


Abstract
Western blot analysis was used to determine the concentration of the aryl hydrocarbon receptor nuclear translocator (ARNT) protein and aryl hydrocarbon receptor (AHR) in 11 mammalian cell culture lines derived from hepatic and nonhepatic tissues. The strategy was to first use Western blot analysis to determine the expression of ARNT or AHR in each cell line relative to its concentration in murine wild-type Hepa-1c1c7 (Hepa-1) cells. Actual ARNT and AHR concentrations in known amounts of total cell lysates were then determined by generating a standard curve with defined amounts of a highly purified ARNT or AHR protein and performing regression analysis. The results show that the level of ARNT expression in each of the cell lines is similar and represents ∼0.001–0.002% of total cellular protein. The range of expression was only ∼3-fold with wild-type Hepa-1 cells expressing the highest level of ARNT (33,000/cell) and canine kidney cells (MDCK line) expressing 14,000 ARNT molecules/cell. In contrast, the concentration of AHR varied by 65-fold over the different cell lines with the wild-type Hepa-1 expressing 323,000 AHR/cell and rat hepatoma cells (H4IIE) expressing 4700. The ratio of AHR to ARNT ranged from 0.3 in H4IIE cells to 10 in the Hepa-1 line with the majority of cells expressing 1–5 times more AHR than ARNT protein. Immunocytochemical staining of each cell line showed that ARNT was exclusively localized to the nuclear compartment and that a conserved nuclear localization signal mapped to the NH-terminal portion of the protein.
The ARNT protein is a bHLH/PAS transcription factor that seems to have a wide range of functions. It was initially discovered as a necessary component of the AHR signal transduction pathway, where it dimerizes with the AHR to mediate many of the biological responses to halogenated aromatic hydrocarbons (1-4). Recently, however, it has been demonstrated that ARNT can bind to E-box motifs as a homodimer (5-7) and can form complexes with the bHLH proteins SIM and HIF-1α to mediate their biological responses (8-13). In addition, recent reports have described (i) the expression of a second form of ARNT from rat and mouse termed ARNT2 (14, 15), (ii) an alternatively spliced ARNT from rainbow trout that has dominant negative function on AHR-mediated signal transduction (16), and (iii) a lethal phenotype occurring in mice knocked out for ARNT expression (17). Collectively, these reports suggest that ARNT is a protein that plays a central role in mediating many important biological responses and that recruitment of ARNT by one pathway could affect the response of another. Indeed, inhibition of AHR-mediated processes by hypoxic conditions have recently been demonstrated in mouse Hepa-1 cells (11). These results highlight the importance of establishing the concentration of ARNT in cells and determining the ratio of this protein to its plethora of binding partners.
As a first step in determining such ratios, Western blot analysis was used to determine the concentration of ARNT and AHR in 11 cell culture lines. This type of assay is especially important for analysis of ARNT because a method to directly quantify this protein in crude samples has not been reported. The cells analyzed in this study were mouse hepatoma 1c1c7 (Hepa-1 wild-type, type I, and type II), mouse skeletal muscle (C2C12), mouse embryonic fibroblast (NIH-3T3, C3H10T1/2), rat hepatoma (H4IIE), rat smooth muscle (A7), canine osteogenic sarcoma (D-17), canine kidney (MDCK), and human breast cancer (MCF-7). The choice of these lines was based on (i) their traditional use in studies of AHR-mediated signal transduction (Hepa-1, H4IIE, MCF-7), (ii) their increased use in recent years (NIH-3T3, 10T1/2), or (iii) their potential as novel models to study AHR-mediated events in nonhepatic cells. The experiments demonstrate that quantitative Western blot analysis can be used to determine the actual concentration of ARNT and AHR in complex cell lysates. The findings reveal that the ratio of ARNT to AHR ranged from 0.3 to 10 depending on the cell line evaluated with the AHR in excess of ARNT in 9 of the 11 cell lines. In addition, immunocytochemical staining of all cells showed that ARNT was exclusively localized to the nuclear compartment and that a putative nuclear localization signal mapped to the NH-terminal portion of the mouse ARNT.
Materials and Methods
Antibodies and reagents.
Specific antibodies against the mouse ARNT (R-1) have been described previously (18). The antibodies raised against the mouse AHR (A-1A) were generated against a bacterial expressed portion of the mouse AHR (amino acids 1–416) essentially as previously described (18). All antibodies are affinity-purified IgG fractions. Antibodies specific to P450 1A1 were a generous gift from Dr. Colin Jefcoate (University of Wisconsin). GAR-HRP was used for Western blot analysis. GAR-TR was used for immunocytochemical studies. Both of these reagents were purchased from Jackson Immunoresearch (West Grove, PA).
Buffers.
Phosphate-buffered saline contains 0.8% NaCl, 0.02% KCl, 0.14% Na2HPO4 and 0.02% KH2PO4, pH 7.4. The 2× gel sample buffer contains 125 mm Tris, pH 6.8, 4% SDS, 25% glycerol, 4 mm EDTA, 20 mm dithiothreitol, and 0.005% bromphenol blue. Tris-buffered saline contains 50 mm Tris and 150 mm NaCl, pH 7.5. TTBS contains 50 mmTris, 0.2% Tween 20, and 150 mm NaCl, pH 7.5. TTBS+ contains 50 mm Tris, 0.5% Tween 20, and 300 mmNaCl, pH 7.5. BLOTTO contains 5% nonfat dry milk in TTBS. The 2× lysis buffer contains 50 mm HEPES, pH 7.4, 40 mm sodium molybdate, 10 mm EGTA, 6 mm MgCl2, and 20% glycerol. The 1× immunoprecipitation buffer contains 50 mm Tris, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 4% BSA, 1% glycerol, and 50 mm l-histidine. The IP wash buffer contains 50 mm Tris, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.1% SDS, and 0.5% deoxycholate.
Cells and growth conditions.
wild-type Hepa-1c1c7 cells, type I variants, and type II variants were a generous gift from Dr. James Whitlock, Jr. (Department of Pharmacology, Stanford University, Stanford, CA). These cells were propagated in DMEM supplemented with 5% FBS. All other cells were obtained from American Type Culture Collection (Rockville, MD). H4IIE, MDCK, and D-17 cells were propagated in DMEM supplemented with 10% FBS. NIH-3T3 cells were propagated in DMEM supplemented with 10% calf serum. 10T1/2 cells were propagated in BME supplemented with 10% heat-inactivated FBS. C2C12 and A7 cells were propagated in minimum essential medium supplemented with 10% FBS. MCF-7 cells were propagated in minimum essential medium supplemented with bovine insulin (10 μg/ml) and FBS (10%).
Preparation of total cell lysates.
Total cell lysates for Western blot analysis were prepared by sonicating cell pellets in 1× lysis buffer and Nonidet P-40 (0.5%) essentially as previously detailed (16, 19). The only modification to the procedure was that before sonication of the cell pellets, the total number of cells was determined. This allowed the determination of the amount of protein in a single cell (Table 1, columns 1 and 2). Protein concentrations of the total cell lysates were determined by the Coomassie Plus Protein assay (Pierce, Rockford, IL). All samples were stored at −20°.
Concentration of ARNT protein in continuous cell lines
Preparation of purified AHR and ARNT standards.
The purified ARNT protein used in these studies was the BEARNT, that has been previously described (18). Expression and purification of the BEARNT protein was carried out as detailed (18). Stock samples of purified BEARNT were stored at 4° in 0.1% trifluoroacetic acid, and protein concentrations were determined by the Coomassie Plus Protein assay and the BCA protein assay (Pierce). Purified BEARNT fusion protein was then diluted to a concentration of 1 ng/μl in 1× gel sample buffer that contained BSA (1 mg/ml). The presence of BSA was necessary to maintain solubility of the BEARNT protein. This stock solution was then used to generate a graded series of BEARNT standards that were used throughout the project. The typical standard curve was 187.5, 125, 62.5, 50, 37.5, 25, and 12.5 pg of BEARNT.
The purified AHR protein used in these studies was BEAR-2, which represents amino acids 1–416 of the mAHR. The expression and purification of the BEAR-2 protein were carried out as detailed (18). Stock samples of purified BEAR-2 were stored at 4° in 0.1% trifluoroacetic acid, and protein concentrations were determined by the Coomassie Plus Protein assay and the BCA protein assay (Pierce). Purified BEAR fusion protein was then diluted to a concentration of 1 ng/μl in 1× gel sample buffer that contained BSA (1 mg/ml). The presence of BSA was necessary to maintain solubility of the BEAR-2 protein. This stock solution was then used to generate a graded series of BEAR-2 standards that were used throughout the project. The typical standard curve was 1.5, 1.35, 1.05, 0.75, 0.45, and 0.15 ng of BEAR-2.
Western blot analysis.
Purified BEARNT or BEAR-2 and total cell lysates were resolved by denaturing electrophoresis on discontinuous polyacrylamide slab gels (SDS-PAGE) and electrophoretically transferred to nitrocellulose as previously described (18). Immunochemical staining was carried out with varying concentrations of primary antibody (see text and figure legends) in BLOTTO buffer supplemented with dl-histidine (20 mm) for 1–2 hr at 22°. Blots were washed with three changes of TTBS+ for a total of 45 min. The blot was then incubated in BLOTTO buffer containing a 1:10,000 dilution of GAR-HRP for 1 hr at 22° and washed in three changes of TTBS+ as described above. Before detection, the blots were washed in Tris-buffered saline for 5 min. Bands were visualized with the enhanced chemiluminescence kit as specified by the manufacturer (Amersham, Arlington Heights, IL). Multiple exposures (autoradiographs) of each blot were produced to ensure linearity.
Immunoprecipitation.
Total cell lysate (500 μg) was incubated with 5 μg of affinity-purified antibody in the presence of 1× immunoprecipitation buffer for 1 hr at 22°. The samples were put into fresh tubes, supplemented with 10 μl of a 50% slurry containing protein A-Sepharose beads (Sigma Chemical, St. Louis, MO), and incubated for 1 hr at 22°. Samples were then centrifuged at 500 × g for 2 min, and the beads were transferred to fresh tubes. The beads were washed for a total of 45 min with three changes of IP wash buffer, mixed with 25 μl of 2× gel sample buffer, heated for 10 min at 95°, and resolved by SDS-PAGE. Gels were blotted to nitrocellulose and stained with appropriate antibodies as detailed above.
Quantification of protein bands.
For the calculation of ARNT and AHR concentration, the BEARNT or BEAR-2 standard curve was resolved alongside duplicate samples of two concentrations of total cell lysate (5–30 μg). The concentration of target protein was determined by computer analysis of the ECL autoradiographs as previously detailed (16, 19, 20). The density of each standard protein was then plotted against its concentration, and regression analysis used to calculate the concentration of ARNT or AHR/mg of total cell lysate. This value was then multiplied by a factor of 1.7 to adjust for the difference in molecular mass between the BEARNT standard (51 kDa) and the endogenous ARNT protein (86 kDa) or 2.0–2.2 to adjust for the difference in molecular mass between the BEAR-2 standard (47.5 kDa) and the endogenous AHR protein (95–106 kDa)
For each cell line, at least two different gels containing a standard curve (r 2 > 0.9), and two different concentrations of total cell lysate were analyzed. To ensure linearity, multiple exposures of each blot were always evaluated. The mean and standard deviation values of the replicates are reported (Tables 1 and2). To calculate the number of ARNT or AHR molecules in each cell, the fmol of ARNT or AHR/mg of total cell lysate was multiplied by 6.023 × 108 molecules/fmol and divided by the number of cells/mg of total cell lysate.
Concentration of AHR protein in continuous cell lines
Immunocytochemistry.
All immunocytochemical procedures (fixation, staining, and photography) were carried out as previously described (18). The cells were observed on a Zeiss Axiophot microscope using the 568-nm filter. On average, 15–20 fields (5–20 cells each) were evaluated on each slip, and three were photographed to generate the raw data. Experiments were repeated at least three times. The concentrations of antibodies used are detailed in the text.
Amino acid sequence analysis.
ARNT protein sequences were compiled and analyzed by Lasergene software (DNASTAR Inc., Madison, WI). Amino acid comparisons were carried out in the MEGALIGN program according to the method of Jotun-Hein (21).
Construction of ARNT expression vectors and transfection of type II Hepa-1 cells.
The polymerase chain reaction was used to amplify a truncated ARNT fragment from mARNT cDNA. The primers used were 5′-ATATAAGCTTGGCACCATGGGGCTGGATTTTG-ATGATGA-3′ (Trunc-ATG) and 5′-ATATGGATCCCTATTCTGAAAA-GGGGGGAAAACATG-3′ (TAG). The ATG start site is bold, and the restriction sites are underlined. The purified cDNA fragment was cut with HindIII andXbaI and ligated into pRc/CMV to generate pΔNLS. The resulting construct codes for amino acids 46–780 of the mARNT. The full-length mARNT expression vector (pMVmARNT) has been described previously (16). pΔNLS and pMVmARNT were transiently transfected into type II Hepa-1 cells, treated with TCDD, and stained for ARNT as previously detailed (16).
Results and Discussion
Strategy.
Previous reports from this laboratory have established the linearity of Western blotting assays for determining relative changes in AHR and ARNT protein concentration in cell extracts (16, 19, 20). The goal of this study was to develop Western blotting protocols that allowed the determination of the actual concentrations of ARNT and AHR in lysates prepared from cultured cells. The strategy for these studies was to first use Western blot analysis to determine the expression of ARNT or AHR in each cell line relative to its concentration in wild-type Hepa-1 cells. The actual concentrations of ARNT or AHR in known amounts of total cell lysates were then determined by regression analysis using a standard curve of highly purified ARNT or AHR protein.
Calculation of ARNT concentration in hepatic and nonhepatic cell culture lines.
Experiments were designed to calculate the concentration of ARNT in total cell lysates. In the first set of experiments, the relative level of ARNT expression was determined in each cell line by resolving identical concentrations of total cell lysates on the same gel, staining for ARNT and comparing the values to that observed in wild-type cells. Fig. 1A shows a representative blot of total cell lysates stained with the R-1 antibody. A predominate band migrating at ∼86-kDa was observed in all cell lines except the Hepa-1 type II cell line that is defective in ARNT protein expression (18, 22). An additional band migrating at ∼60 kDa was also observed in every cell line except those derived from canine cells. This protein does not represent an ARNT degradation product as it is observed in the type II cells and shows no correlation to the level of ARNT expression. This may represent an additional ARNT isoform or nonspecific reactivity to a highly expressed protein and was not considered in the analysis. The 86-kDa bands were then quantified by computer densitometry as detailed in Materials and Methods, and all values were compared with the level of expression in wild-type Hepa-1 cells. The values are reported in Table 1 (column 4) and show a range of ARNT expression of ∼3-fold (compare the A7 with the wild-type).

Western blot analysis of ARNT and BEARNT expression. A, 10 μg of the indicated total cell lysates was resolved by SDS-PAGE, blotted to nitrocellulose, and stained with 1.0 μg/ml R-1 followed by GAR-HRP (1:10,000). WT, wild-type.Numbers on right, migration of molecular mass standards (kDa). B, Graded concentrations of BEARNT were resolved by SDS-PAGE, blotted to nitrocellulose, and stained with 1.0 μg/ml R-1 followed by GAR-HRP (1:10,000). Bands were quantified by densitometry and plotted against the dilution starting with the 250-pg sample (lane 1). Inset, Western blot. BEARNT concentrations are 250 pg (lane 1), 187.5 pg (lane 2), 125 pg (lane 3), 62.5 pg (lane 4), 50 pg (lane 5), 37.5 pg (lane 6), 25 pg (lane 7), and 12.5 pg (lane 8).
The actual concentration of ARNT was then determined in each cell line by comparing the ARNT concentration in total cell lysates with that of a highly pure ARNT protein standard. The ARNT protein used as the standard for these experiments was a highly pure bacterially expressed mARNT (BEARNT) that corresponded to amino acids 318–773. This is the same protein fragment that was used as an antigen to generate the R-1 antibodies (18). Because the integrity of the analysis was based on the concentration of the BEARNT standard, the stock sample of BEARNT was quantified by a Coomassie protein assay and by the BCA protein assay with BSA as the standard. The BCA assay produced values that were on average 25% higher than the Coomassie dye-based assay. To determine the nature of this observation, the purity of the BEARNT was evaluated by SDS-PAGE. The purified BEARNT migrated as a single band of ∼47 kDa.1 However, the intensity of the Coomassie-stained bands were 25% lower than identical amounts of BSA.1 These results are consistent with the protein assay data and suggest that the BEARNT does not bind the same amount of Coomassie dye as BSA. Because of this, the BEARNT concentration determined by the BCA method was used for all analyses.
Fig. 1B shows a Western blot of a representative BEARNT standard curve. The curve was linear over a 20-fold range of BEARNT protein with a correlation coefficient of .96. The R-1 antibody could detect as little as 12.5 pg of BEARNT when used at a concentration of 1 μg/ml BLOTTO. Similar results were observed when stock BEARNT samples were repeatedly used over a 6-week period. The concentration of ARNT in the various cell lines was then determined by resolving at least two concentrations of total cell lysate on the same gel with a BEARNT standard curve, blotting to nitrocellulose, and staining with the R-1 antibody. The resulting autoradiographs were then analyzed by densitometry, and regression analysis was performed as detailed in Materials and Methods. Each cell line was evaluated on at least three different blots, and the results are presented in Table 1. The actual concentration of ARNT correlated well (r 2 = .78) with the values determined when all lysates were resolved on the same gel and expressed as a percentage of ARNT in wild-type Hepa-1 cells (compare Table 1,columns 4 and 5). The majority of the variation between the two assays was due to the two canine cell lines.
The results show that the level of ARNT expression in each of the cells lines is similar and represents ∼0.001–0.002% of total cellular protein. The range of expression was only ∼3-fold, with wild-type Hepa-1 cells expressing the highest level of ARNT (33,000/cell), whereas the MDCK cells express ∼14,000 ARNT molecules/cell. This level of expression is consistent with certain hormone receptors and other transcription factors (23) and is also consistent with a previous report that measured the concentration of ARNT in Hepa-1 cells (24). However, there are some caveats within the experimental design that deserve comment. First, although the R-1 antibody is polyclonal to a large portion of mARNT (464 amino acids) that shows high homology among rat, mouse, and human (1, 15, 25), it is possible that the ARNT from each species does not bind to an identical amount of R-1 antibody. Second, our recent report on ARNT expression in rainbow trout has shown that alternative RNA splicing can produce distinct ARNT isoforms with divergent COOH-terminal ends (16). If similar mechanisms are functioning in these mammalian cells, then the R-1 antibody may not detect the additional forms of ARNT. Finally, the contribution of the ARNT2 isoform to the total pool of ARNT in each cell has not been determined, and the results are expressed in relation to the conventional ARNT isoform.2Therefore, it is important to note that although all attempts have been made to maximize the integrity of the analysis, the total pool of ARNT isoforms may be higher than that reported here.
Calculation of AHR concentration in hepatic and nonhepatic cell culture lines.
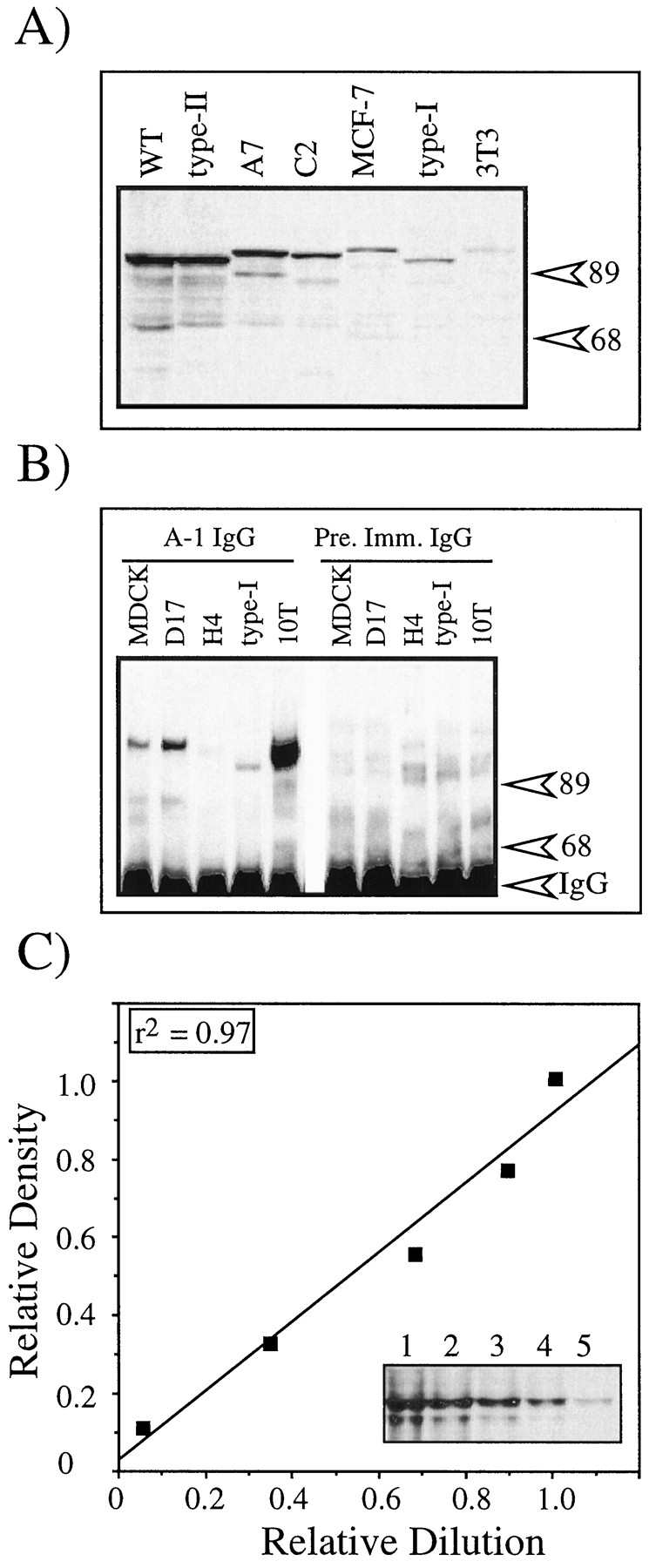
Having established the feasibility of the Western blotting approach for calculating levels of ARNT, we focused on the use of this method to determine the concentration of the AHR. Identical cell lysates to those used for analysis of ARNT were resolved by SDS-PAGE, blotted to nitrocellulose, and stained with the A-1A antibody as detailed in Materials and Methods. Fig.2A shows a representative Western blot containing 20 μg of total cell lysate from seven of the cell lines. A single band is prominently observed in wild-type Hepa-1, type II, C2C12, A7, and MCF-7 cells that correlates with the deduced molecular mass of the AHR in the mouse (26), rat (27), and human (28). A weakly stained band can be observed in 3T3 and type I cells that also correlates with the expected size of the AHR. Because the AHR expression in 20 μg of total cell lysates of MDCK, D-17, H4IIE, and type-I cells were difficult to visualize on standard Western blots, 500 μg of total cell lysate was immunoprecipitated with the A-1A antibody, resolved by SDS-PAGE, blotted, and then stained with the A-1A antibody to confirm the expression of the AHR in these cells. Fig.2B shows a representative blot of the immunoprecipitated AHR from MDCK, D-17, H4IIE, type-I, and 10T1/2 lysates (as high expression control). The low level of AHR expression relative to 10T1/2 cells can be observed. The intensity of each AHR band was then quantified by computer densitometry as detailed for ARNT, and the results are expressed as a percentage of AHR in Hepa-1 cells. These values are shown in Table 2 and range from 3% in type I and 3T3 cells to 72% in the Hepa-1 type II cell line. The 30-fold range of AHR expression is in sharp contrast to the small range of ARNT expression (Fig. 1A).

Western blot analysis of AHR and BEAR-2 expression. A, 20 μg of the indicated total cell lysates was resolved by SDS-PAGE, blotted to nitrocellulose, and stained with 1.0 μg/ml A-1A followed by GAR-HRP (1:10,000). WT, wild-type.Numbers on right, migration of molecular mass standards (kDa). B, 500 μg of the indicated total cell lysates was immunoprecipitated with either A-1A or preimmune IgG and analyzed by Western blots as detailed in Material and Methods. Numbers on right, migration of molecular mass standards (kDa) and the location of the immunoreactive IgG. C, Graded concentrations of BEAR-2 were resolved by SDS-PAGE, blotted to nitrocellulose, and stained with 1.0 μg/ml A-1A followed by GAR-HRP (1:10,000). Bands were quantified by densitometry and plotted against the dilution, starting with the 1.35-ng sample (lane 1). Inset, Western blot. BEAR-2 concentrations are 1.35 ng (lane 1), 1.05 ng (lane 2), 0.75 ng (lane 3), 0.45 ng (lane 4), and 0.15 ng (lane 5).
The next set of experiments were carried out to establish a quantitative Western blotting procedure for the determination of the actual concentration of AHR in each cell line. The experiments were done in the exact manner as detailed for ARNT. Briefly, the AHR protein used as the standard for these experiments was highly pure BEAR-2 that corresponded to amino acids 1–416 of the mouse sequence. This region of the AHR contains the bHLH and PAS domains and is highly conserved in all mammalian AHRs (26-28). Therefore, differences in the specificity of the A-1A antibody to mouse, rat, and human AHR should be minimal. The integrity of the BEAR-2 protein sample was evaluated by Coomassie-stained gels as detailed for BEARNT, and a final stock was prepared as detailed in Materials and Methods. Fig. 1C shows a representative Western blot of a BEAR-2 standard curve. The assay was linear over a 10-fold range of BEAR-2 protein (0.15–1.5 ng) and had a correlation coefficient of .97. Similar results were observed when stock BEAR-2 samples were repeatedly used over a 6-week period. The concentration of AHR in the various cell lines was then determined by resolving at least two concentrations of total cell lysate on the same gel with two concentrations of wild-type Hepa-1 cell lysate and the BEAR-2 standard curve. In the case of lysates with low AHR expression, the amount of total lysate loaded was set so that the intensity of the AHR band was within the linear range of the assay. Gels were then blotted to nitrocellulose and stained with the A-1A antibody. The resulting autoradiographs were then analyzed by densitometry and regression analysis performed as detailed in Materials and Methods. The results are presented in Table 2. As observed with the ARNT data, the actual concentration of AHR correlated well (r 2= .92), with the values determined when all lysates were resolved on the same gel and expressed as a percentage of AHR in Hepa-1 cells (compare Table 2, columns 2 and 3).
The wild-type and type II Hepa-1 cells express the highest number of AHRs per cell (∼300,000), whereas an H4IIE cell contains a modest 4763 AHRs. It is interesting to note that there is a 67-fold range of AHR concentration when the protein is expressed on a per-cell basis and a ∼30-fold range when expressed per mg of total cell lysate. This is in sharp contrast to the 3-fold range of ARNT expression in the identical cell lines. In general, the values of AHR concentration reported here are 10–15-fold higher than values previously published for Hepa-1 and H4IIE cells that were based on ligand binding assays (22, 29). There are limited data on most of the other cell types. It is important to note, however, that the ability to directly compare AHR concentrations produced in different laboratories is difficult due to the different methods of sample preparation (cytosol versus total lysates), the use of different types of AHR ligand binding assays (that may not detect the entire AHR pool), the use of different protein assays, and the fact that AHR concentration is generally not reported for an individual cell. The concentration of AHR in an individual cell is a useful calculation because the number will be important in establishing the ratio to other proteins and for determining the feasibility of detecting the receptor with immunocytochemical methods.
In summary, these results show that Western blot analysis can be used to accurately estimate the concentration of AHR in complex cellular lysates. This method should prove useful in future analyses because (i) it is rapid, (ii) the samples that are evaluated can be easily produced and are denatured, (iii) the assay does not rely on a functional AHR, (iv) the total pool of AHRs in all cellular compartments can be measured, (v) the assay is not affected by low affinity receptors, and (vi) the assay does not require the use of radiolabeled compounds.
The ratio of AHR to ARNT shows a high level of variation.
Because the AHR and ARNT associate to from a functional DNA binding transcription factor, the ratio of these two proteins is critical for determining their interplay and ability to affect additional signaling pathways. The results presented in Table3 show that AHR is equal to or in excess of the conventional ARNT in 9 of the 11 cell lines evaluated.2 There appears to be three classes of cells: (i) those with an AHR/ARNT ratio of >5 (Hepa-1, A7, and 10T), (ii) those with essentially an equal amount of AHR and ARNT (type I, C2C12, MDCK, D-17, and MCF-7), and (iii) those with 2–3-fold more ARNT than AHR (H4IIE, 3T3). The high ratio of AHR to ARNT in Hepa-1 cells is consistent with a previous report that also evaluated the concentration of each protein using a Western-based protocol (24). The >30-fold variation in AHR/ARNT ratio between the cell lines suggests that there is no correlation between the concentration of AHR and ARNT but implies that ARNT may be the limiting protein in AHR-mediated events in certain cells or tissues. This is consistent with results showing that Hepa-1 cells have higher levels of TCDD-mediated gene expression when ARNT is overexpressed in transfected cells (25).
Ratio of AHR protein to ARNT protein in continuous cell lines
ARNT is a nuclear protein in cell lines derived from nonhepatic tissues.
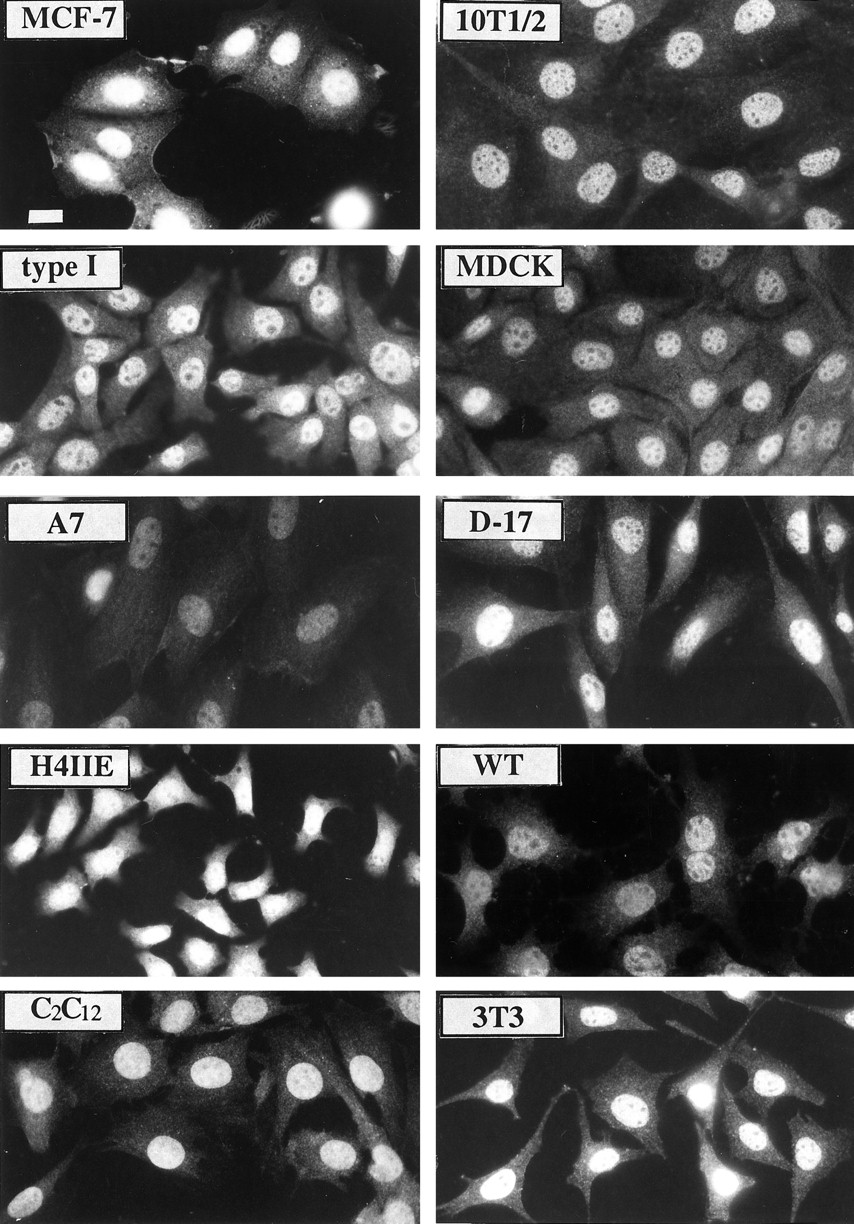
Previous studies have determined that ARNT is expressed predominately in the nucleus of Hepa-1 cells (18, 19, 30), but there is limited information about the location of this protein in nonhepatic cells (29). Identification of ARNT location is critical to understanding the ability of this protein to interact with the AHR, SIM, and HIF-1α in diverse tissues, especially if it is the limiting binding partner. To determine the subcellular location of ARNT, cells were fixed and stained with the R-1 antibody as detailed in Materials and Methods. The specificity of the R-1 for each cell type is demonstrated in Fig. 2B. Fig. 3 shows that the expression of ARNT is predominately nuclear in nonhepatic cell lines derived from rat, mouse, human, and canine tissues. In addition, the exposure of each cell to TCDD (2 nm) for ≤72 hr had no affect on the intensity or location of the staining in any cell type.3 These results confirm the description of ARNT as a nuclear transcription factor in both hepatic and nonhepatic cells and indicate that physical interaction with ARNT requires entry into the nucleus.

Immunofluorescence microscopy of ARNT protein expression in cultured cells. Cells were grown onto glass coverslips, fixed, and stained with 1.0 μg/ml R-1 IgG followed by GAR-TR (1:750) as previously detailed (20). The magnification of each micrograph is identical, and the identity of each cell line is indicated. Cells were not stained at the same time; therefore, the intensity of staining cannot be compared between different cell lines. Bar, 10 μm.
ARNT contains a putative nuclear localization signal within the NH-terminal region that is conserved in all ARNT proteins.
Having established that ARNT was a nuclear protein in cell lines derived from seven different tissues in four different species, we performed amino acid sequence analysis on mARNT, mARNT2, rARNT, rARNT2, hARNT, and rainbow trout ARNTb sequences to determine whether an NLS could be identified. A domain with the sequence RXXKRR is precisely conserved in the NH-terminal portion of six different ARNT proteins (Fig. 4). This sequence is consistent with other NLS domains that are short stretches of amino acids containing four or five basic residues (31, 32). To begin to evaluate the functionality of the RXXKRR sequence, PCR was used to generate an mARNT construct in which the nucleotide sequence encoding the first 45 amino acids (including the RXXKRR) was deleted and replaced by an ATG codon and Kozak sequence. The resulting construct, pΔNLS, or a construct containing full-length ARNT (pMVmARNT) was then transiently transfected into the type II Hepa-1 cell line that lacks functional ARNT protein (16, 18, 22). The location of the expressed proteins was then determined by immunocytochemical staining as previously detailed (18).

Identification of putative nuclear localization domain in ARNT. The amino acid sequences of rARNT and rARNT2, mARNT and mARNT2, hARNT, and rainbow trout ARNTb were analyzed as detailed in Materials and Methods. Numbers, location of the amino acids relative to the first residue (methionine).
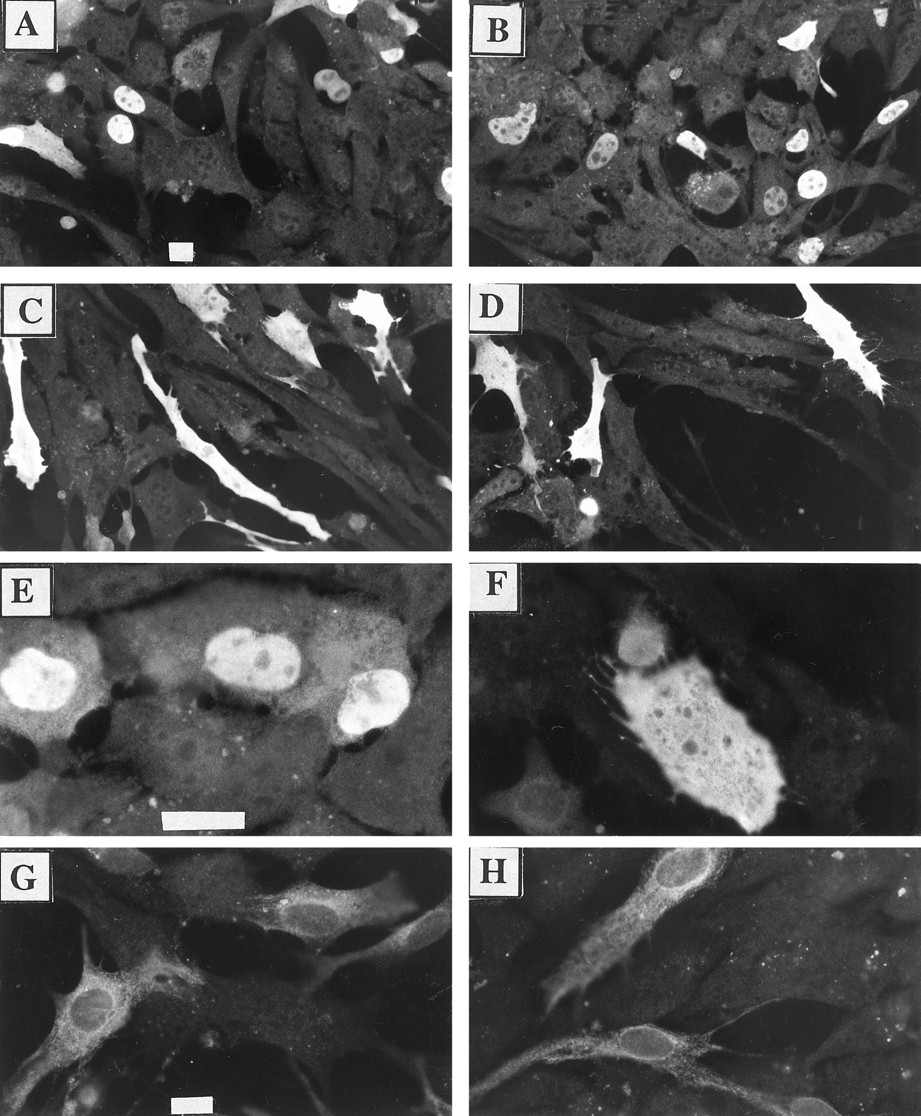
The results show that type II cells transfected with pMVmARNT exhibit intense staining for ARNT within the nuclear compartment (Fig.5, A, B, and E). These results are consistent with the location of ARNT in wild-type Hepa-1 cells (Refs.18 and 19 and Fig. 3). In contrast, type II cells transfected with pΔNLS show intense ARNT staining predominately in the cytoplasmic compartment (Fig. 5, C, D, and F). To determine whether the presence of ΔNLS in the cytoplasm was due a higher level of expression than mARNT, triplicate plates of type II cells were transfected with pΔNLS or pMVmARNT, and total cell lysates were evaluated by Western analysis. Fig. 6 shows that the levels of ΔNLS and mARNT expression are similar in three independent transfections. The transfection efficiency of both plasmids was similar3(also note the similar number of stained cells in Fig. 5, A–D).

Immunofluorescence microscopy of mARNT protein expression in type II Hepa-1 cells. A–F, Type II Hepa-1 cells grown onto glass coverslips were transfected with 1.5 μg of pΔNLS or 1.5 μg pMVmARNT and fixed 48 hr later. Coverslips were incubated with 1.0 μg/ml R-1 IgG followed by GAR-TR (1:750). G and H, Type II Hepa-1 cells grown onto glass coverslips were transfected with 1.5 μg of pΔNLS or 1.5 μg pMVmARNT and 24 hr later, cells were treated with TCDD (2 nm final concentration) and fixed after 20 hr. Coverslips were incubated with anti-P450 1A1 IgG (1:750) followed by GAR-TR (1:750). A, B, and E, Cells transfected with pMVmARNT and stained with R-1. C, D, and F, Cells transfected with pΔNLS and stained with R-1. G, Cells transfected with pMVmARNT and stained with anti-CYP1A1. H, Cells transfected with ΔNLS and stained with anti-P450 1A1. The specificity of the antibodies is shown by the lack of staining in nontransfected cells. Bar, 10 μm.
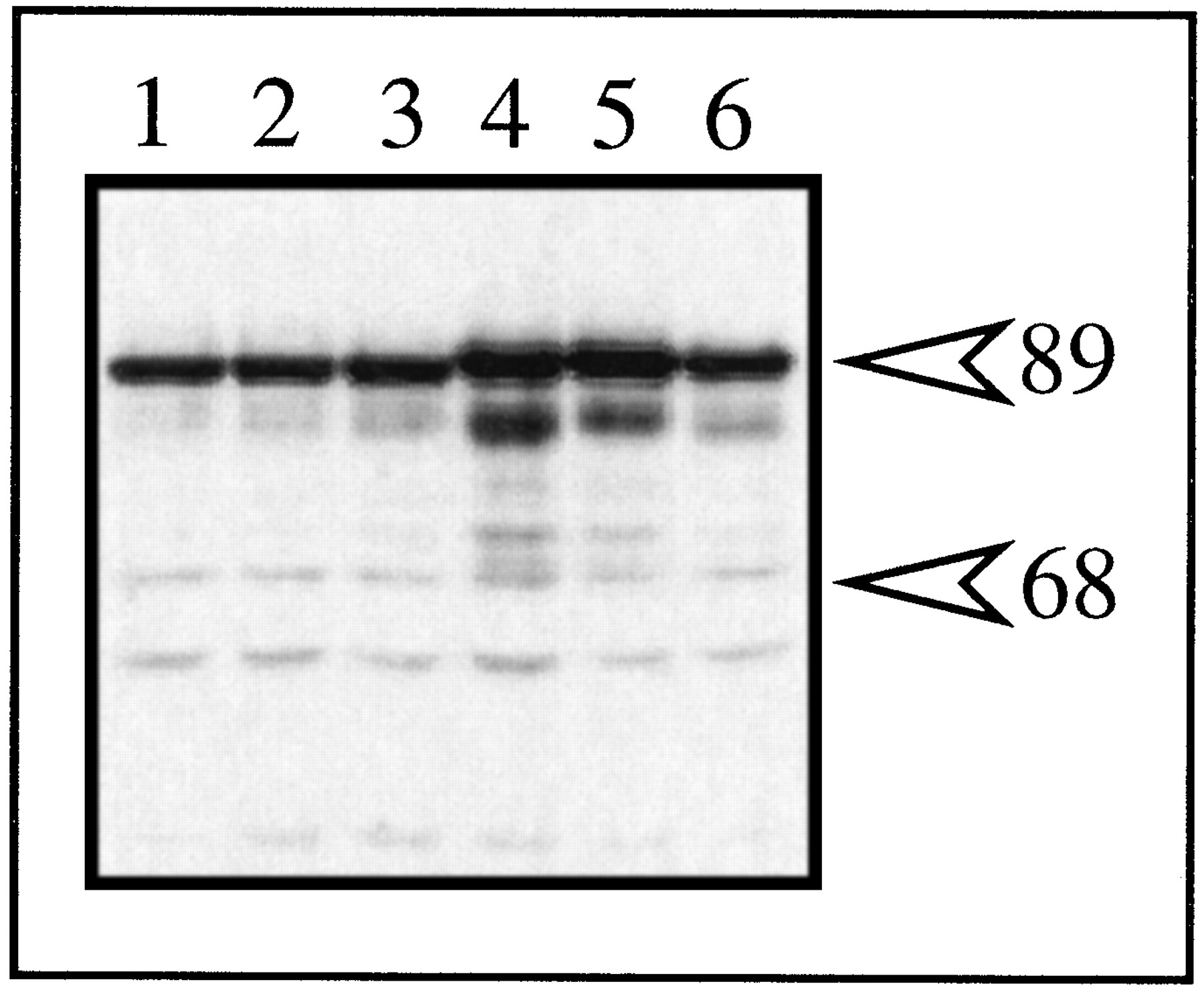
Western blot analysis of mARNT and ΔNLS expression in transfected type II cells. Triplicate plates of type II Hepa-1 cells were transfected with 1.5 μg of pΔNLS or 1.5 μg of pMVmARNT, and total cell lysates were prepared after 48 hr. Identical amounts of lysate were resolved by SDS-PAGE, blotted to nitrocellulose and stained with 1.0 μg/ml R-1 followed by GAR-HRP (1:10,000).Lanes 1–3, ΔNLS. Lanes 4–6, mARNT.Numbers on right, migration of molecular mass standards (kDa).
To further evaluate ΔNLS, type II cells were transfected with pΔNLS or pMVmARNT as detailed above, treated with TCDD (2 nm) for 24 hr before fixation, and stained with an antibody against P450 1A1. Interestingly, P450 1A1-specific perinuclear staining was observed in cells expressing either mARNT or ΔNLS (Fig. 5, G and H). These results suggest that ΔNLS can complement AHR-mediated signal transduction even though it is predominately cytoplasmic. These results can be explained by the high level of protein expression in transiently transfected cells (16) and the presence of some of the protein in the nuclear compartment. Because the AHR is in large excess in the Hepa-1 cells, a high level of nuclear ARNT may not be required to obtain AHR/ARNT dimerization and gene activation. Alternatively, the ΔNLS may dimerize with the AHR in the cytoplasm and be transported into the nucleus by the AHR. Studies are in progress to further evaluate these possibilities. Collectively, these results provide compelling evidence that ARNT has at least one functional NLS that is localized within the 45 NH-terminal amino acids of the protein.
Conclusions and implications.
The AHR-mediated signal transduction pathway and ARNT function have become increasingly complex as additional bHLH/PAS proteins and pairing strategies have been defined (5-13). Therefore, the determination of the concentration and location of all the bHLH/PAS proteins will be critical for assessing the interaction of these protein in vivo and the physiological relevance of dimerization data generated in vitro. Such studies are made more relevant by the recent report showing that an ARNT knock out in mice is lethal (17).
The results presented in this report clearly show that ARNT is a nuclear protein in both hepatic and nonhepatic cells and identify a putative NLS signal in the NH-terminal region of all published ARNT sequences. These results are consistent with previous studies that identified ARNT as a nuclear protein in Hepa-1 cells (18, 19, 30) and suggest that physiological interactions require a binding partner to enter the nuclear compartment or be a nuclear protein. From a signal-transduction point of view, it is intriguing to consider the finding that the AHR is generally in excess of ARNT. The implication of this result is that the liganded-AHR might “recruit” sufficient ARNT from functions it might be performing in an AHR-independent manner. However, several pieces of data seem to argue against this hypothesis. First, a recent study shows that the liganded AHR does not affect ARNT dimerization with HIF-1 α in the Hepa-1 cells (10:1 ratio of AHR/ARNT), whereas hypoxic conditions can partially affect AHR-mediated signaling (11). Second, it has been demonstrated that the AHR but not ARNT is rapidly depleted by ≤90% in vitro andin vivo after exposure to AHR agonists and that the quantity of ARNT sequestered by the liganded AHR is a fraction of the total ARNT pool in Hepa-1 cells (19).4Collectively, these results suggest that the AHR may in fact be the limiting partner in dimerization with ARNT and that there may be regulatory events affecting the dimerization of these proteins. The development of quantitative Western blot procedures for analysis of these proteins and the characterization of cell lines and tissues with a wide range of AHR/ARNT ratios should provide novel models for continued study of the bHLH/PAS proteins and their function.
Acknowledgments
We thank Dr. Gary Perdew (Pennsylvania State University, State College, PA) for helpful discussions concerning this project and the reviewers of this manuscript for pertinent advice.
Footnotes
- Received January 31, 1997.
- Accepted April 30, 1997.
-
Send reprint requests to: Dr. Richard S. Pollenz, Dept. of Biochemistry and Molecular Biology, Medical University of South Carolina, 171 Ashley Avenue, Charleston, SC 29425-2211. E-mail:pollenzr{at}musc.edu
-
↵1 J. Holmes, unpublished observations.
-
↵2 The term “conventional” ARNT refers to all ARNT isoforms except ARNT2.
-
↵3 R. S. Pollenz, unpublished observations.
-
↵4 Preliminary results show that AHR but not ARNT protein is depleted for ≤7 days in numerous tissues of the rat after a single oral dose of TCDD.
-
This project was funded in part by the Medical University of South Carolina Institutional Research Funds for 1995–1996.
Abbreviations
- ARNT
- aryl hydrocarbon nuclear translocator
- AHR
- aryl hydrocarbon receptor
- bHLH
- basic helix-loop-helix
- BCA
- bicinchoninic acid
- BEAR-2
- bacterial expressed aryl hydrocarbon receptor
- BEARNT
- bacterial expressed aryl hydrocarbon nuclear translocator
- hARNT
- human aryl hydrocarbon nuclear translocator
- rARNT
- rat aryl hydrocarbon nuclear translocator
- mARNT
- mouse aryl hydrocarbon nuclear translocator
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- PAS
- periodicity-aryl hydrocarbon nuclear translocator-simple-minded, GAR-HRP, goat anti-rabbit horseradish peroxidase
- GAR-TR
- goat anti-rabbit Texas red
- TTBS
- Tris-buffered saline/Tween 20
- BSA
- bovine serum albumen
- DMEM
- Dulbecco’s minimum Eagle’s medium
- FBS
- fetal bovine serum, SDS, sodium dodecyl sulfate
- NLS
- nuclear localization signal
- PAGE
- polyacrylamide gel electrophoresis
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- P450
- cytochrome P450
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}