


Abstract
Two of the common mechanisms regulating G protein-coupled receptor (GPCR) signal transduction are phosphorylation and sequestration (internalization). Agonist-mediated receptor phosphorylation by the β-adrenergic receptor kinase (βARK) facilitates subsequent interaction with an arrestin protein, resulting in receptor desensitization. Studies of the β2-adrenergic receptor (β2AR) receptor in human embryonic kidney (HEK) 293 cells indicate that βARK and arrestin proteins (β-arrestins) also regulate sequestration. Consistent with this notion, we show in HEK 293 cells that reduction in or removal of the ability of the β2AR to be phosphorylated by βARK or to interact normally with β-arrestin substantially reduces agonist-mediated sequestration. To evaluate βARK and β-arrestin regulation of β2AR sequestration, we examined the relationship between βARK and/or β-arrestin expression and β2AR sequestration in a variety of cultured cells, including HEK 293, COS 7, CHO, A431, and CHW. COS cells had both the lowest levels of endogenous β-arrestin expression and β2AR sequestration, whereas HEK 293 had the highest. Overexpression of β-arrestin, but not βARK, in COS cells increased the extent of wild-type β2AR sequestration to levels observed in HEK 293 cells. However, a βARK phosphorylation-impaired β2AR mutant (Y326A) required the simultaneous overexpression of both βARK and β-arrestin for this to occur. Among all cell lines, sequestration correlated best with the product of βARK and β-arrestin expression. Moreover, an agonist-mediated translocation of wild-type β2AR and endogenous β-arrestin 2 to endocytic vesicles prepared from CHO fibroblasts was observed. These data suggest not only that the complement of cellular βARK and arrestin proteins synergistically regulate β2AR sequestration but also that β-arrestins directly regulate β2AR trafficking as well as desensitization.
β2AR sequestration is a reversible agonist-stimulated process in which plasma membrane β2AR binding activity decreases while total cell receptor binding activity remains constant (1, 2). β2AR sequestration, first observed during the study of receptor desensitization in frog erythrocytes (3), results from receptor internalization (1, 4). Agonist-dependent desensitization of β2AR responsiveness is primarily a consequence of receptor phosphorylation by GRKs or cAMP-dependent protein kinase, but only GRK-mediated receptor phosphorylation increases β2AR affinity for arrestin proteins (5-10). Recently, some of the mechanisms that regulate β2AR sequestration in HEK 293 cells have been determined; they apparently involve the same GRKs (βARK 1 or 2) and arrestins (β-arrestin 1 or 2) that regulate agonist-mediated receptor phosphorylation and homologous desensitization (11-14).
β2AR sequestration and βARK phosphorylation have previously been considered to be independently regulated processes (6,15, 16). This hypothesis was based on the facts that β2ARs sequester in the combined presence of protein kinase A and βARK phosphorylation inhibitors (16), phosphorylation site-deficient β2AR mutants sequester as well as the wild-type receptor in CHW cells (6, 15), and receptor phosphorylation may occur in the absence of sequestration (16). However, recent observations with the m2 muscarinic acetylcholine receptor and the β2AR (11, 13) are not consistent with this hypothesis and provide direct evidence for a role of βARK-mediated phosphorylation in GPCR sequestration. For example, overexpressed βARK 1 rescued sequestration (11, 14) of a βARK-phosphorylation and sequestration-impaired β2AR mutant (β2AR-Y326A) (17, 18) in HEK 293 cells.
The recent discovery that β-arrestins intimately regulate β2AR sequestration in HEK 293 cells provides a basis for these apparently incompatible observations (12). In these cells, β-arrestin overexpression rescues β2AR-Y326A sequestration but not its impaired phosphorylation. In contrast, overexpression of dominant negative β-arrestin blocks sequestration of either the normally phosphorylated wild-type β2AR or the β2AR-Y326A phosphorylated in the presence of overexpressed βARK 1 (12). At least in HEK 293 cells, βARK and β-arrestin have a dual role. They mediate β2AR homologous desensitization and direct sequestration by their combined interaction with the receptor. Thus, insofar as sequestration of the β2AR presumably represents the cellular pathway by which receptors are dephosphorylated (18-22) and recycled to the plasma membrane as competent receptors, βARK and β-arrestin activities both desensitize the signaling machinery and provide the trigger for its resensitization.
Although agonist-mediated sequestration of the β2AR occurs in all cell types, the extent of sequestration varies appreciably from one type to another. To determine whether an interplay between βARK and β-arrestin in receptor sequestration might underlie these differences and to assess whether these effects occur in cell types other than HEK 293, we examined sequestration of the wild-type β2AR and mutant β2ARs in different cell lines (HEK 293, CHO, A431 human adenocarcinoma cells, CHW, and COS). Our data indicate that the synergistic regulation of β2AR sequestration by βARK and β-arrestin is probably a general phenomenon and that the extent of agonist-mediated sequestration in various cells correlates with the product of the complement of βARK and β-arrestin. In addition, the simultaneous redistribution of the β2AR and β-arrestin 2 to a light vesicular fraction suggests that they remain associated, at least during the initial steps of internalization.
Experimental Procedures
Materials.
The sources for the different reagents have been described previously (12, 17, 18). HEK 293 cells, African Green Monkey fibroblasts COS-7 cells, CHO-K1 cells, and human adenocarcinoma A431 were obtained from American Type Culture Collection (Rockville, MD). CHW#1102 Chinese hamster fibroblasts were from Coriell Cell Repositories (Camden, NJ). Minimal essential medium and Hanks’ balanced salt solution were purchased from Life Technologies (Grand Island, NY). Bioluminescent detection reagents were from DuPont (Wilmington, DE). Normal goat serum and horseradish peroxidase-coupled goat anti-rabbit antibodies were from Jackson Immunoresearch (West Grove, PA). All other chemicals were of reagent grade.
Cell culture.
All cells were grown in media containing 10% fetal bovine serum and either a 1:100 dilution of penicillin/streptomycin or 50 μg/ml gentamicin. A431 and COS cells were grown in Dulbecco’s modified Eagle’s medium plus serum. HEK 293 and CHW cells were grown in minimal essential medium with serum, and CHO cells were cultured in Ham’s F-12 with serum.
Generation of plasmid constructs.
The β2AR and Y326A mutant were each epitope tagged at their amino termini with the 12CA5 (HA) peptide sequence as previously described (11, 17). The 12CA5 epitope-tagged PKA, βARK phosphorylation site-deficient β2AR mutant was obtained by replacing theStuI/AccI cassette containing all the potential βARK phosphorylation sites of the epitope-tagged, PKA site-deficient mutant in pBC (6, 15) with the same cassette from the βARK site-deficient β2AR in pBC. The construct was transferred to pcDNA1/Amp as previously described (11). The generation of the K220M βARK 1 point mutant and subcloning of β-arrestin 1 and 2 cDNAs were performed as previously described (11, 12).
Cell transfection.
Stable transfection of the β2AR in CHW and the β2AR and Y326A mutant in CHO cells has been previously described (6, 17). COS and HEK 293 cells were transiently transfected with wild-type and mutant β2ARs mutant together with the appropriate GRK and/or β-arrestin constructs as previously described (11).
Sequestration assay.
The fraction of sequestered receptor was determined by radioligand binding using CGP-12177 (150 nm final), 125I-pindolol (350–550 pm), and/or 10 μm propranolol or by flow cytometry analysis as previously described (6, 17).
Whole-cell phosphorylation.
COS cells were seeded 1 day after transfection at a density of 0.25–0.75 million cells/25-mm well. Cell labeling with [32PO4]phosphoric acid, immunoprecipitation, protein resolution on 12.5% polyacrylamide gels, and phosphorylation quantification were performed as previously described (11, 14).
Immunoblotting.
The expression levels for βARK 1/2 or β-arrestin 1/2 were determined by immunoblotting using specific antibodies. The generation of anti-βARK 1/2 and anti-β-arrestin 1/2 polyclonal antisera has been previously described (23-25). Equivalent amounts of protein were electrophoresed on polyacrylamide gels (7.5% for βARK 1 and 2 and 12.5% for β-arrestin 1 and 2, respectively) and transferred as previously described (11, 14). The incubation procedure was as previously described (11, 14) except that the membrane was blocked for 1 hr with 5% (v/v) normal goat serum, 1% (w/v) nonfat dried milk, and 0.05% Tween-20 in phosphate buffer saline, pH 7.4.
Subcellular cell fractionation.
CHO cells permanently expressing transfected 12CA5 epitope-tagged β2AR or nontransfected naive cells were grown in 150-mm dishes and stimulated with 10 μm isoproterenol in 100 μmascorbate buffer or with ascorbate alone for 30 min at 37°. The cells were washed twice with ice-cold PBS and incubated for 20 min on ice in PBS containing 0.25 mg/100 ml of concanavalin A (16). The cells from three dishes were each resuspended by gentle scraping after a 10-min incubation with 5 ml of ice-cold PBS containing 5 mm EDTA. They next were centrifuged at 800 × g for 10 min. The cell pellet was resuspended in 2 ml of cold buffer A (10 mmTris·HCl, pH 7.4, 2 mm EDTA) and incubated for 20 min on ice. Cells were homogenized using a Dounce homogenizer, and nuclei were removed by centrifugation at 400 × g for 10 min. The supernatant was loaded on a stepwise sucrose cushion (4 ml each of 60% and 35% sucrose in buffer A) and centrifuged at 150,000 ×g for 90 min at 4°. After the centrifugation, the supernatant was removed, and the 35% (light) and 35%/60% (heavy) sucrose interface fractions containing the receptor were collected, diluted with buffer A, and centrifuged at 150,000 × gfor 60 min at 4°. The pellets for each fraction were resuspended in Tris-EDTA. Protein from the supernatant was precipitated with 20% trichloroacetic acid for 60 min on ice and centrifuged for 15 min at 14,000 × g, and the remaining trichloroacetic acid was removed with ether. The pellet was resuspended in SDS sample buffer; 25 μg for each protein sample was loaded for SDS-PAGE.
Protein determination.
Protein levels were determined using the BioRad (Richmond, CA) protein assay with bovine serum albumin as the standard.
Steady state distribution of sequestered receptors as a function of βARK and β-arrestin concentrations.
The model presented below results from and adheres to our qualitative observations of β2AR sequestration. It is intended to provide a simplified means to explain and quantify the kinetics of this sequestration and perhaps the sequestration of other GPCRs that use the same regulatory proteins. It asserts that the most important regulation of β2AR sequestration occurs from four processes: (i) βARK phosphorylation represented by rate coefficientk 1, (ii) βARK facilitated β-arrestin binding, which occurs at a rate k 2, (iii) subsequent β-arrestin mediated translocation of the receptor to and internalization via clathrin-coated areas of membrane, represented by rate coefficient k 3, and (iv) externalization of receptor back to the plasma membrane at rate k 4. Further assumptions of the model are listed below.
Assumption a states that the agonist concentration is much greater than the concentration required for 50% receptor occupancy, and the cellular β-arrestin complement is much greater than the cell receptor complement.2
Assumption b states that the rate coefficients for βARK or β-arrestin interaction with the appropriate form of the receptor are proportional to their respective intracellular concentrations and correspond to irreversible processes.
Assumption c states that the time to reach a steady state receptor distribution during a sequestration experiment is short compared with other processes that change total cell receptor number or redistribution.
With these assumptions and R*, the agonist-occupied receptor; R*phos, the phosphorylated receptor; R*phos-
β
arr, the β-arrestin-bound form of the receptor; and Rseq
phos-
β
arr, the sequestered receptor, the rate equations are as follows:
Because
This approach to Seqmax for a given receptor is initially quasilinear as the βARK-β-arrestin product increases if thek 1·k 2/Seqmaxterm is less than the [Externalization rate]·(k 1 + k 2) term. For different receptors in the same cell, the approach may be nearly linear if [Externalization rate]·(k 1 +k 2) remains relatively constant ask 1·k 2 increases.
The relative affinity of a receptor for β-arrestin (i.e., its desensitization rate k c 2) may be calculated by determining its rate of phosphorylation,k 1; externalization, k 4; and maximal sequestration. These parameters can be readily determined experimentally. Analysis of published data for time-dependent sequestration experiments in the literature suggests that the externalization rates of the cell types used here are essentially equal. Results obtained for k 4 from the sequestration data for the respective cell lines were consistent with externalization rates for β2AR in HEK 293 (0.075/min) (12), A431 (0.075/min) (22), CHW (0.085/min) (28), and CHO (0.085/min) (17) and for m2 muscarinic acetylcholine receptor in COS 7 of (0.075/min) (13). Thus, k 3 also can be determined.
Receptor desensitization, sequestration, and resensitization are simultaneously regulated by the expression by a cell of βARK and β-arrestin protein and the inherent affinity of the receptor for these proteins.
Results
Effect of removal of phosphorylation sites in the β2AR on sequestration.
Fig. 1demonstrates the effect that impairment of β2AR/β-arrestin interactions (12) has on receptor sequestration. Removal of the phosphorylation sites in the carboxyl tail of the β2AR has been shown to prevent its βARK-mediated but not PKA-mediated phosphorylation in HEK 293 cells (11). As indicated in the figure, this mutation also reduces relative sequestration by approximately half, from 39 ± 5% for wild-type receptor to 22 ± 4% for the β2AR- Phos− mutant. Similarly, overexpression of dominant negative β-arrestin 1-V53D reduces relative sequestration of wild-type β2AR receptor by approximately one third to 28 ± 5% and the Phos− mutant sequestration by an additional 45% to 12 ± 4%. In contrast, overexpression of β-arrestin rescues the sequestration of the β2AR-Phos− mutant to 41 ± 8%, which is comparable to wild-type levels of 46 ± 4%.

Effects of the dominant negative β-arrestin/V53D mutant on β2AR and the β2AR phosphorylation site-deficient mutant (β2AR-Phos−) in HEK 293 cells. HEK 293 cells were transfected with the respective receptor cDNA and either empty vector, β-arrestin-V53D, or β-arrestin cDNA. Sequestration was measured by flow cytometry (see Experimental Procedures) and is expressed as a percentage loss of cell surface receptor. A significant difference was observed between wild-type receptor and β2AR-Phos− sequestration in the absence (Mock, p = 0.004) and presence (p = 0.006) of overexpressed β-arrestin/V53D. β-Arrestin/V53D also reduced sequestration of wild-type receptor (p = 0.05) and the mutant receptor (p = 0.04) by ∼30–45%. No significant difference was seen between sequestration of each receptor in the presence of overexpressed β-arrestin (p = 0.29). Results are mean ± standard deviation of three or four separate experiments.
Effect of overexpression of βARK and β-arrestin on the sequestration of the β2AR and Y326A mutant receptor in COS cells.
As shown in Fig. 2 (top), the β2AR sequestered poorly (8 ± 4%) in COS cells in the presence of endogenous levels of βARK or β-arrestin, whereas overexpression of wild-type βARK 1 slightly increased it to 14 ± 2%. In contrast, with overexpression of β-arrestin (1 or 2), β2AR sequestration increased to the range observed in HEK 293 cells (>30%) in the absence (34 ± 6%) or presence (39 ± 6%) of overexpressed βARK 1.

Effect of overexpression of βARK and β-arrestin on the sequestration of β2AR and β2AR-Y326A in COS cells. Cells were transfected with the β2AR or Y326A mutant without (Mock) or with 1 μg of plasmid containing βARK and/or β-arrestin cDNA. Sequestration was determined by flow cytometry analysis as described in Experimental Procedures. Sequestration is expressed as the percentage loss of cell surface receptor. Results are mean ± standard deviation of three to five experiments.
The βARK phosphorylation-impaired Y326A receptor does not sequester in COS cells (1.5 ± 11%; Fig. 2, bottom), a behavior that is also observed in CHO and HEK 293 cells. Overexpression of βARK 1 resulted in a sequestration of 9 ± 6%, and overexpression of β-arrestin alone only increased it to 15 ± 7%. The full rescue of Y326A mutant sequestration (39 ± 7%) was observed only when βARK 1 and β-arrestin were coexpressed. Coexpression of the dominant negative βARK 1/K220M and β-arrestin (data not shown) promoted sequestration no better than did β-arrestin alone (16 ± 3%), indicating that the catalytic activity of βARK 1 is required to promote the full potency of β-arrestin to rescue the Y326A sequestration.
Effect of overexpression of βARK and β-arrestin on the phosphorylation of the β2AR and Y326A receptors in COS cells.
In COS cells, the Y326A mutant was phosphorylated 10% as well as the wild-type receptor (Fig. 3, leftand right). Overexpression of βARK 1 alone or in combination with β-arrestin 1 increased the phosphorylation level of the mutant to that observed for the wild-type receptor. Overexpression of β-arrestin 1 alone did not significantly affect the phosphorylation level of the Y326A mutant but decreased that of the wild-type β2AR significantly, suggesting that β-arrestin 1 may be competing for the ability of endogenous kinases to interact with receptor or facilitating sequestration before kinase interaction. Expression of both βARK 1 and β-arrestin produced the same phosphorylation level as βARK 1 alone and overcame the apparent inhibiting effect of β-arrestin alone (Fig. 3, left andright).

Effects of βARK and β-arrestin on the phosphorylation of the β2AR and β2AR/Y326A mutants. Cells were transfected with either the β2AR or Y326A mutant without (MOCK) or with 1 μg of plasmid containing βARK (BARK1) and/or β-arrestin (BARR1) cDNA. Cells were metabolically labeled with [32PO4], the receptors were immunoprecipitated with 12CA5 antibody directed against the epitope (see Experimental Procedures) and resolved on polyacrylamide gels, and the radioactivity migrating at the position of the glycosylated receptor (molecular mass, 50–80 kDa) was quantified using a PhosphorImager. Data for β2AR and Y326A were normalized to the increase in radioactivity above basal values obtained with the β2AR without any additional kinases (3.6 ± 1.0-fold above basal = 100%). Left, autoradiogram from a representative experiment. Right, quantitation of the relative phosphorylation levels. Results are mean ± standard deviation of three to five experiments.
Subcellular localization of β-arrestin during receptor sequestration.
To gain better insight into how β-arrestin regulates β2AR sequestration, we determined the subcellular localization before and after agonist stimulation. The β2AR has been shown to “accumulate” in a light vesicular fraction after agonist exposure. These vesicles contain endosomal markers and are believed to represent the population of endocytosed receptors (2, 29, 30). After exposure to agonist, as shown in Fig. 4, β-arrestin 2 (but not β-arrestin 1) levels increased 500-1000% in the light membrane fraction as opposed to a 50–100% increase in the heavy (membrane) fraction of CHO cells permanently transfected with the wild-type β2AR. No increase was seen in the untransfected control cells or in cells transfected with the sequestration-defective Y326A mutant. The CHO cells were the only permanent line that expressed epitope-tagged receptor for which monoclonal antibodies were available. Attempts to duplicate this experiment in transiently transfected HEK 293 cells or COS cells containing epitope-tagged receptor were unsuccessful, with the gels showing no consistent shift of β-arrestin among fractions.

Agonist-induced subcellular distribution of βARK 1 and β-arrestin in CHO cells. Permanently transfected CHO cells expressing the β2AR were fractionated on sucrose gradients as described in Experimental Procedures. Twenty-five micrograms of the supernatant, light vesicular (35%) and membrane fractions (60%) sucrose interface was resolved on PAGE, transferred to nitrocellulose, and probed with GRK 1/2 and β-arrestin antisera. Most of the cell β-arrestin resides in the soluble fraction (left band). No agonist-induced colocalization of βARK with receptor was observed (data not shown). The gel of β-arrestin immunoreactivity is representative of three experiments. β-Arrestin 1 migrates more slowly than β-arrestin 2 (markers on right of gel).
β-Arrestin expression, βARK 1 expression, and β2AR sequestration in various cell lines.
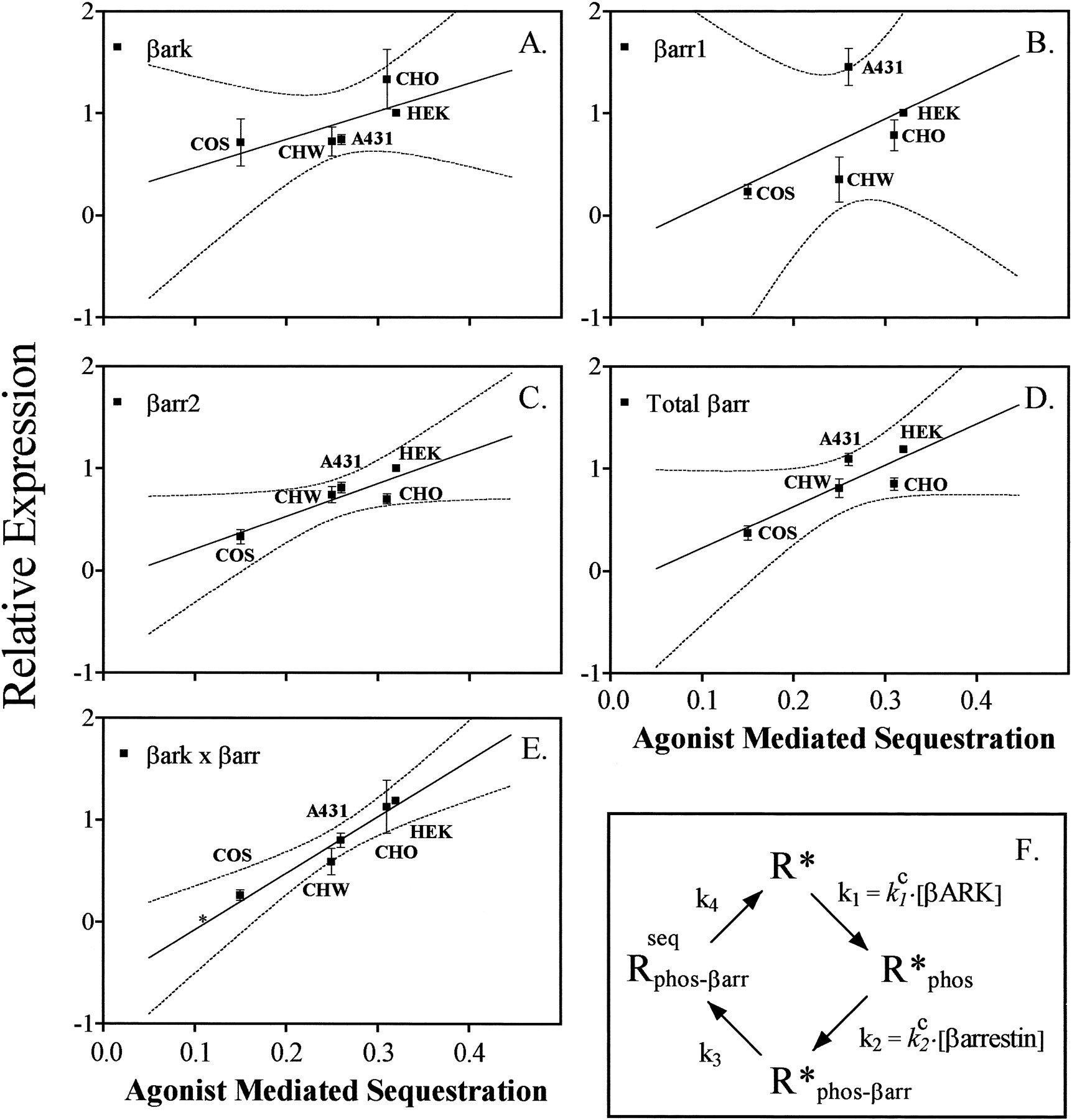
Both βARK and β-arrestin affect β2AR sequestration in HEK 293 cells. This suggests that sequestration is a function of the content of cellular βARK and/or β-arrestin. The relative expression of β-arrestin 1 and 2 or βARK 1 and 2 was assessed using specific rabbit antisera (Fig. 5), and the steady state sequestration of the β2AR in each of the cell lines (Fig.6) was measured by radioligand binding. The expression levels of βARK 1 and β-arrestin 2 in HEK 293 cell were each arbitrarily assigned a value of 1. The relative expression levels for these same proteins in other cell types were scaled to these arbitrary unit definitions. Agonist-induced sequestration varied from a high of 32% in HEK 293 cells to a low of 15% in COS cells. The relative levels of βARK 1 (Fig. 5) were greatest in CHO (1.33 ± 0.29) cells and least in COS cells (0.71 ± 0.03), which contained nearly 50% less βARK 1. HEK 293 cells expressed the most total β-arrestin/mg of protein (1.19, in arbitrary units), and COS cells expressed the least (0.37 ± 0.07). COS cells also had the lowest levels of β-arrestin 1 (0.23 ± 0.07), whereas A431 cells had the highest (1.45 ± 0.18). The correlation between sequestration in the different cell types and the content of these proteins (Fig. 6) was determined by linear regression analysis. Individually, only β-arrestin 2 had a significant correlation with sequestration among the cell types (r 2 = 0.78). However, sequestration had the best correlation with the product of βARK 1 and total β-arrestin concentration (r 2 = 0.94).

Expression of βARKs and β-arrestins and sequestration of the β2AR in different cell lines. Sixty micrograms of total cellular protein was resolved by SDS-PAGE and transferred onto nitrocellulose. The relative amounts of βARK 1 (BARK), β-arrestin 1 (Barr 1), and β-arrestin 2 (Barr 2) expressed in each of the cell types were determined using rabbit polyclonal antibody raised against each respective protein and horseradish peroxidase-conjugated secondary antibodies using the ECL system (see Experimental Procedures). Photograph is a composite of representative 10% gels developed using each antibody. Bars on right, position of the proteins. For all cell lines, no βARK 2 was observed above the background as a separate band from βARK 1 on 7.5% gels (two experiments), which is in contrast to the βARK 2 standard, which ran faster than that for βARK 1. βARK 2 would not be resolved as a separate band on the 10% gel. β-Arrestin 1 antibody (middle) is capable of resolving the endogenous cellular quantities of both β-arrestin subtypes (subtype 1 runs slower than 2), whereas anti-β-arrestin 2 antibody (bottom) has much lower affinity for β-arrestin 1. The amounts of total βARK, β-arrestin 1, or β-arrestin 2 were quantified relative to their expression in HEK 293 cells (right column).

Sequestration of the β2AR in different cell lines relative to expression of βARKs and β-arrestins. Relative amounts of BARK, β-arrestin 1, or β-arrestin 2 among the different cells were determined as described in the legend for Fig. 5. The absolute amount of β-arrestin 1 relative to β-arrestin 2 was determined for HEK 293 cells using the known relative affinity of anti-β-arrestin 1 antibody for β-arrestin 1 and β-arrestin 2 (3.6 ± 0.13 more sensitive to β-arrestin 1) and the measured expression of β-arrestin 1 to β-arrestin 2 in this cell line (0.68 ± 0.13). The total amount of β-arrestin for each individual cell line relative to HEK 293 cells could then be computed as: total β-arrestin = β-arrestin 2 + 1/α × β-arrestin 1, where α = (3.6/0.68) = 5.3 ± 0.19. The amount of βARK 2 in each cell type was below the level of sensitivity of the anti-βARK antibody, so total βARK is reported as βARK 1. The amount of β-arrestin 1 was < 20% of the total amount of β-arrestin in all cell types, except for A431 cells, where it was approximately one third of the total. Sequestration and β2AR expression levels were assessed by ligand binding. Expression levels were 1.0 pmol/mg of protein for HEK 293 cells, 1.0 pmol/mg for CHO, 0.5 pmol/mg for CHW, and 200 fmol/mg for A431 and COS cells. Results are mean of three experiments. The correlations between sequestration and either β-arrestin 1, β-arrestin 2, total βARK, total β-arrestin, or the product of total βARK and total β-arrestin (▪) are shown in A–E. The degree of correlation is reflected by the r 2 value (range, 0–1) determined by linear regression analysis (GraphPAD Prism), with 0 signifying no correlation, and 1 signifying perfect correlation. Only β-arrestin 2 (r 2 = 0.78) and the product of total βARK and total β-arrestin (r 2 = 0.94) are significantly related to sequestration (i.e., the regression lines have nonzero slope, p = 0.047 andp = 0.006, respectively). Dotted lines, 95% confidence intervals. E, Position for sequestration of wild-type β2AR in a cell without βARK (∗, not used in the analysis) (L. Barak, unpublished observations). F, Simple model relating the amount of sequestered receptors at equilibrium, Rseq phos- β arr, with the total amount of surface receptors, R* + R*phos+ R* phos- β arr, in an experiment performed with a large excess of agonist. The rate-limiting steps for sequestration are assumed to be due to βARK phosphorylation, rate constant k 1; β-arrestin receptor binding with internalization, rate constantk 2; downstream internalization events, rate constant k 3; and receptor externalization, rate constant k 4. Under these conditions, the sequestered fraction Rseq phos- β arr/(R* + R*β ARK + R*phos- β arr + Rseq phos- β arr) =k 1 k 2/[k 2 k 3+ k 1 k 3 +k 1 k 2 (1 +k 4/k 3)].
Discussion
The data presented in this work suggest a fundamental role for βARK and β-arrestin in the regulation of agonist-mediated β2AR sequestration and extend the initial observations made with the m2 muscarinic acetylcholine receptor and β2AR in HEK 293 cells (11-14). Our conclusion stems from studies of the sequestration of the β2AR-Phos− mutant in HEK 293 cells, the behavior of the β2AR and Y326A mutant in COS cells in the absence and presence of overexpressed βARK and β-arrestin, the colocalization of β2AR and β-arrestin 2 in the light vesicular fraction in CHO cells, and the correlation between sequestration and the product of βARK and β-arrestin expression in five different cell types.
The impaired sequestration of the β2AR-Phos−mutant in HEK 293 cells demonstrates the importance of phosphorylation and the necessity of β-arrestin for normal agonist-mediated receptor sequestration. Removal of βARK sites in the Phos− mutant decreases its sequestration to about one half of wild-type, suggesting that other factors are also involved. Overexpression of the V53D β-arrestin mutant similarly reduces wild-type β2AR sequestration to one half, suggesting that interaction with β-arrestin is important for sequestration. Moreover, the V53D mutant substantially reduces Phos− receptor sequestration, suggesting that β-arrestin interaction is an obligatory step for sequestration. Even though GRKs may not be absolutely necessary for sequestration (11) as the β-arrestins apparently are, in the absence of GRK and without a compensatory increase in β-arrestin expression, agonist-mediated receptor internalization will be markedly reduced. Thus, the role of GRKs in enhancing β-arrestin/receptor complex formation (8, 9) may in certain cells be a critical, rate-limiting component, not only of desensitization but also of receptor trafficking and resensitization.
A seemingly unusual result of these studies is that β-arrestin 2 and not β-arrestin 1 was colocalized into the light vesicular fraction along with the β2AR in CHO cells. Possible explanations for this include the differential affinity of the two β-arrestins (β-arrestin 2 > β-arrestin 1) for the β2AR (9), the 4-fold greater content of β-arrestin 2 in CHO cells than β-arrestin 1, or that β-arrestin 2 seems to have a 5-fold greater affinity for clathrin than does β-arrestin 1 (31). We were unable to study this colocalization phenomenon in the other permanent lines, perhaps because in the CHO cells, the receptors were not only epitope tagged but also expressed in a 5-fold greater amount.
In COS cells, the wild-type β2AR and the βARK phosphorylation-impaired Y326A mutant sequester poorly. Interestingly, these cells contain the lowest endogenous levels of βARK and by far the lowest endogenous levels of β-arrestin of the cells tested. The sequestration of the wild-type receptor is fully enhanced to levels comparable to HEK 293 cells by overexpression of β-arrestin alone (but not βARK), whereas rescue of phosphorylation and sequestration for the Y326A mutant requires βARK and β-arrestin. In HEK 293 cells, overexpressed βARK was sufficient to rescue the phosphorylation and sequestration of the Y326A mutant receptor (11, 12,14). These results demonstrate that the sequestration impairment of COS cells is mainly due to a low β-arrestin complement, but as in HEK 293 cells, βARK-mediated phosphorylation also plays a regulatory role in β2AR sequestration.
The inhibition of β2AR sequestration by dominant negative β-arrestin (12) suggests that β-arrestins direct receptor trafficking by functioning as adaptor proteins and targeting β2AR to clathrin-coated vesicles or other internalization pathways (26). The colocalization and agonist-mediated translocation of β-arrestin and β2AR to the light vesicular fraction of CHO fibroblasts support this proposed function of β-arrestin and further suggest that receptor resensitization may require endosomal β-arrestin dissociation in addition to receptor dephosphorylation by endosomal phosphatases (21).
β-Arrestin, when present in sufficient concentrations in the absence of βARK phosphorylation, is sufficient for β2AR sequestration (12), but under normal physiological conditions, the absence of either protein may be rate limiting. This interdependence between βARK and β-arrestin in regulating sequestration is reflected by the data shown in Fig. 6, in which the steady state sequestration seems to be best correlated with the product of total endogenous βARK and β-arrestin concentrations rather than with either one alone. Possibly, a more direct approach to this problem would be to measure the initial internalization rates (near zero time) rather than the steady state sequestration. However, uncertainties in the relative amount of receptor initially internalized would preclude obtaining significantly greater accuracy for the βARK-β-arrestin product (i.e., internalization rates) compared with steady state experiments, in which receptor externalization rates are required (see Experimental Procedures).
The simple model shown in Fig. 6f provides a qualitative and a semiquantitative basis for this correlation. Its purpose is to demonstrate that the experimental correlation of the βARK-β-arrestin product to sequestration is consistent with the general phenomenological observations made concerning βARK, β-arrestin, and sequestration. Although its simplicity implies that it may not necessarily be the best description for a series of complicated processes resulting in sequestration, the model provides a useful formalism for their interpretation. For a sequestration experiment represented by the model and performed in the presence of large amounts of agonist, receptor probably becomes stabilized in an activated form R* (32), in which βARK phosphorylation rather than agonist binding becomes the rate-limiting step. Phosphorylation, R* → R*phos, promotes β-arrestin binding, R*phos → R*phos- β arr, and β-arrestin-directed internalization, R*phos- β arr → Rseq phos- β arr. The receptor and β-arrestin migrate to endosomes, where β-arrestin presumably dissociates and the receptor is dephosphorylated (33). The receptor subsequently recycles back to the plasma membrane, where it again encounters saturating concentrations of agonist, Rseq phos- β arr→ R* (it initially goes to state R, but in the presence of saturating amounts of agonist, the concentration of unbound, free receptor R quickly approaches zero). The rate coefficients for each step are defined as k 1, k 2,k 3, and k 4, respectively, and the total amount of receptor RT = R* + R*phos + R*phos- β arr + Rseq phos- β arr. The steady state fraction of sequestered receptor, (Rseq phos- β arr)/RT=k 1 k 2/[k 2 k 3+ k 1 k 3 +k 1 k 2(1 +k 4/k 3)]. Under conditions of normal cellular protein expression, one might expect the kinetic rate coefficients for phosphorylation and β-arrestin-directed activity to be proportional to their respective concentrations (i.e.,k 1 =k 1 c × [βARK] andk 2 =k 2 c × [β-arrestin]), suggesting that sequestered receptor is proportional tok 1·k 2 =k 1 c·k 2 c× [βARK]·[β-arrestin] (see Experimental Procedures). Thus, in this model, the sequestration kinetics of a particular GPCR (e.g., the Y326A mutant β2AR) are regulated not only by the concentrations of βARK or β-arrestin but also by the affinity of that receptor for each protein, as reflected by thekc j values.
We examined the contributions of βARK and β-arrestin to the sequestration of the β2AR in different cell types and demonstrate that βARK and β-arrestin synergistically regulate β2AR sequestration, supporting an adjunct role for βARK in β-arrestin-directed β2AR sequestration. This view explains how it could have been concluded based on the results of previous investigations that sequestration was independent of receptor phosphorylation (1). These earlier studies preceded the realization of the general involvement of arrestin proteins in the mechanisms of GPCR regulation, so it was not possible to consider their relationship to receptor sequestration and the interdependence of β-arrestin and GRK in receptor behavior.
Given the ubiquity of both GRKs and arrestin proteins in mammalian cells, these two families of proteins probably regulate the endocytosis of a large number of GPCRs.3 This can be further supported by studies with the angiotensin II type 1A receptor, another prototypic GPCR. It does not seem to use βARK/β-arrestin dependent sequestration but can be coerced to use this pathway in cells in which β-arrestin is increased (26). Because efficient GPCR resensitization may require internalization, the synergy between GRKs and arrestin proteins may be equally important in the regulation of GPCR resensitization as it is believed to be in the dampening of GPCR signaling. These considerations may be important in understanding the pathophysiology of diseases involving GPCRs, such as congestive heart failure (34, 35), in which GRKs are elevated and receptor desensitization is persistent.
Acknowledgments
We thank Lucie Bertrand for technical support, Dr. Kathy Krueger for help in standardizing β-arrestin antibody, and Dr. Terry Kenakin for comments concerning the manuscript.
Footnotes
- Received November 21, 1996.
- Accepted February 12, 1997.
-
Send reprint requests to: Dr. Larry Barak, Box 3287, Department of Cell Biology, Duke University, Durham, NC 27710. E-mail:lbarak{at}cellbio.duke.edu
-
↵1 Current affiliation: BioSignal Inc., Montreal, Quebec, Canada H3J IR4.
-
↵2 The use of purified β-arrestin in preliminary experiments indicates endogenous cell β-arrestin is in excess of receptor by 100-fold, which is in agreement with the observations of others (35).
-
↵3 J. Zhang and M. G. Caron, unpublished observations.
-
This work was supported in part by National Institutes of Health Grant NS19576, a Bristol Myers Squibb Unrestricted Grant Award (M.G.C.), a K-08 award HL03422 (L.S.B.), and a Michael Smith Postdoctoral Fellowship from the MRC Canada (S.S.G.F.).
Abbreviations
- β2AR
- β2-adrenergic receptor
- GPCR
- G protein-coupled receptor
- βARK
- β-adrenergic receptor kinase
- PKA
- protein kinase A
- PBS
- phosphate-buffered saline
- HEK
- human embryonic kidney
- CHO
- Chinese hamster ovary
- CHW
- Chinese hamster fibroblasts
- COS
- African Green Monkey fibroblasts
- SDS
- sodium dodecyl sulfate
- PAGE
- polyacrylamide gel electrophoresis
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}