. For example, the level of immunostaining resulting from transfection of 10 μg of the D122S dLoop construct was similar to that given by 3 μg of the wild-type sequence (Fig. 3, a and b), but the [3H]NMS binding activity was only 10% of the level produced by this amount of wild-type DNA (Fig. 3c). This implies that most of the immunoreactive material generated by translation of the mutant sequence was inactive in the [3H]NMS binding assays. Similar conclusions could be drawn from experiments with the D122A dLoop and D122L dLoop constructs. The behavior of these Asp122 mutants contrasts with that of the Tyr124 mutants, whose binding activities underwent much larger increases.
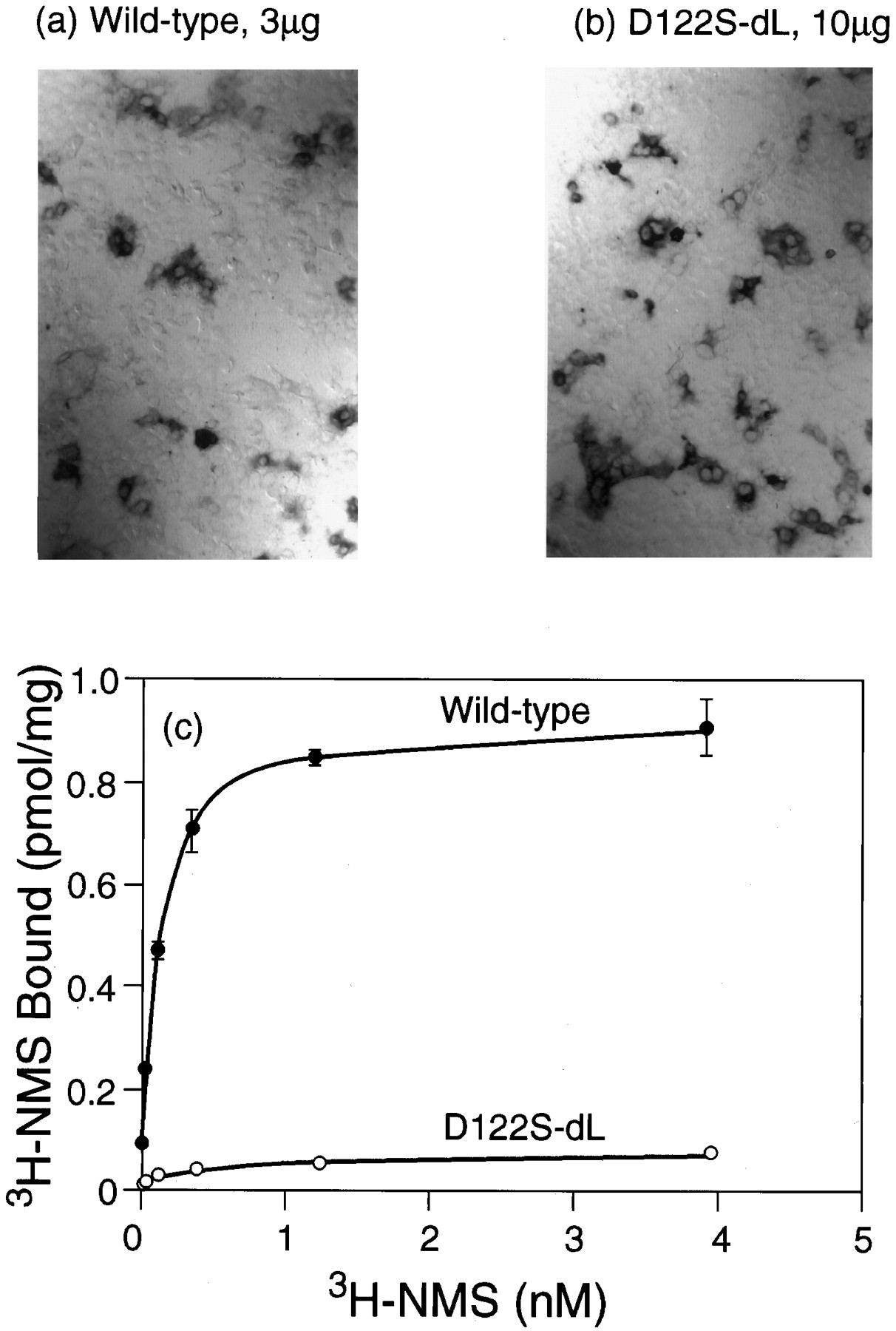
Expression of the wild-type receptor and of the D122S dLoop mutation by immunocytochemistry (a and b) and binding of [3H]NMS (c). a, Wild-type, 3 μg of DNA transfected. b, D122S dLoop, 10 μg of DNA transfected. c, Saturation binding curves with [3H]NMS; full curves are fits to a one-site model of binding.
Discussion
In this study, we examined the effects of a number of mutations of Asp122 and Tyr124 on the expression, binding, and function of m1 mAChRs after transient expression in COS-7 cells. We replaced these residues with most of the naturally occurring variants, including glutamic acid, asparagine, and histidine at position 122 and phenylalanine, tryptophan, histidine, and cysteine at position 124. In addition, we made several other changes that encompass a range of amino acid properties (e.g., small/large, polar/nonpolar, charged/uncharged). Combined with our previous study of Arg123, this represents a comprehensive survey of the functions of the amino acids of the Asp-Arg-Tyr triad in the m1 mAChR.
The results of these experiments show the functions of these three amino acids to be highly differentiated. There were large inhibitory effects of mutations at positions 122 and 124 on the expression of m1 receptors. These were shown by both binding of the antagonists [3H]NMS and [3H]quinuclidinyl benzilate, which detect the presence of fully folded binding sites, and, less dramatically, immunocytochemical measurements using an antibody directed against a carboxyl-terminal epitope, which can visualize both fully folded and misfolded full-length translation products. In contrast, a range of substitutions for Arg123 had little effect on levels of expression of the m1 mAChR (Fig. 4a).

Effects of mutations of Asp122, Arg123, and Tyr124 on (a) receptor expression level (see Table 1) and (b) coupling efficiency, relative to the wild-type. The method of calculation is explained in Experimental Procedures. For serine, alanine, and leucine mutants, the values were calculated relative to the dLoop construct because these mutants were not expressed in the absence of this deletion. Data for the Arg123 mutations are from Jones et al. (6).
Measurements of function showed the opposite pattern. Mutation of Arg123 strongly disrupts PI signaling (6), but all mutants of Asp122 and Tyr124 yielding measurable expression of binding sites were able to mediate stimulation of PI breakdown by ACh; these included the variants D122A, D122S, and D122L, which gave no functional binding sites in the context of the full-length sequence but were partially rescued by deletion of the central portion of the i3 loop of the receptor. Several of the Asp122 mutations caused moderate increases in ACh affinity, extending previous reports on the D3.49N mutation in the m1 mAChR and the β-adrenoceptor (8, 9).
Blockade experiments with the irreversible antagonist PrBCM indicated that differences in functional potency were largely attributable to variations in expression levels of the different mutants. These experiments demonstrated a receptor reserve that was substantial (≥10-fold) for the PI response mediated by the wild-type receptor, smaller for the D122H mutant, and small or absent for the D122N and D122S dLoop mutants, which were all expressed at lower levels. It should also be noted that alteration of receptor expression by transfection of different amounts of DNA affected only the proportion of the cells that expressed receptor and changed the maximum but not the EC50 value of the PI dose-response curve. This is consistent with the properties of the pCD vector in COS cells and shows that the transfection of different amounts of DNA is not an appropriate control for differences in expression levels in experiments involving the transient transfection of COS cells with expression vectors containing a simian virus 40 origin.
The use of a simple version of the ternary complex model allowed the estimation of a parameter that is a measure of the relative efficacies of the different mutant receptors. The calculation is described in Experimental Procedures, and the results are summarized in Fig. 4b.
The apparent efficacy of the i3 loop deletion variant was diminished by ∼10-fold. Thus, the residues in this sequence are not entirely dispensable for efficient signaling to the PI pathway. In fact, the deletion used in the current study is slightly larger than those previously reported to have no effect on PI signaling by the m1 mAChR (24, 25).
For the mutations at positions 122 and 124, only the D122H variant had significantly reduced signaling capability (∼20%) compared with the appropriate parent sequence. This is in striking contrast to the highly deleterious effects of the substitution of Arg123 (6) (Fig. 4b). These observations argue against direct, specific contacts between positions 122 or 124 and the G protein but would be consistent with such contact in the case of position 123. The unaltered activation characteristics and the apparent lack of constitutive activity of mutant receptors with neutral substitutions at position 122 do not support the proposition that a proton-acceptor group at this position is necessary for ligand-dependent activation (13).
The extreme sensitivity of m1 receptor expression to substitution of smaller residues at position 122 is unusual. Cysteine-scanning mutagenesis of TM helix 3 of the D2 dopamine receptor, excluding the Asp-Arg-Tyr triad, did not reduce expression levels below 25% of wild-type (26). In agreement with this, we have found that alanine substitution of the 10 residues amino terminal to Asp122 in the m1 mAChR reduces expression to below 20% only in the case of Leu116, which shows a sensitivity to mutagenesis comparable to that of Tyr124.1 Multiple substitutions in the second intracellular loops of the m1 and m2 mAChRs have also been made without large effects (27, 28). In addition, cysteine-scanning mutagenesis of the i2 loop of rhodopsin failed to abolish the expression of protein capable of binding retinal, although it is interesting that substitution of Tyr3.51 produced a poor yield, which is in agreement with the current findings for the equivalent position in the m1 mAChR (15). Thus, the exceptional intolerance of smaller side chains that characterizes position 122 is not a property shared by neighboring residues in the m1 mAChR or related receptors. The effect of mutation of Asp122 seems to be more extreme than that reported for mutation of the highly conserved prolines in TM helices 5–7 of the m3 mAChR, whose integrity is believed to be important for efficient receptor folding (29).
It is striking that the only substitutions at position 122 that were compatible with measurable levels of expression were the naturally occurring variants histidine, glutamic acid, and asparagine, all of which have hydrogen bond-accepting side chains of similar size. Position 124 was class specific, displaying a strong preference for the aromatic amino acids phenylalanine and tryptophan, whereas the other naturally occurring variants histidine and cysteine, as well as methionine, gave much lower levels of expression. Other substitutions abolished binding activity.
Position 122 and, to a lesser extent, position 124 are highly conserved in all GPCRs. Their mutation strongly affects the total level of expression of m1 mAChR binding sites but has minimal effects on the signaling capability of those sites that finally achieve a state capable of ligand binding. A hypothesis that could account for the strong conservation of these residues is that they participate in intramolecular contacts that are important for the generation of a fully folded receptor structure. The comparison of immunocytochemical with binding studies suggests that if they are altered, misfolded receptors accumulate. As far as can be determined, these misfolded forms, like the wild-type receptor, accumulate in the cell-surface membrane, as reported for inactive m2/m3 mAChR hybrids (30).
In GPCRs such as the mAChRs, which have a long third intracellular loop, TM domains 1–5 and 6–7 may fold as independent subunits that subsequently collide and associate to form a complete helical bundle (31). In general agreement with this hypothesis, we found that the deletion of 129 amino acids (residues 225–353) from the i3 loop increases the expression of the m1 mAChR in COS-7 cells (Table4), SF9 cells, and Escherichia coli.2 This may reflect an increased frequency of productive encounters between the putative amino- and carboxyl-terminal domains, as well the removal of protease-sensitive sites, and kinase target sequences (32), which may prolong the lifetime of incompletely folded intermediates.
Deletion of the greater part of the i3 loop partially rescued Asp122 and Tyr124 mutants that were expressed at low levels. It may be relevant that the i3 loop is short in the majority of naturally occurring sequences in which deviations occur from these two consensus residues. In contrast, in the cationic amine receptors, in which the i3 loop is long, there are no deviations from the optimum Asp-Arg-Tyr(Phe) motif (33). Interestingly, although the i3 loop deletion increased expression of the Tyr124 mutants by ≤20-fold, it had smaller (<3-fold) effects on the expression of Asp122 mutants. This suggests that a future search for residues postulated to contact Tyr124 might commence in a part of the receptor strongly affected by, or within, TM domains 6 and 7, whereas those contacted by Asp122 may be sought in a region more remote from these elements. Such intramolecular contacts may represent conserved elements in the structure of the rhodopsin-like GPCRs. As in the case of smaller proteins (34), their perturbation may have a large effect on the kinetics of receptor folding.
Footnotes
- Received July 15, 1996.
- Accepted October 28, 1996.
-
Send reprint requests to: Dr. E. C. Hulme, Division of Physical Biochemistry, National Institute for Medical Research, Mill Hill, London NW7 1AA, U.K. E-mail:e-hulme{at}nimr.mrc.ac.uk
-
↵1 Z.-L. Lu, unpublished observations.
-
↵2 E. C. Hulme and C. A. Curtis, unpublished observations.
-
This work was supported by the Medical Research Council (UK) and by a Wellcome Travelling Fellowship (Z.L.).
Abbreviations
- ACh
- acetylcholine
- mAChR
- muscarinic acetylcholine receptor
- GPCR
- G protein-coupled receptor
- NMS
- N-methylscopolamine
- PrBCM
- propylbenzilylcholine mustard
- PI
- phosphoinositide
- TM
- transmembrane
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}