


Abstract
Phenobarbital elicits pleiotropic effects in the liver, including induction of enzymes involved in xenobiotic metabolism. The spectrum of this response was analyzed by differential display of a large population (∼7500) of mRNAs in chicken embryo liver treated in vivo with phenobarbital. We identified 29 cDNA fragments that reproducibly and significantly changed in intensity after a 48-hrin ovo treatment. Eighteen of these (62%) were increased, whereas 11 (38%) were decreased. Twenty strongly regulated cDNA fragments were subcloned and further analyzed. Nucleotide sequence analysis revealed three types of genes: (a) those previously described to be regulated by phenobarbital, including CYP2H1, glutathioneS-transferase, and uridine diphosphate-glucuronosyltransferase; (b) genes reported herein for the first time to be regulated by phenobarbital, including fibrinogen β-chain and γ-chain, retinal glutamine synthetase, apolipoprotein B, two gene products with homologies to elongation factor 1δ and complement factor H, respectively, and (c) several novel genes with hitherto unknown functions. If these data are extrapolated to the entire population of mRNAs of a liver cell, phenobarbital seems to significantly modulate the expression of more than 50 different genes. Our results also demonstrate that a large fraction of genes is negatively regulated by drug treatment.
Several classes of environmental and therapeutic chemicals are recognized to modulate gene expression in liver but also in other tissues. In particular, the induction of genes coding for enzymes involved in the biotransformation and detoxification of drugs and chemicals is well known (1, 2). Among the inducing agents are polyaromatic and polychlorinated hydrocarbons, peroxisome proliferators such as clofibrate, glucocorticoids and antiglucocorticoids, alcohol, and numerous drugs and chemicals (3-5). Phenobarbital is the prototype inducer for a variety of other xenobiotic compounds called “phenobarbital-like,” which exhibit inductive effects on different biotransformation enzymes including GSTs, UDP-glucuronosyl transferase, epoxide hydrolases, NADPH-cytochrome P450 reductase, aldehyde dehydrogenases, and several cytochrome P450 monooxygenases (2, 6, 7).
The response to phenobarbital is dose-dependent, reversible, and accompanied by an increase in liver weight and proliferation of the smooth endoplasmatic reticulum. More recent studies indicate that genes not involved in drug metabolism may also be regulated by phenobarbital-like compounds. These include α1-acid glycoprotein, apolipoprotein apo-A1, cholesterol 7α-hydroxylase, and a rat canalicular glutathione transporter (8-11). The inductive response can have a major impact on drug-drug interactions, toxicity, and carcinogenicity of exogenous and endogenous compounds (for review, see Refs. 2 and 4).
The molecular mechanisms of this induction phenomenon are still poorly understood. The best understood system is the phenobarbital induction of two fatty acid hydroxylases in Bacillus megaterium, in which induction involves inhibition of repression of the controlled genes by a repressor protein (12-15). Major obstacles to intensive investigation of mammalian mechanisms have been the apparent lack of a phenobarbital receptor as well as the lack of appropriate cell culture systems. The response seems to be largely controlled at the level of transcription, and, more recently, molecular analysis of phenobarbital regulated promoters and enhancer regions has been performed (16-20). Despite these recent advances, the way in which the cell recognizes phenobarbital and phenobarbital-like compounds and the way in which this information is conveyed to the transcriptional machinery remains a mystery.
The pleiotropic response to phenobarbital and phenobarbital-like compounds suggests that, in addition to drug-metabolizing enzymes, the expression of numerous other genes may be affected. The spectrum of altered gene expression, however, has not been studied systematically. Therefore, we investigated the extent and character of phenobarbital-mediated changes of gene expression in the liver. As a model system, we used the chick embryo because previous studies in our laboratory (21, 22) have demonstrated reliable induction of cytochromes P450 by phenobarbital and other inducers in primary cultures of chick embryo hepatocytes maintained under chemically defined conditions. These results have been confirmed by other investigators (23, 24). This system also allows investigations in vivo by direct injection of phenobarbital into the fluids surrounding the embryo (in ovo) (21).
In the present study, we used the recently developed differential display technique (25, 26) to analyze the effect of phenobarbital on the hepatic mRNA population of chicken embryos treated in ovo. We describe the isolation and sequences of 20 cDNAs influenced by phenobarbital detected by this method. Genes not previously known to be regulated by phenobarbital were discovered, and, surprisingly, a large number of genes were found to be down-regulated by phenobarbital.
Materials and Methods
White Leghorn fertilized eggs were obtained from Geflügelzucht Gallipor AG, Ottenbach, Switzerland. Phenobarbital (sodium salt) was obtained from the Hospital Pharmacy, University Hospital, Zurich, Switzerland. Redivue [α-35S]dATP (1000 Ci/mmol), [α-32P]dATP (3000 Ci/mmol), and [γ-32P]dATP (5000 Ci/mmol) were purchased from the Radiochemical Center, Amersham International, Bucks, England. All other chemicals were from standard commercial sources. Moloney murine leukemia virus reverse transcriptase was purchased from Gibco BRL/Life Technologies, Basel, Switzerland. Recombinant ribonuclease inhibitor was obtained from Promega, Zurich, Switzerland. All other enzymes were purchased from New England Biolabs, BioConcept, Allschwil, Switzerland. PCR reactions were performed in a Perkin Elmer Cetus (Rotkreuz, Switzerland) thermocycler using AmpliTaq DNA polymerase also obtained from Perkin Elmer. PCR primers were purchased from Intron Laboratory for Molecular Biology, Kaltbrunn, Switzerland. Ten-mer oligonucleotides (Table 1) were selected from a list of random sequences and checked for absence of stem loops and self-complementarity of more than two nucleotides. A liver cDNA library in λ Zap Vector from a 7-day-old male broiler breeder chicken was obtained from Stratagene, La Jolla, CA.
List of arbitrary 10-mers used for differential display
Induction in vivo.
For induction studies in vivo (in ovo), direct injections of phenobarbital into the fluids surrounding the embryo were performed. Phenobarbital (5 mg per egg) was dissolved in 0.2 ml of 0.9% saline. Control animals were injected with the same amount of 0.9% saline without phenobarbital. For injection, the orientation of the embryo was marked using a small intensive source of light. Injection was then performed with a 0.9-mm needle and a 1-ml syringe into the yolk sac. Chicken embryo livers were perfused through the heart with 20 ml of ice-cold 0.9% NaCl and 2 mm EDTA to remove blood cells. Livers were taken out and frozen immediately in liquid nitrogen. If not used for immediate RNA isolation, livers were stored at −80°.
RNA isolation.
Isolation of total RNA was performed by the classical guanidinium thiocyanate method of Chomczynski and Sacchi (27). For each isolation, two livers were pooled (total of approximately 1 g of tissue) to overcome interindividual differences in differential display analysis.
Differential display.
One microgram of total RNA was reverse transcribed using free nucleotides at a concentration of 200 μm. cDNA templates were diluted 1:10 for subsequent PCR analysis. Differential display PCR was performed in the presence of 10 μCi [α-35S]dATP, 1.6 μm random 10-mer and oligo-dTMN primer using 1 unit of Taq polymerase in 1 × reaction buffer containing 2 mm Mg2+, 2 μm free nucleotides, under the following conditions: 50 cycles of 94° for 30 sec, 40° for 60 sec, and 72° for 30 sec, and one cycle of 72° for 5 min, followed by cooling to 4°. PCR products were ethanol-precipitated and resuspended in 10 μl of 1× loading buffer containing 90% formamide, 10% 10× Tris/borate/EDTA (1 × Tris/borate/EDTA is 90 mm Tris-borate, 2 mmEDTA), bromphenol blue, and xylenecyanol. Samples were displayed on a 6% denaturing polyacrylamide gel. PCR fragments identified as differentially regulated were confirmed by reverse transcription and PCR using RNA isolated from independent induction experiments. PCR fragments of interest were electro-eluted and used for PCR reamplification using the same primer set and the same conditions as described for differential display except that the free nucleotide concentration was increased to 20 μm and no isotope was added. PCR products were subcloned in pBluescript SK+vector and sequenced (Sequenase Version 2.0; United States Biochemical, Cleveland, OH).
Northern analysis.
Twenty micrograms of total RNA was subjected to electrophoresis on a formamide-containing 1% agarose gel. RNA was transferred to a nylon membrane (GeneScreen; New England Nuclear Research Products, Boston, MA) by blotting overnight in 20× SSC (1× = 150 mm NaCl, 15 mm sodium citrate). The membrane was baked in a vacuum oven for 2 hr at 80°. Hybridization was carried out in buffer containing 50% deionized formamide, 5× SSC, 5× Denhardt’s solution, 1% SDS and 10% (w/v) dextransulfate. Before being added to the hybridization solution, the random labeled probe was boiled for 5 min in 500 μl of 10 mg/ml salmon sperm DNA and quickly chilled on ice. Hybridization was carried out for 16–20 hr. Washes were performed in 2× SSC/0.1% SDS at room temperature for 30 min and 2× SSC/1% SDS at 65° for 20 min. Membranes were exposed to X-ray film using intensifying screens or to PhosphorImager screens for 12–48 hr.
cDNA library screening.
All manipulations were carried out according to the suppliers’ protocols. Typically, 300,000 phages were plated on six large 15-cm Petri dishes. Replicas were made using nitrocellulose filters (BA85; Schleicher & Schuell, Riehen, Switzerland). After denaturing and neutralizing, the filters were baked in a vacuum oven at 80° for 2 hr. Prehybridization was performed for 2 hr at 42° in 0.02 m PIPES, pH 6.5, 0.8 mNaCl, 50% deionized formamide, 0.5% SDS, and denatured salmon sperm DNA at a final concentration of 100 μg/ml. The random-labeled probe was added directly to the prehybridization solution. Depending on the length of the probe, hybridization was carried out at different stringencies: 42° for longer fragments, 38° for shorter fragments. Washing conditions were set up as follows: 0.1 × SSC/0.1% SDS at 60° for 2 × 30 min for high stringency and 2 × SSC/0.1% SDS at 45° for low stringency. Washing conditions were for high stringency: 0.1 × SSC/0.1% SDS 2 × 30 min at 60° and for low stringency: 2 × SSC/0.1% SDS 2 × 30 min at 45°. Filters were exposed to X-ray film using intensifying screens for 12–24 hr. Positive clones were isolated and further purified in a secondary and tertiary screening. The pBluescript phagemid was excisedin vivo using the EXASSIST/SOLR system provided with the cDNA library kit.
Computer sequence analysis.
Nucleotide sequences were analyzed by comparison with the GenBank/EMBL nucleotide databases using the FASTA program (28) of the GCG sequence analysis package, version 7.0 (Madison, WI).
Results
Design and optimization of an internal control.
Because it is known that phenobarbital strongly induces CYP2H1 mRNA in chick embryo liver (21, 29), we first designed a nonrandom DDRT-PCR primer pair for this gene product. Observation of the predicted 220-bp PCR product allowed an efficient initial optimization of a number of parameters, including length of the 5′ primer, structure of the 3′ oligo-dTMN primer, RNA preparation, and PCR parameters (data not shown). Indeed, the CYP2H1 product appeared as a strongly inducible signal under optimized conditions (Fig. 1, lane 1). Induction was highly reproducible when RNA from independent experiments was used. The PCR product was excised from the polyacrylamide gel, electro-eluted, reamplified, and subcloned. Sequence analysis confirmed its identity. Furthermore, Northern blotting of liver mRNA using the amplified product as a probe revealed an mRNA size of 3.5 kb and an induction rate of ∼55-fold (Fig. 2), which is in close agreement with previously published data (30). This analysis confirmed the validity of the technique to isolate phenobarbital-responsive genes.

Differential display gel. Chick embryo liver RNA fingerprints. RNA from control (−) and phenobarbital induced (+) chicken embryo livers was reverse transcribed and the resultant cDNA used for PCR amplification with different primer pairs in the presence of [α-35S]dATP. PCR products were resolved on a 6% denaturing polyacrylamide gel and visualized by autoradiography.Lane C, control amplification with a primer pair designed to detect chicken CYP2H1; other lanes,combinations of different oligo-dTMN primers (indicated by the two ultimate bases) and random 10-mers (indicated by numbers).Dots, significantly regulated PCR fragments.
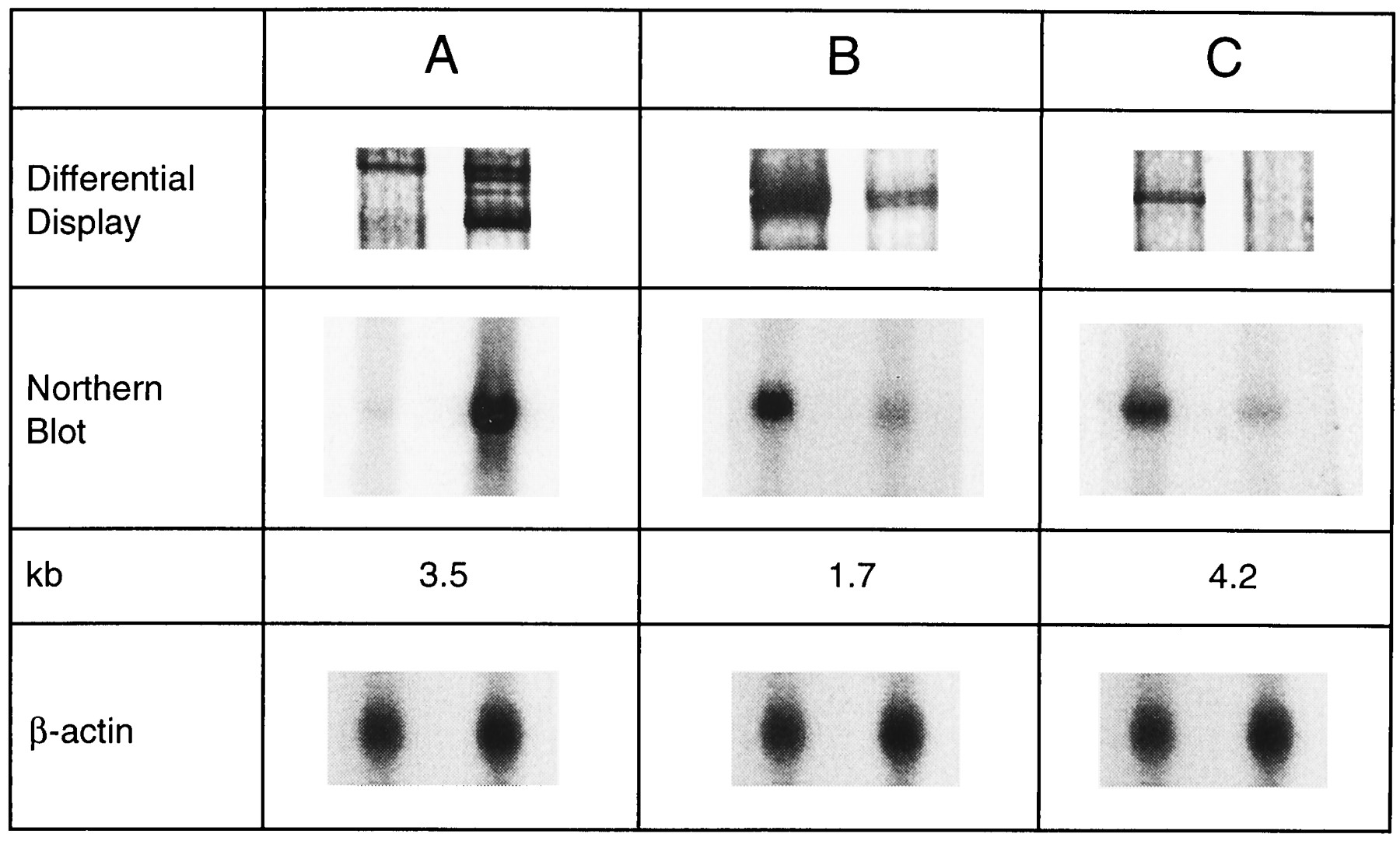
Northern blot analysis of selected genes.Lane A, internal control CYP2H1; lane B,complement factor H; and lane C, fibrinogen. RNA was isolated from chicken embryo liver 48 hr after phenobarbital treatment (+) or from controls (−). Northern blots were performed using the subcloned fragments obtained from differential display as probes.
Effect of phenobarbital on chick embryo liver mRNA.
A total of 25 arbitrary 10-mers combined with 6 of 12 possible oligo-dTMN primers were used to scan approximately 7500 mRNA species, given that an average of approximately 50 individual gene products were amplified and visualized by each pair of primers. The patterns obtained were highly specific for the primer pair used, and little variation was seen in repeated independent experiments. No bands were observed in negative control experiments when reverse transcription was omitted and PCR was performed as usual (data not shown). On the basis of reproducible and visually easily detectable intensity changes, a total of 29 DNA fragments were finally identified as being significantly regulated by the drug treatment. Of these, 18 fragments strongly increased, whereas 11 fragments decreased in intensity (Table 2).
Summary of results for differentially regulated mRNAs of chick embryo liver after a 48-hr treatment with phenobarbital
Identification of regulated gene products.
Of the 29 regulated cDNA fragments, 20 fragments were reamplified and subcloned for further analysis. Nucleotide sequencing revealed that all of them were flanked by sequences complementary to the PCR primers used. Only one product (clone 12) was apparently generated by amplification with a single 10-mer priming at 5′ and 3′ positions, whereas all other products originated as intended. Sequence analysis revealed that three isolated fragments were identical (clones 4, 5, and 13; Table 2). These PCR fragments had been amplified by the same random 10-mer (primer 6) in combination with the three oligo-dTMA primers. This was the only observed case of degeneracy of oligo-dTMN primers, whereas all other isolated DNA fragments had distinct sequences and did not overlap with each other.
Three of the identified genes have already been described in chicken: of these, GST is also known to be inducible by phenobarbital in other species (2), whereas retinal glutamine synthetase and apolipoprotein B have not yet been recognized as being regulated by phenobarbital. Two sequences showed high homologies to genes identified and characterized in other species. Both sequences belong to coding regions of the corresponding genes. Clone 7 shows 72% homology to a coding region of human and murine complement factor H, and clone 16 shares 82%, 78%, and 76% sequence homology to human, Xenopus laevis, and rabbit elongation factor 1δ, respectively. Homologies of all other fragments were either not significant or restricted to short fragments within the PCR products.
Regulation of mRNA expression in vivo.
An important consideration is the verification of the regulation of the identified cDNA fragments by alternative methods (31). It was assumed that Northern blots using these subcloned fragments as probe would permit us to confirm the increase or decrease in the corresponding mRNA. However, when fragments that were positioned directly in front of the polyalanine tail were used, no signals were obtained on Northern blots. It is possible that aminotransferase-rich sequences and the relatively short fragments used as probes are at the root of this observation. We therefore tried to circumvent this problem by directly screening a chicken liver cDNA library using some of the DDRT-PCR fragments as probes. If the isolated cDNA clones contained mainly coding sequences, Northern blotting confirmed the regulation of the respective gene (e.g., for UDP glucuronosyl-transferase). Thus, the differential display method is indeed very sensitive for detecting regulated genes, but confirmation by Northern blotting is not always possible. We also observed that some of the PCR amplification products were generated from annealing of the 3′ primer within the coding sequence of a gene or at a position upstream of the polyalanine tail in the 3′ noncoding region (data not shown). Northern blotting reliably confirmed the effect of phenobarbital on CYP2H1 mRNA by using the subcloned 220-bp cDNA fragment as probe (Fig. 2, left lane). Regulation of fibrinogen β-chain and complement factor H was also confirmed by Northern analysis (Fig. 2, middle and right lanes). The decrease in their expression after 48 hr in vivo treatment by phenobarbital was determined to be 2.5- and 5.5-fold, respectively.
Screening of a chicken liver cDNA library.
Some of the regulated PCR products were isolated, subcloned, and used to screen a chicken liver cDNA library. Three hundred thousand recombinant phages were plated and screened using these PCR fragments as probe. Sequence analysis of isolated positive clones further identified one gene (isolated with fragment 2; Table 2) as the β-chain of chick fibrinogen and another clone (isolated with fragment 19) showed 65% homology to rat fibrinogen γ-chain. The cDNA isolated with fragment 12 showed 78% homology to human, murine, and rat UDP glucuronosyl-transferases, enzymes known to be induced by phenobarbital in other organisms (2, 32, 33). In all of these cases, the original fragments produced by DDRT-PCR were located within the 3′ noncoding region but were located upstream of the polyalanine tail. These sequences could not be identified directly because no sequence information on these noncoding regions was contained in the databases. Several cDNA clones representing other PCR products did not show homology to known genes, which indicated that they were derived from novel genes with as yet undefined functions. Partial sequences of these new genes are presented in Figure 3.

Sequences with no homologies to sequences in database. Sequences representing the primers used for DDRT-PCR are underlined. Underlined letters, polyadenylation signals.
Discussion
The present study provides new information on the extent and complexity of the overall cellular response to the xenobiotic phenobarbital. Our data indicate that phenobarbital significantly affects the expression of presumably more than 50 genes in a liver cell at the transcriptional and/or post-transcriptional level. These data were generated by applying the mRNA differential display technique to chicken embryo liver treated with phenobarbital.
Only cDNA fragments reproducibly amplified in at least two independent experiments and showing at least a 2-fold change in density after phenobarbital treatment were considered for further analysis. Thus, if smaller changes are included, an even greater number of genes will emerge as being influenced by this drug. By using 150 primer combinations, of which each resulted in the display of at least 40–60 distinct bands, approximately 7,500 mRNA species were covered. This represents ∼50% of the estimated 10,000–20,000 cellular mRNAs (34). Of the 27 different genes regulated by phenobarbital, 16 genes were up-regulated and 11 down-regulated.
Our first analysis was performed 48 hr after phenobarbital treatment because chicken CYP2H1 is maximally induced at this time and we wanted to analyze a sustained change in response. This time course of induction may not apply to other genes influenced by phenobarbital and, in particular, to those coding for transcription factors.
In addition to the CYP2H1 used as internal control, two genes already known to be induced by phenobarbital were identified: GST and UDP glucuronosyl-transferase. This finding demonstrated the suitability of the DDRT-PCR technique and of our system. In addition, the identification of genes already known but not described to be regulated by phenobarbital opens the possibility to study their regulation in comparison to genes whose regulations have been studied for many years (e.g., cytochromes P450). Of major interest for systems affected by phenobarbital are two clones with sequences revealing high homology to human, rabbit, rat, and X. laevis sequences—complement factor H and elongation factor 1δ. The reported property of these proteins to bind DNA (35, 36) indicate that the induction mechanism of phenobarbital could involve regulation at different steps and that probably more than a single trans-acting protein might be involved. Complement factor H is a negative regulator of the alternative pathway of the complement system. Earlier studies (35-37) suggest that complement factor H and complement factor B are the two major DNA-binding proteins of human serum. The question is whether factor H may be one of the phenobarbital-modulated transcription factors that trigger the induction of the other genes or just part of the pleiotropic response. Elongation factor 1δ catalyzes the GTP-dependent binding of aminoacyl-tRNA to ribosomes during the elongation step of prokaryotic protein synthesis. It has been shown recently (38) that elongation factor 1δ in Artemia sp. andX. laevis possesses a leucine zipper, which suggests that this motif is conserved and has a special function. It has also been shown that elongation factor 1α is present in the nucleus (39) and has a role in DNA replication (40). Our identification of known genes not yet recognized to be regulated by phenobarbital and the identification of new genes points to a more general effect of phenobarbital on gene expression than previously recognized. The regulated genes are not only involved in metabolic activities (cytochromes P450, GST, UDP-glucuronosyl transferase) but are also part of cellular defense mechanisms (complement factor H), translation activities (elongation factor 1δ), and blood clotting (fibrinogen). Interestingly, the cellular response reveals not only induction but also repression of a considerable number of genes. In fact, almost half of the identified fragments (38%) were significantly repressed. These results underline the complex and very subtle cellular response to phenobarbital, which indicates a regulatory cascade at many different levels of gene regulation.
In conclusion, our data reveal a much more profound change in gene expression in response to phenobarbital than that previously envisioned as restricted to a few drug-metabolizing enzymes. The further identification of the spectrum and time course of the extensive reported down-regulation of genes should allow insight into the initial molecular events associated with mediating the pleiotropic response to phenobarbital.
Acknowledgment
We are grateful to Ms. D. Marek for technical assistance.
Footnotes
-
Send reprint requests to: Urs A. Meyer, Ph.D., Department of Pharmacology, Biozentrum, Klingelbergstrasse 70, University of Basel, CH-4056 Basel, Switzerland. E-mail:meyer2{at}ubaclu.unibas.ch
-
↵1 Current affiliation: Department of Molecular Pharmacology, School of Medicine, Stanford University, Stanford, California 94305-5332.
-
This work was supported by the Swiss National Science Foundation.
- Abbreviations:
- PCR
- polymerase chain reaction
- GST
- glutathione S-transferase
- SSC
- standard saline citrate
- SDS
- sodium dodecyl sulfate
- PIPES
- piperazine-N,N′-bis(2-ethanesulfonic acid)
- bp
- base pair(s)
- dTMN
- oligodeoxythymidine, where M is guanosine, cytidine, or adenosine and N is guanosine, cytidine, adenosine, or thymidine
- dTMA
- oligodeoxythymidine, where M is guanosine, cytidine, or adenosine and A is adenosine
- DDRT-PCR
- differential display of reverse transcribed mRNA amplified by the polymerase chain reaction
- Received August 28, 1996.
- Accepted November 11, 1996.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}