


Abstract
PD98059 [2-(2′-amino-3′-methoxyphenyl)-oxanaphthalen-4-one] is a flavonoid and a potent inhibitor of mitogen-activated protein kinase kinase (MEK). Concentrations of PD98059 of ≤20 μm were not cytotoxic to cultures of the immortalized human breast epithelial cell line MCF10A. The agent was weakly cytostatic at concentrations of ≥10 μm. In vivo exposure of cultures to ≤20 μm PD98059 for 2–22 hr did not affect overall extracellular signal-regulated kinase contents; however, exposure to PD98059 resulted in a rapid loss (>95%) of the dually phosphorylated forms of extracellular signal-regulated kinase (IC50 = 1 μm). Treatment of cultures with PD98059 of ≥1 μm either at the time of addition or up to 48 hr before the addition of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) suppressed in a concentration-dependent manner the accumulation of induced steady state CYP1A1, CYP1B1, and NQO1 mRNAs. The addition of PD98059 to rat liver cytosol just before the addition of TCDD suppressed TCDD binding (IC50 = 4 μm) and aryl hydrocarbon receptor (AHR) transformation (IC50 = 1 μm), as measured by sucrose gradient centrifugation and electrophoretic mobility shift assays. Flavone and flavanone, two closely related structural analogs of PD98059, inhibited AHR transformation by TCDD with IC50 values similar to that obtained with PD98059. However, neither analog was as potent as PD98059 in inhibiting MEK (IC50 ∼ 190 μm for both). These results suggest that PD98059 is a ligand for the AHR and functions as an AHR antagonist at concentrations commonly used to inhibit MEK and signaling processes that entail MEK activation.
The halogenated hydrocarbon TCDD is a widespread environmental pollutant with apoptotic, teratogenic, immunomodulating, tumor-promoting, and antiestrogenic activities (Holsapple et al., 1991;Hankinson, 1995; Schmidt and Bradfield, 1996). It also is a potent modulator of the expression of several phase I and II biotransforming enzymes such as CYP1A1 (nomenclature for P450 genes, mRNAs, and proteins is as recommended by Nelson et al., 1996), CYP1B1, AHD4, and NQO1 (Nebert, 1994; Hankinson, 1995; Schmidt and Bradfield, 1996). Biochemical, genetic, and molecular approaches have demonstrated that these activities are mediated by the interaction of TCDD with the AHR protein.
In its ligand-free form, the AHR is a cytosolic protein complexed to two heat shock 90 molecules (hsp90) and a 37–43-kDa protein recently identified as an immunophilin homolog (Hankinson, 1995; Schmidt and Bradfield, 1996; Ma and Whitlock, 1997). On binding TCDD, the AHR translocates to the nucleus, where it forms a dimer with the ARNT protein. At some undetermined step in this process, the AHR loses its two molecules of hsp90 and the immunophilin-like protein (Wilhelmssonet al., 1990; Perdew, 1991; Ma and Whitlock, 1997). The resulting AHR/ARNT heterodimers interact with specific enhancer sequences in target genes called DREs. This binding to DREs stimulates the transcriptional activation of target genes (Nebert, 1994;Hankinson, 1995; Schmidt and Bradfield, 1996).
A variety of agents and physiological conditions modulate AHR function. For example, some flavonoids suppress the transcriptional activation of target genes by TCDD (Wilhelmsson et al., 1994; Gasiewiczet al., 1996; Lu et al., 1996). This suppression reflects their functioning as AHR ligands and forming complexes that are unable to bind to DNA (Gasiewicz et al., 1996; Luet al., 1996) or act as transactivators (Wilhelmssonet al., 1994). Hence, some flavonoids modulate TCDD-mediated processes by functioning as AHR antagonists. In contrast, neither TPA nor EGF is a ligand of the AHR; however, they also suppress the transcriptional activation of TCDD-responsive genes in some cell types (Hohne et al., 1990; Reiners et al., 1992;Berghard et al., 1993). The introduction of an oncogenicras expression vector into cultured human breast cells also suppresses the transcriptional activation of several members of theAh battery by TCDD (Reiners et al., 1997). A similar suppression occurs in the skins of transgenic mice with a targeted expression of a v-Ha-ras oncogene in their keratinocytes (Reiners et al., 1997).
The mechanisms by which exposure to TPA or EGF, or the introduction and expression of an oncogenic ras gene, modulates TCDD-mediated processes are not known. Members of the ras gene family encode a 21-kDa polypeptide (p21-ras) that links the EGF receptor with its distal effector kinases (Satoh et al., 1992; Cano and Mahadevan, 1995). Specifically, the binding of EGF to the EGF receptor stimulates the transient activation of components of a cascade involving p21-ras, raf-1 kinase, MEK, and ERK (Satoh et al., 1992; Cano and Mahadevan, 1995). A similar transient activation of this cascade (referred to as the MAPK cascade) also occurs in several cell types after exposure to TPA (Troppmair et al., 1994; Alessiet al., 1995). In contrast to the effects of EGF or TPA, a constitutive activation of components of the MAPK cascade distal to p21-ras is achieved in several cell types expressing oncogenic p21-ras (Leevers et al., 1992; Shibuya et al., 1992). This constitutive activation of the MAPK cascade is a consequence of a mutation in the p21-ras protein that locks it in an active conformation (Grand et al., 1991).
Although circumstantial, the common effects of EGF, TPA, and the expression of oncogenic p21-ras on TCDD-mediated processes suggest that AHR function might be affected by processes involving the MAPK cascade. Assessment of the contributions of the MEK/ERK components of the MAPK pathway to transformation, proliferation, and reactive oxygen signaling has been greatly facilitated by the availability of PD98059 [2-(2′-amino-3′-methoxyphenyl)-oxanaphthalen-4-one], an inhibitor of MEK (Alessi et al., 1995; Dudley et al., 1995;Guyton et al., 1996; Cook et al., 1997). The usefulness of this agent stems from its solubility, specificity for MEK as opposed to other kinases, and ability to cross cell membranes (Alessi et al., 1995; Dudley et al., 1995). The original intention of the current study was to use PD98059 as a tool to assess the relationship between the activation status of MEK/ERK and AHR function in MCF10A-NeoT cells. This cell line was derived by transfection of an Ha-ras oncogene into the normal human breast epithelial MCF10A cell line (Basolo et al., 1991). Expression of oncogenic p21-ras in these cells down-regulates AHR function and suppresses the transcriptional activation ofCYP1A1, CYP1B1, and NQ01 by TCDD (Reiners et al., 1997). An unexpected finding of our studies was that concentrations of PD98059 that inhibited MEK also suppressed TCDD-activated, AHR-dependent processes in MCF10A cells, a cell line that lacked an Ha-ras oncogene. Subsequent structure-activity analyses demonstrated that this suppression was related to PD98059 functioning as an AHR antagonist, as opposed to an MEK inhibitor.
Experimental Procedures
Materials.
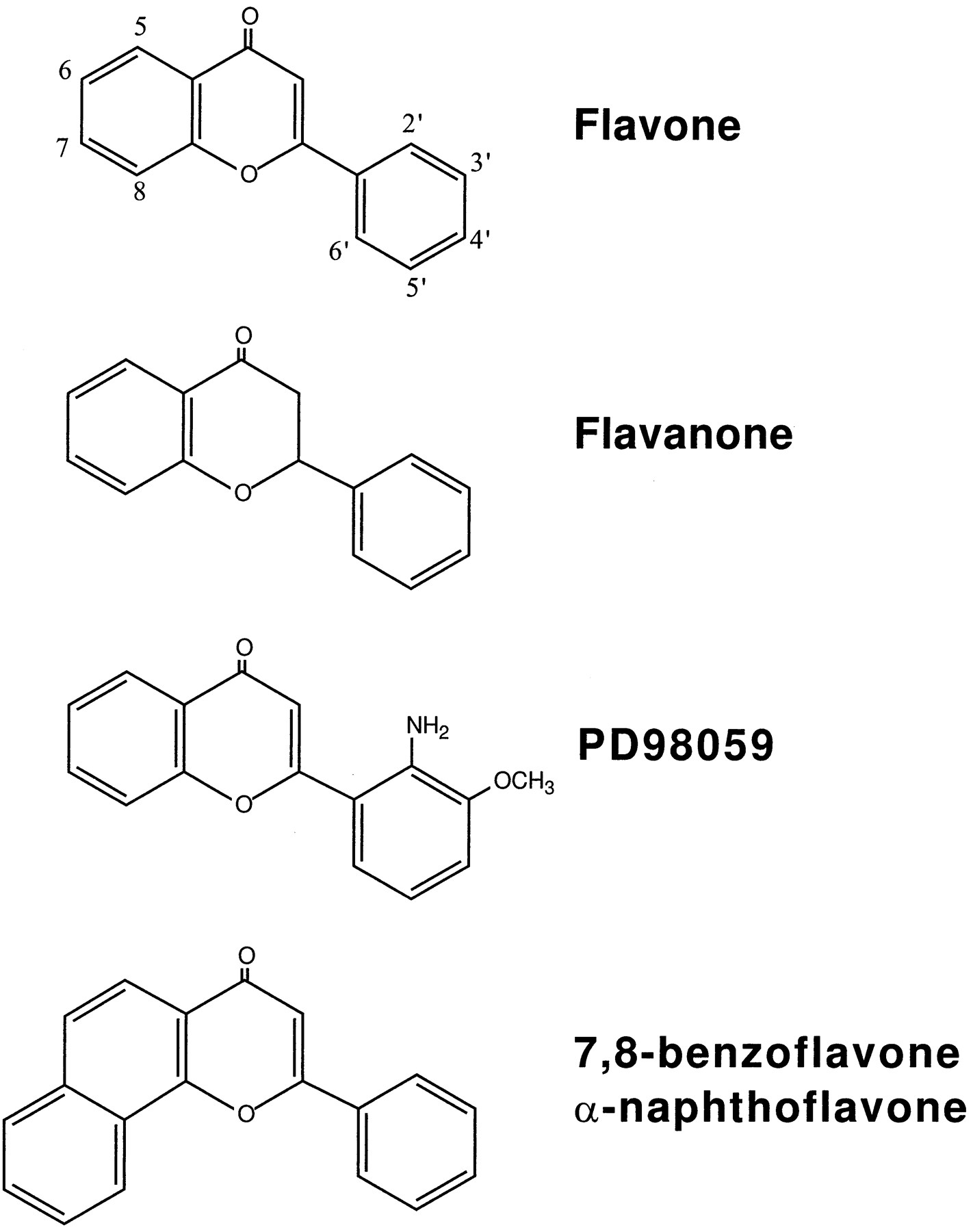
PD98059 was purchased from New England Biolabs (Beverly, MA). Flavone and flavanone, two structural analogs of PD98059 (Fig. 1), were obtained from Aldrich Chemical (Milwaukee, WI). [3H]TCDD (29 Ci/mmol), TCDD, and 2,3,7,8-tetrachlorodibenzofuran were generous gifts of Dr. S. Safe (Texas A&M University, College Station, TX). Trypsin, EGF, penicillin/streptomycin solution, and horse and bovine sera were purchased from GIBCO BRL (Gaithersburg, MD). [γ-32P]dATP and [α-32P]dCTP were purchased from DuPont-New England Nuclear (Boston, MA). MBP was purchased from Sigma Chemical (St. Louis, MO). Homogeneous preparations of GST fusion proteins containing 44-kDa ERK1 or the 45-kDa MEK1 were provided by Parke-Davis Pharmaceutical Research (Ann Arbor, MI). The MEK1-GST fusion protein is constitutively activated as a consequence of serine-to-glutamate mutations at positions 218 and 222 (Dudley et al., 1995). Antibodies specific for the dually phosphorylated (active) forms of ERK1 and ERK2 were purchased from Promega (Madison, WI). Antibodies recognizing both phosphorylated and nonphosphorylated forms of ERK1 and ERK2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).

Structures of PD98059 and related flavonoids.
Cell culture and treatment.
The MCF10A, MCF10A-Neo, and MCF10A-NeoT cell lines were obtained from the Cell Lines Resource (Karmanos Cancer Center, Detroit, MI). The MCF10A-Neo and MCF10A-NeoT lines were derived by transfection of the MCF10A cell line with the pHo6 plasmid and the pHo6 plasmid containing an Ha-rasoncogene derived from the human T24 bladder carcinoma cell line, and subsequent selection for resistance to G418. The transfected lines represent pooled survivors, as opposed to clonal lines. The derivation and characterization of these cell lines have been described elsewhere (Soule et al., 1990; Basolo et al., 1991). With the exception of the EGF content being increased from 10 to 20 ng/ml, the cells were cultured in supplemented Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium as described by Basolo et al. (1992) in a humidified atmosphere of 95% air/5% CO2 at 37°. Subconfluent cultures were treated with varying concentrations of chemicals dissolved in DMSO (absolute volume of solvent, <0.1% of medium volume). Details of the treatment are provided in the text. Viability of cells after treatment was assessed by ability to exclude trypan blue. Cultures earmarked for RNA isolation were washed twice with phosphate-buffered saline (2.7 mm KCl, 1.5 mmKH2PO4, 137mm NaCl, 8 mm Na2HPO4, pH 7.2) at harvesting and stored at −80°.
RNA preparation and Northern blot analyses.
Total cellular RNA was isolated according to the acidic phenol extraction method ofChomczynski and Sacchi (1987). RNA was resolved on 1.2% agarose/formaldehyde gels and transferred to nylon membranes as described previously (Reiners et al., 1997). The probes used for the detection of CYP1A1, CYP1B1, NQO1, and 7S RNAs and the conditions used for hybridization have been described in detail (Reiners et al., 1997).
Sucrose density gradient centrifugation.
Rat liver cytosol was prepared as described by Elferink and Whitlock (1994) and incubated with 1 nm [3H]TCDD in the presence or absence of competitor for 2 hr at 4° as described by Harperet al. (1991). At the end of the incubation, the extract was adjusted to 0.4 m KCl and treated with dextran/charcoal to remove nonspecifically bound [3H]TCDD as described by Harper et al. (1991). The resulting supernatant was centrifuged on sucrose gradients (made in 0.4 m KCl) for 20 hr at 225,000 × g. We adjusted the ionic strength of the reaction mixture to convert any 9S AHR/[3H]TCDD complex to a ∼5S AHR/[3H]TCDD complex. At low ionic strength, we routinely obtained both 9S and ∼5S peaks, and conversion of all of the complex into the ∼5S form facilitated quantification. Gradients were siphoned from the top and collected as 200-μl fractions in scintillation vials. Radioactivity was detected by liquid scintillation counting. The 14C-labeled bovine serum albumin was added to some homogenates immediately before centrifugation to calibrate the gradients. The sedimentation position of catalase (11S) in the gradients was determined by enzymatic analyses using the assay described by Reiners et al. (1988).
EMSA.
The conditions reported by Shen et al. (1991) were used for the EMSA. Complementary oligonucleotides 5′-GATCCGGCTCTTCTCACGCAACTCCGAGCTCA-3′ and 5′-GATCTGAGCTCGGAGTTGCGTGAGAAGAGCG-3′ (single-core recognition sequence is underlined) were annealed and used to detect activated AHR/ARNT complexes.
Kinase cascade assay.
The conditions used for the MEK-dependent activation of ERK1 and the subsequent phosphorylation of myelin basic protein were similar to those reported by Dudley et al. (1995). Kinase reactions were performed in 50-μl reaction volumes and contained 50 mm Tris, pH 7.4, 10 mmMgCl2, 2 mm EGTA, 10 μmATP (containing 1 μCi of 3000 Ci/mmol [γ-32P]ATP), 7.6 μg of GST-MEK1, 7.2 μg of GST-ERK1, and 20 μg of MBP. PD98059 and other flavonoids were added to the reactions mixtures immediately after the addition of GST-MEK1 but before the addition of GST-ERK1 and ATP. Control reactions contained ERK1 and MBP but no MEK. Reaction mixtures were incubated at 30° for 15 min before being stopped by the addition of Laemmli’s SDS sample buffer. Proteins were separated on SDS-15% polyacrylamide gels, according to the method of Laemmli (1970). After vacuum drying of the gel, radioactivity was detected by autoradiography on X-ray film or phosphoimaging using a BioRad (Hercules, CA) GS-525 Molecular Imager.
Western blot analyses of ERK1/ERK2.
Culture dishes were washed twice with cold phosphate-buffered saline (containing 1 mm NaVO4) before the addition of cold lysis buffer (70 mm NaCl, 50 mm glycerol phosphate, 10 mm HEPES, pH 7.4, 1% Triton X-100, 1 mm NaVO4, 1 μmaprotinin, 1 μm leupeptin, and 1 μmphenylmethylsulfonyl fluoride). Insoluble material was removed by centrifugation, and the supernatant was boiled in Laemmli’s sample buffer. Equal amounts of protein were resolved on SDS-10% polyacrylamide gels and then transferred to nylon membranes. The resulting protein blot was blocked overnight at 4° with TBST (0.9% NaCl, 10 mm Tris, pH 7.5, 0.1% Tween-20) containing 1% bovine serum albumin and 1% ovalbumin. The blots were washed with TBST and probed for 3 hr at room temperature with primary antibodies diluted into TBST containing 0.5% bovine serum albumin. After a brief wash with TBST, blots were incubated with goat anti-rabbit Ig conjugated with horseradish peroxidase for 1 hr at room temperature. Blots then were washed five times in TBST over the course of an hour and developed using Amersham (Arlington Heights, IL) enhanced chemiluminescence reagents. ERK-immunoglobulin conjugates were recorded on X-ray film and quantified with a Molecular Dynamics (Sunnyvale, CA) densitometer. Antibody dilutions and enhanced chemiluminescence development was conducted according to manufacturer specifications.
32P Quantification.
32P-Labeled nucleic acids and proteins were quantified using the BioRad GS-525 Molecular Imager and Molecular Dynamics software.
Results
In vitro suppression of MEK and ERK activities.
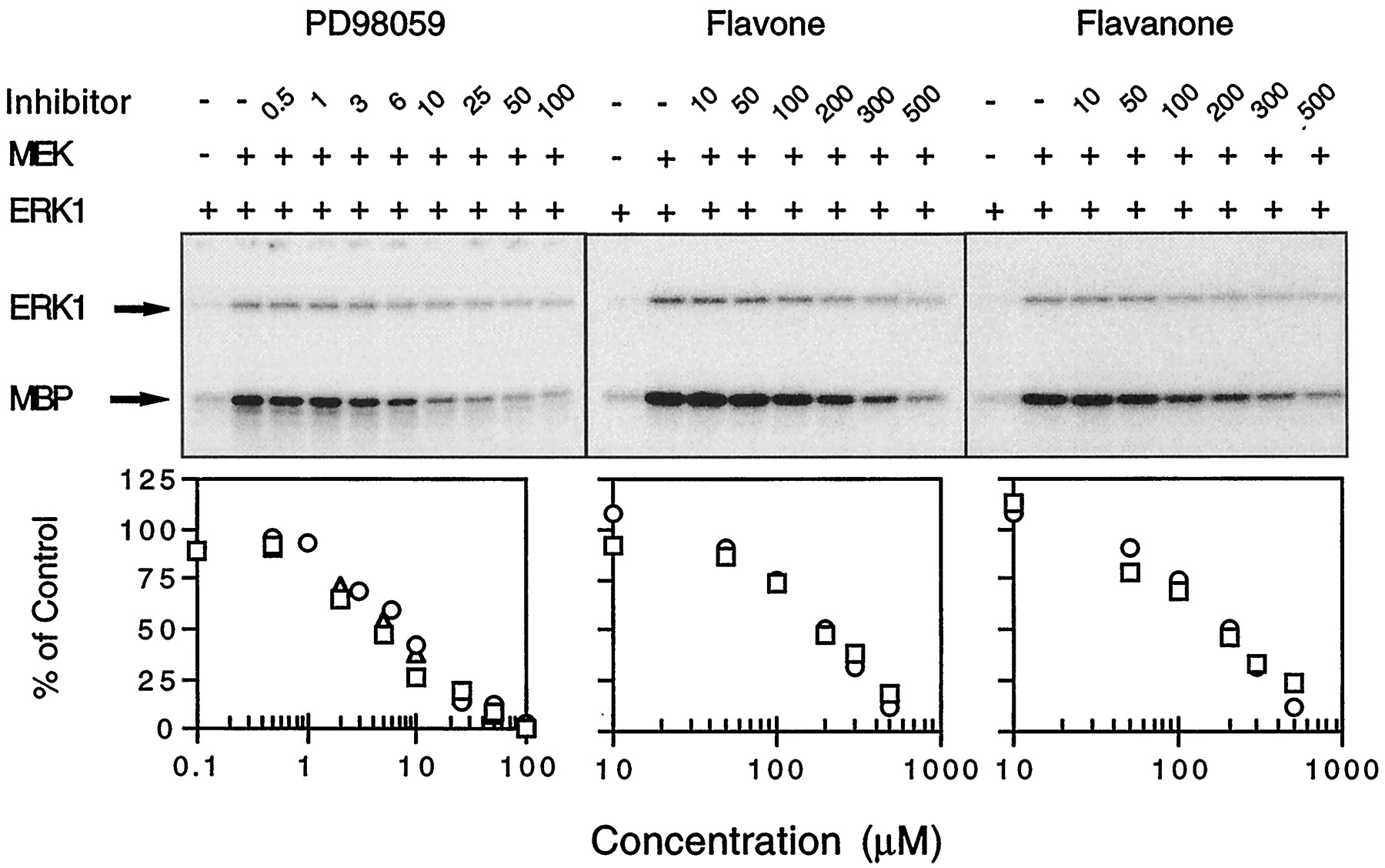
Interest in the use of PD98059 as a tool in signal transduction research is derived from the specificity with which it inhibits MEK but no other kinases (Alessi et al., 1995; Dudley et al., 1995). A kinase cascade assay was used to quantify inhibition of MEK by PD98059 (Fig. 2). This assay uses a mutated, constitutively activated form of MEK to phosphorylate and activate ERK1, which in turn phosphorylates MBP. Hence, the activity of MEK can be monitored by measuring either ERK1 or MBP phosphorylation. Supplementation of this kinase cascade system with PD98059 resulted in a concentration-dependent suppression of MEK activity (Fig. 2). The IC50 value for suppression of MBP phosphorylation was ∼5 μm (Table1). This value is very similar to that reported by Dudley et al. (1995) using a similar in vitro assay and an in vivo assay with Swiss 3T3 fibroblasts (Dudley et al., 1995).

Suppression of MEK activity by PD98059 and its analogs. Kinase cascade assay mixtures were incubated with varying concentrations of PD98059, flavone, or flavanone before being analyzed by SDS-PAGE. Top, data generated in a representative experiment. Top and bottom bands, GST-ERK1 fusion protein and MBP, respectively. Bottom, each symbol type represents an experiment in which MBP signals have been corrected for signals measured in the absence of exogenous MEK addition. The 100% control value represents signals obtained in the absence of flavonoid.
PD98059 suppression of MEK activities and TCDD binding to the AHR, transformation of the receptor, and transcriptional activation of members of the Ah battery
Flavone and flavanone, two structural analogs of PD98059 (see Fig. 1), also were examined in the kinase cascade assay for their abilities to inhibit MEK (Fig. 2). Both agents inhibited MEK. However, the IC50 values for flavone and flavanone inhibition of MEK (∼190 μm) were minimally 38-fold greater than the value determined for PD98059 (Table 1).
In vivo suppression of MEK/ERK activities.
Activation of ERK1 and ERK2 by MEK entails a dual phosphorylation of the threonine and tyrosine residues in the TEY motif located in the catalytic core of the two enzymes (Boulton et al., 1991). Antibodies generated to a ERK polypeptide containing the dually phosphorylated TEY motif recognize only the catalytically active, dually phosphorylated form of the enzyme. Immunodetection of the dually phosphorylated forms of ERK1 and ERK2 specifically versus both phosphorylated and nonphosphorylated forms of the ERK, has proved to be a very convenient and sensitive method of assessing the activation status of the two kinases (Cook et al., 1997).
Western blot analyses of lysates prepared from untreated MCF10A-Neo and MCF10A-NeoT cultures using antibodies recognizing both phosphorylated and nonphosphorylated forms of the ERKs demonstrated the presence of ERK1 and ERK2 in both cell lines (Fig. 3,zero concentration lanes on top). However, although the relative amount of ERK2 was quite similar in both cell lines, the ERK1 content of untreated MCF10A-NeoT cells was markedly less than that detected in the MCF10A-Neo line. Untreated cultures of both cell lines also contained the dually phosphorylated forms of ERK1 and ERK2 (Fig.3, zero concentration lanes on bottom). Although the signal corresponding to the dually phosphorylated form of ERK1 in MCF10A-NeoT cells was much less than that detected in MCF10A-Neo cultures, the difference paralleled the relative content of ERK1 in the two cell lines.

In vivo suppression of MEK by PD98059 in MCF10A cell lines. Subconfluent cultures of MCF10A-Neo and MCF10A-NeoT cells were treated with DMSO or varying concentrations of PD98059 for either ∼2 or ∼22 hr before harvesting and processing of extracts for Western blot analyses. Samples (7 μg of protein) were separated on SDS-10% polyacrylamide gels, transferred to nylon membranes, and analyzed as described in the text using antibodies to ERK1, ERK2, and phospho-ERK1 (ERK1-P) and -ERK2 (ERK2-P).
Concentrations of PD98059 of ≤20 μm were not cytotoxic to cultured MCF10A, MCF10A-Neo, and MCF10A-NeoT cells. However, PD98059 was weakly cytostatic to all three lines at concentrations of ≥10 μm (Clift RE and Reiners JJ Jr., unpublished observations). Treatment of MCF10A-Neo and MCF10A-NeoT cultures with concentrations of PD98059 up to 20 μm for 2–22 hr did not alter the total ERK content (Fig. 3, top). However, treatment with PD98059 did result in concentration-dependent reductions in the dually phosphorylated forms of ERK1 and ERK2 (Fig. 3,bottom). Within 2 hr of a 10-μm treatment, phosphorylated ERK contents were reduced ∼74% and ∼86% in MCF10A-Neo and MCF10A-NeoT cultures, respectively (IC50 ∼ 1 μm). Within 22 hr of treatment, phosphorylated ERK forms were almost completely eliminated in both cell lines (Fig. 3).
Results similar to those presented in Fig. 3 were obtained in a second independent concentration-dependence study (Dudley DT, unpublished observations).
Suppression of transcriptional activation of members of theAh battery.
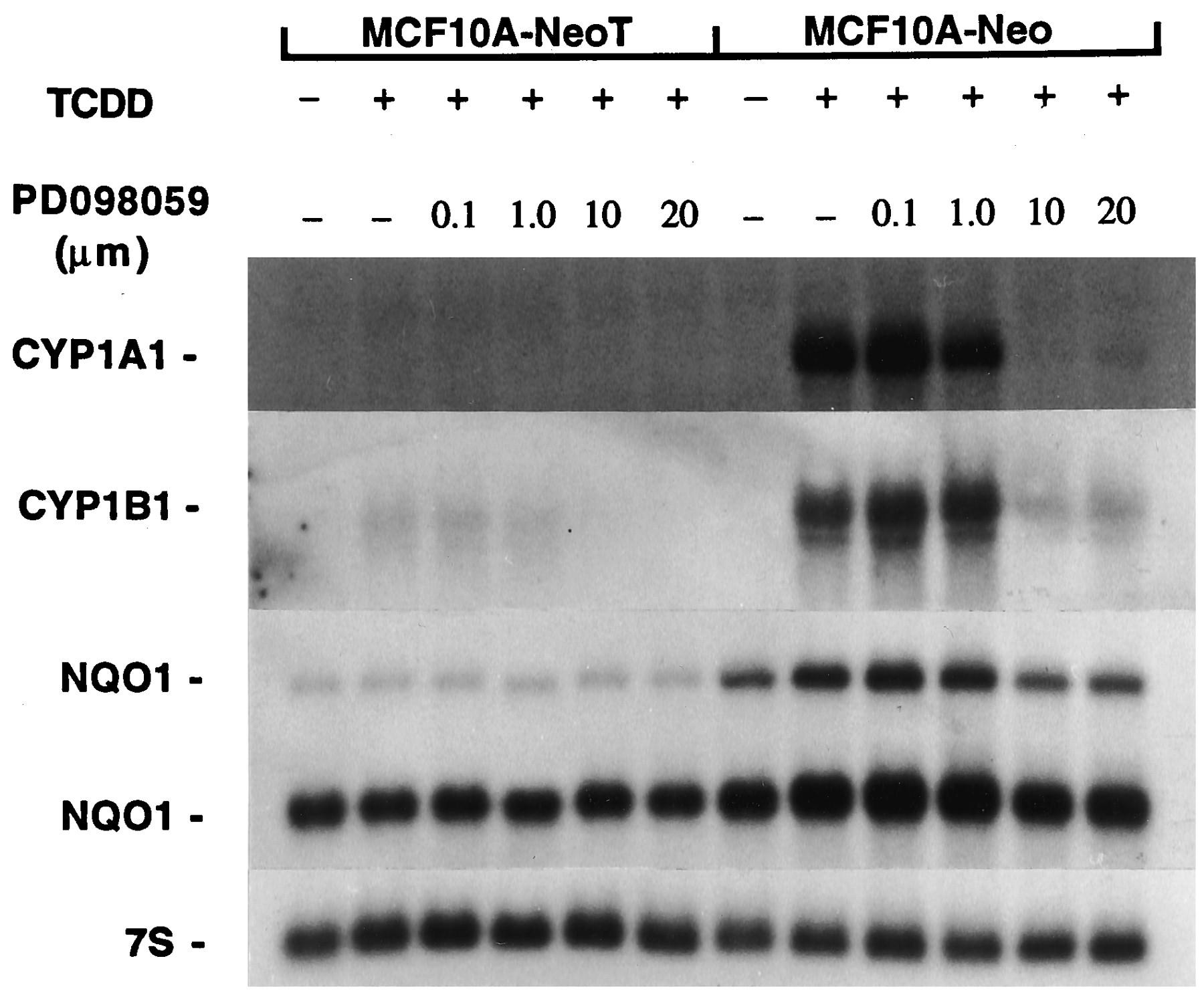
CYP1A1 and CYP1B1 mRNAs were not detected in asynchronous, DMSO-treated MCF10A-Neo or MCF10A-NeoT cultures (Fig. 4). However, constitutive levels of NQO1 mRNAs were detected in both cell lines. The two prominent NQO1 mRNAs reflect splicing variants (Pan et al., 1995). Exposure of MCF10A-Neo cells to TCDD elevated markedly steady state levels of CYP1A1 and CYP1B1 mRNAs and, to a lesser extent, NQO1 (Fig. 4). In marked contrast, but in agreement with our recent report (Reiners et al., 1997), similar accumulations of these mRNAs did not occur in MCF10A-NeoT cells after exposure to TCDD (Fig. 4).

Suppression of Ah battery induction by TCDD in MCF10A cell lines. Cultures of MCF10A-Neo and MCF10A-NeoT cells were exposed to varying concentrations of PD98059 for 1 hr before the addition of TCDD (10 nm). Cultures were harvested 6 hr after TCDD addition for isolation of RNA and analyses of CYP1A1, CYP1B1, NQO1, and 7S RNAs.
Treatment of MCF10A-NeoT cells with varying concentrations of PD98059 1 hr before the addition of TCDD did not enhance the accumulation of CYP1A1, CYP1B1, or NQO1 mRNAs (Fig. 4). Indeed, similar treatment of MCF10A-Neo cells with concentrations of PD98059 of ≥10 μm totally suppressed TCDD-mediated accumulations of CYP1A1, CYP1B1, or NQO1 mRNAs (Fig. 4). A weak suppressive effect was obvious with a concentration as low as 1 μm. A study similar to that reported in Fig. 4, but using murine hepatoma 1c1c7 cells, also demonstrated a concentration-dependent PD98059 suppression of CYP1A1 induction that was identical to that seen with the MCF10A-Neo cell line (Lee J-Y and Reiners JJ Jr., unpublished observations). These data suggest that PD98059 inhibits the transcriptional activation of several TCDD-responsive genes.
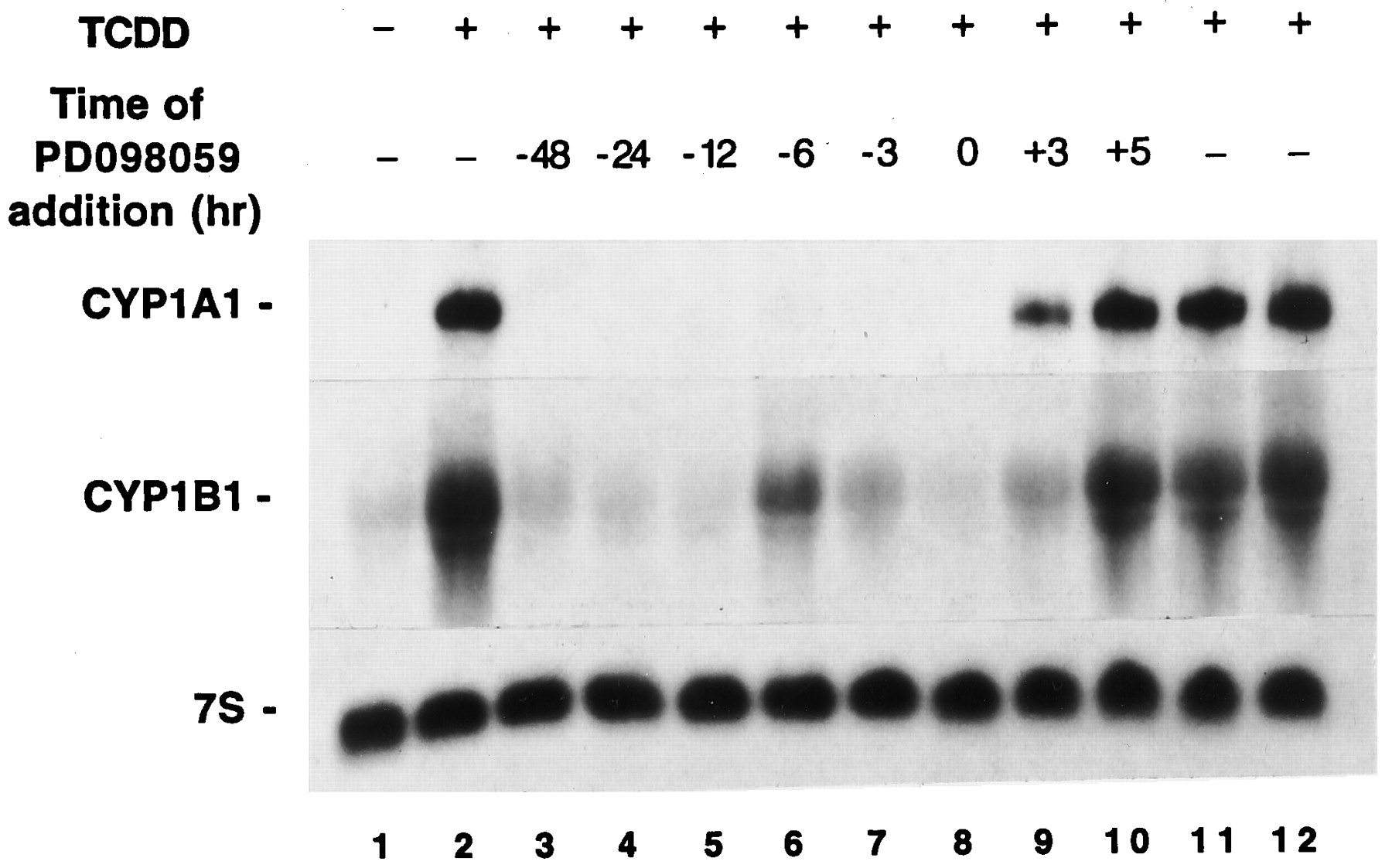
The effects of PD98059 noted in Fig. 4 were obtained in a protocol in which it was added to cultures 1 hr before the addition of TCDD. Pretreatment of cultures with PD98059 as much as 48 hr before the addition of TCDD also inhibited the subsequent accumulation of CYP1A1 and CYP1B1 mRNAs (Fig. 5). Hence, the inhibitory effects of PD98059 pretreatment were sustained for ≥48 hr.
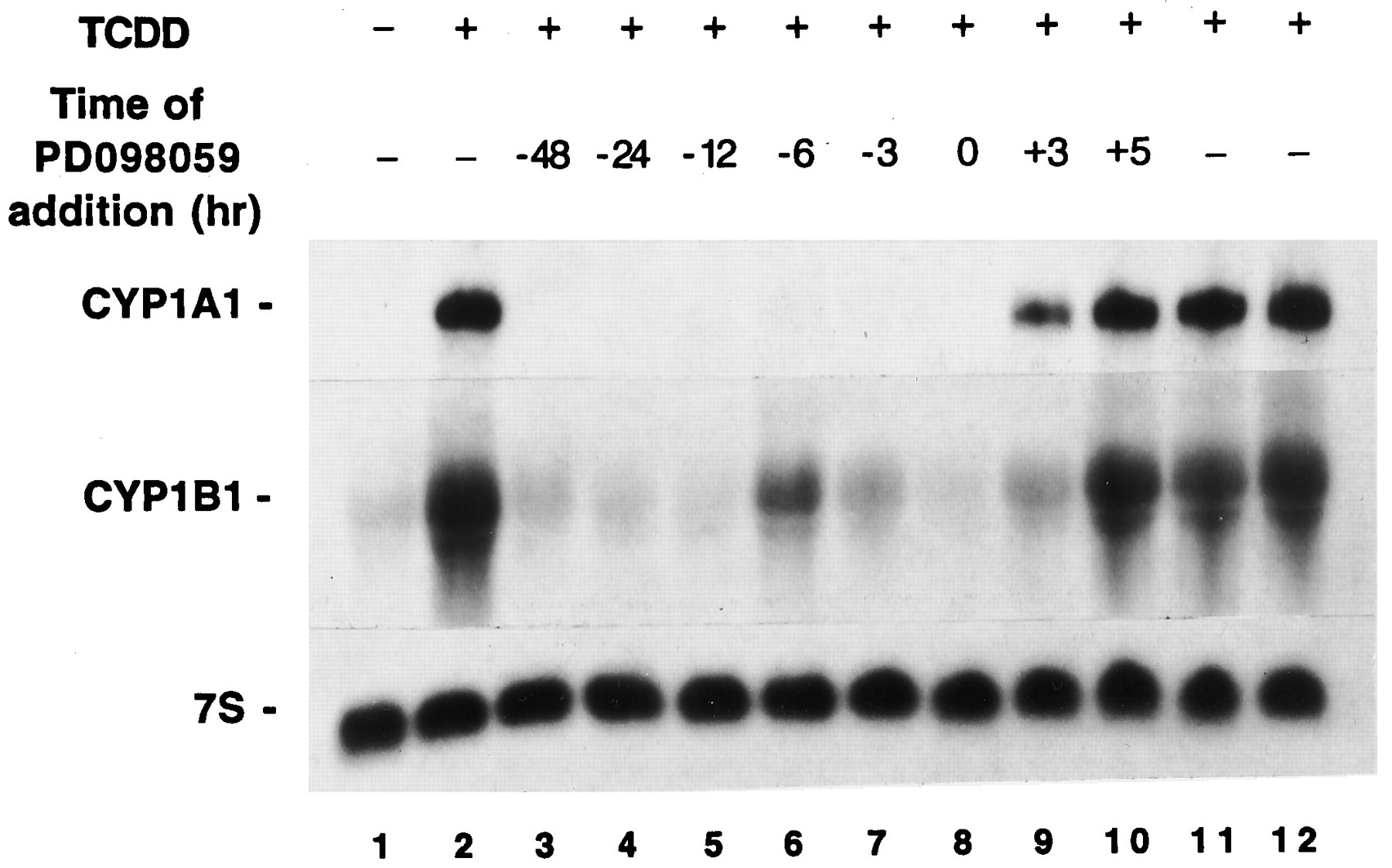
Relationship between time of PD98059 and TCDD additions and the level of suppression. Cultures of MCF10A cells were treated with 10 μm PD98059 at various times either before or after the addition of TCDD (10 nm). Cultures were treated at time zero with either DMSO (lane 1) or TCDD (lanes 2–12). Cultures were pulled for isolation of RNA 3 (lane 11), 5 (lane 12), or 6 (lanes 1–10) hr after the addition of TCDD or DMSO.
Steady state levels of CYP1A1 and CYP1B1 mRNAs in MCF10A-Neo cells were near their maximum levels within 3 hr of TCDD addition (Fig. 5, comparelanes 2, 11, and 12). Cultures treated with PD98059 3 hr after TCDD induction, and harvested 3 hr later, had steady state CYP1A1 and CYP1B1 RNA contents less than those observed in cultures harvested after 3 hr of TCDD exposure (Fig. 5, compare lanes 9 and 11). In contrast, cultures treated with PD98059 5 hr after TCDD induction, and harvested 1 hr later, had steady state CYP1A1 and CYP1B1 RNA contents comparable to those observed in cultures harvested after either 5 or 6 hr of TCDD exposure (Fig. 5, compare lanes 10 and 12). It is conceivable that the difference noted in mRNA contents (lanes 9 and 11) reflect the consequences of mRNA turnover during a 3-hr period. If so, CYP1A1 and CYP1B1 mRNA half-lives are of sufficient length that changes in abundance would be very subtle during the span of 1 hr; this could explain why relative mRNA abundances seemed similar in Fig. 5, lanes 10 and 12.
AHR transformation and DNA binding.
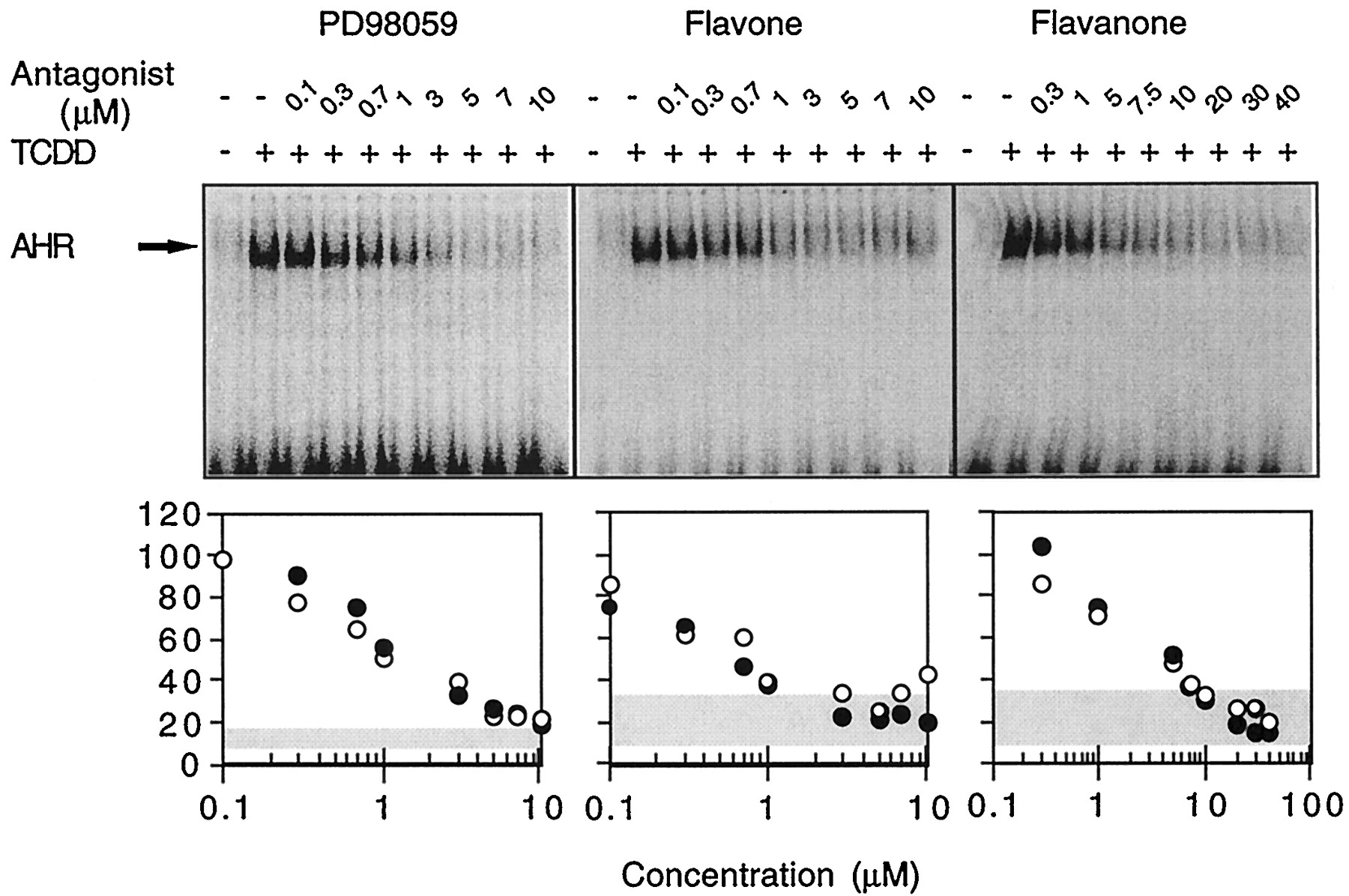
In vitrotransformation of the AHR into a DNA-bound complex with ARNT is a well characterized phenomenon (Bank et al., 1992). We used the EMSA to examine the capacity of PD98059 and related flavonoids to inhibit AHR-DNA binding in response to TCDD (Fig.6). These studies used rat liver cytosol instead of MCF10A cytosol because the latter, in the absence of TCDD, displayed a high level of spontaneous AHR-DNA complex formation (Reiners et al., 1997). Incubation of rat liver extracts with TCDD transformed the AHR so that it bound ARNT and formed a complex capable of binding to an oligonucleotide containing a DRE sequence (Fig. 6). DNA-complex formation mediated by TCDD exposure was suppressed in a concentration-dependent fashion by coincubation with PD98059 (Fig. 6, IC50 = 1 μm); a similar IC50 value was determined when extracts of murine hepatoma 1c1c7 cells were used as the source of AHR forin vitro transformation studies (Myrand SP and Reiners JJ Jr., unpublished observations).

In vitro suppression of TCDD-induced AHR/DNA complex formation by PD98059 and its analogs. Rat liver extract was transformed in vitro with 10 nm TCDD in the presence of varying concentrations of PD98059, flavone, or flavanone before being incubated with labeled DRE oligonucleotide and analyzed in a gel retardation assay. Bottom, ○ and • represent two independent experiments. The 100% values represent the amount of AHR/DNA complex generated after transformation in the presence of only TCDD. Shaded area, values for the amount of AHR/DNA complex, relative to the TCDD control, generated in liver samples treated with DMSO.
Concentration-dependent inhibitions of AHR/DRE complex formation also occurred when TCDD-treated rat liver extracts were cotreated with flavone or flavanone (Fig. 6). Both flavone and flavanone inhibited AHR/DNA complex binding with IC50 values similar to that of PD98059 (Table 1). The IC50 value for flavone inhibition of AHR transformation reported in Table 1 is similar to the value (e.g., 0.2 μm) reported in a recent study that also used rat liver extract (Lu et al., 1996).
Although a few flavonoids behave as pure AHR antagonists, most flavonoids that bind to the AHR function as either an AHR antagonist or agonist, with the latter activity occurring at concentrations higher than it takes to function as an antagonist (Gasiewicz et al., 1996; Lu et al., 1996). Flavone exhibited agonist activity in a gel retardation assay at concentrations of >20 μm (Fig. 7). Maximum agonist activity occurred at ∼80 μm. The amount of AHR/DRE complex formed at concentrations of flavone of ≥80 μm was comparable to that detected with TCDD. In contrast, flavanone was an extremely poor agonist and exhibited only a weak response at concentrations as high as 200 μm (Fig.7). PD98059 also exhibited a concentration-dependent agonist activity for the AHR that became obvious at concentrations of >60 μm (Fig. 7).

In vitro transformation of the AHR by PD98059 and its derivatives. Rat liver extract was transformedin vitro with either TCDD or varying concentrations of PD98059, flavone, or flavanone before being incubated with labeled DRE oligonucleotide and analyzed in an EMSA.
Competition by PD98059 with [3H]TCDD for the AHR.
The suppression of AHR transformation occurring in TCDD plus PD98059-treated liver extracts could reflect suppression of ligand binding, inhibition of AHR-ARNT heterodimerization, or suppression of heterodimer binding to DNA. The first of these possibilities was examined by sucrose gradient analyses of AHR/[3H]TCDD complex formation (Fig.8). Exposure of rat liver extract to TCDD and varying concentrations of PD98059 resulted in a concentration-dependent suppression of [3H]TCDD binding (IC50 = 4 μm). The IC50 value for this parameter was very similar to the IC50 value determined for the suppression of AHR/DNA complex formation scored in the EMSA. Hence, the effects detected in Fig. 6 probably reflect PD98059-mediated suppression of TCDD binding to the AHR.
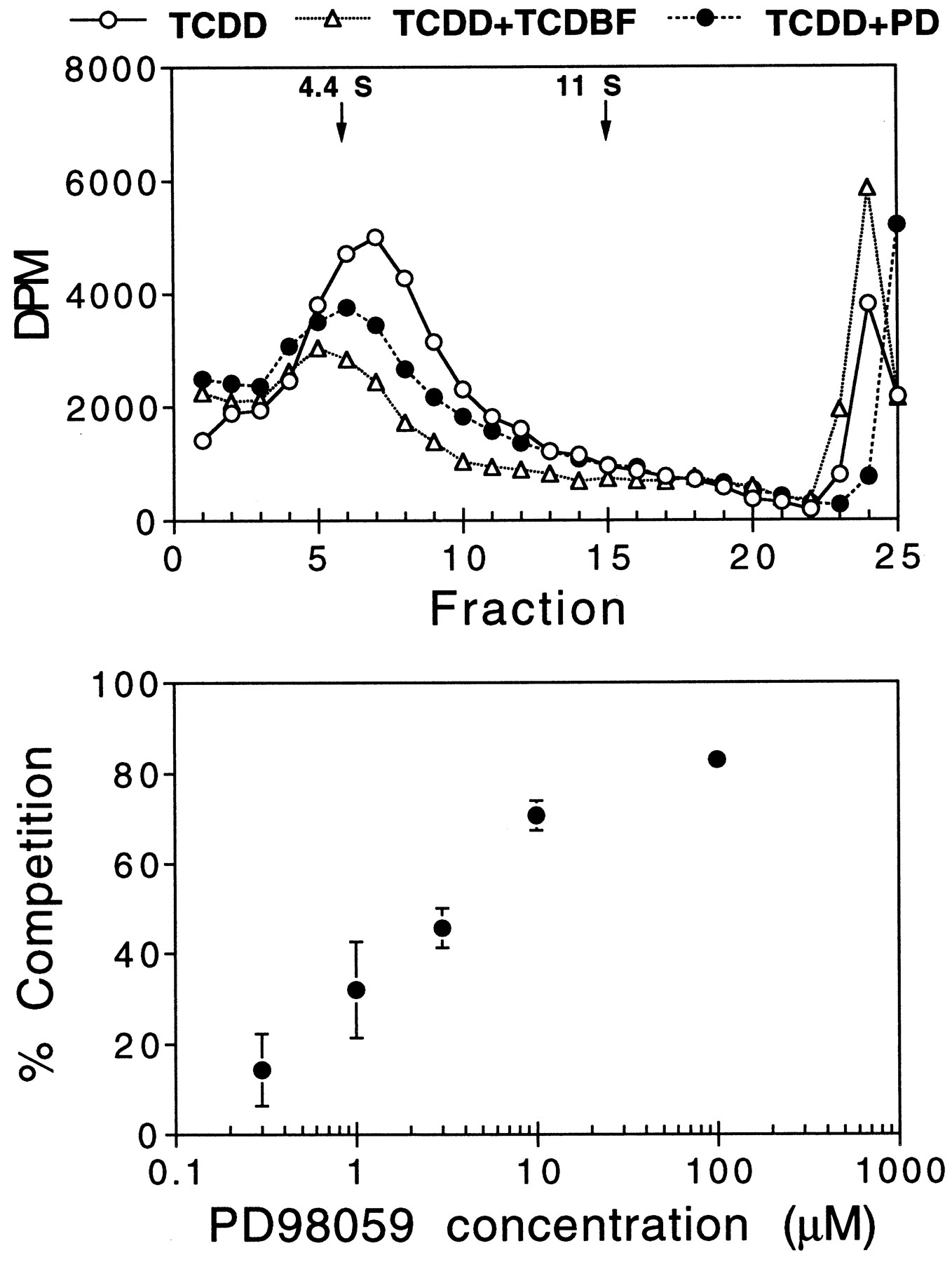
Sucrose gradient analyses of PD98059 (PD) suppression of TCDD binding to the AHR. Rat liver extract was incubated in vitro at 4° for 2 hr with 1 nm [3H]TCDD in the absence or presence of TCDBF (1 μm) or PD98059 (top, 10 μm; bottom, varying concentrations). At the end of 2 hr, the mixtures were treated with dextran/charcoal, adjusted to 0.4 m KCl, and loaded onto sucrose gradients (top). After 20 hr of centrifugation, the gradients were eluted and counted. The 4.4S and 11S markers correspond to bovine serum albumin and catalase, respectively. Bottom, mean ± standard error of a minimum of three experiments (only one experiment for 100 μm).
Discussion
Treatment of MCF10A and MCF10A-Neo cultures with PD98059 before TCDD addition strongly suppressed the subsequent accumulation of CYP1A1, CYP1B1, and NQ01 mRNAs. Concentrations of PD98059 affecting mRNA accumulation also suppressed in vitro transformation of the AHR by TCDD into a DRE-binding species. This latter observation, coupled with the finding that PD98059 inhibited the binding of TCDD to the AHR, strongly suggests that PD98059 is an AHR antagonist. Presumably, the reduced mRNA levels noted in TCDD-treated cultures reflect a PD98059-mediated suppression of the transcriptional activation of CYP1A1, CYP1B1, andNQ01.
Numerous natural and synthetic flavonoids are AHR antagonists. The IC50 values for PD98059 suppression of TCDD binding and transformation of the AHR are very similar to those estimated for flavanone (current study) and flavone and 3′-methoxy-4′-aminoflavone (Gasiewicz et al., 1996; Luet al., 1996). Similarity to the latter chemical is particularly important because it is an isomer of PD98059 that differs only in the positions of the amino and methoxy groups on the phenyl ring. Suppression of AHR transformation by PD98059 occurred at concentrations optimal for MEK inhibition. Flavone and flavanone also suppressed AHR transformation but did so at concentrations ∼100-fold lower than needed for MEK inhibition. Similarly, we estimated that 3′-methoxy-4′-aminoflavone and 3′-amino-4′-methoxyflavone inhibit AHR transformation at concentrations minimally 20-fold lower than that needed for MEK inhibition (Dudley DT, unpublished observations). Such structure-activity relationships do not rule out the possibility that the MEK/ERK cascade modulates AHR function. However, these data do suggest that the ability of PD98059 to affect TCDD-mediated, AHR-dependent processes in vivo can be attributed to its functioning as an AHR antagonist.
A variety of agents and physiological conditions that invoke a transient or protracted activation of the MAPK cascade suppress the transcriptional activation of CYP1A1 by AHR agonists (Hohneet al., 1990; Reiners et al., 1992, 1997;Berghard et al., 1993). Several studies have reported that ERK activities are significantly elevated in cultured cells after transfection and expression of oncogenic p21-ras (Leevers and Marshall, 1992; Shibuya et al., 1992). Hence, we were initially surprised that the phosphorylated (e.g., activated form) ERK content of the MCF10A-Neo line was similar to, if not greater than, that detected in the p21-ras transformed MCF10A-NeoT cell line. It is conceivable that our ERK data are the consequence of culturing in a medium supplemented with serum and growth factors. Such agents are known to stimulate membrane-bound receptors linked to the MAPK cascade and may stimulate ERK activities to levels far greater than that contributed by oncogenic p21-ras and thus obscure its contribution to ERK activation. There is precedent for this explanation because attempts to demonstrate agent-mediated ERK activation commonly use cells cultured in serum/growth factor-depleted medium (Leevers and Marshall, 1992;Troppmair et al., 1994). Indeed, we have found that the dually phosphorylated forms of ERK are undetectable in cultured MCF10A-Neo cells shifted to serum/growth factor-depleted medium. In contrast, although the relative content of dually phosphorylated ERK2 is decreased in MCF10A-NeoT cells after a similar shift, it is detectable (Dudley DT and Reiners JJ Jr., unpublished observations). Regardless of the basis for the basal ERK activities in the two cell lines, the finding that the TCDD-responsive MCF10A-Neo line has activated ERK contents comparable to or greater than the MCF10A-NeoT line strongly suggests that the constitutive down-regulation of AHR function observed in the MCF10A-NeoT cell line is not directly related to its ERK activities.
PD98059 is widely used in the signal transduction field as a tool for dissecting the contributions of distal members of the MAPK cascade to biological processes (Alessi et al., 1995; Dudley et al., 1995; Cook et al., 1997; Pumiglia and Decker, 1997). The results of the current study clearly demonstrate that concentrations of PD98059 that inhibit MEK also modulate AHR function as a consequence of functioning as an AHR antagonist. This latter property of PD98059 could compromise its usefulness as a MEK inhibitor in those situations in which the process being investigated is also influenced by the AHR (as in the current investigation). Recent studies suggest that the AHR may play a role in cellular processes distinct from its function as a ligand-activated transcription factor. Specifically, analyses of AHR-containing and -deficient hepatoma cell lines suggest that the AHR, in the absence of exogenous ligand, influences cell cycle progression, cell shape, and differentiation (Ma and Whitlock, 1996; Weib et al., 1996). Given the expanding scope of AHR functions, the dual activities of PD98059 must be considered in experimental design and data interpretation.
Acknowledgments
We thank Drs. Thomas Kocarek and Cornelis Elferink for their critical reading of this manuscript.
Footnotes
- Received September 10, 1997.
- Accepted December 1, 1997.
-
Send reprint requests to: John J. Reiners, Jr., Ph.D., Institute of Chemical Toxicology, Wayne State University, 2727 Second Avenue, Room 4000, Detroit, MI 48201. E-mail:john.reiners.jr{at}wayne.edu
-
This work was supported by National Institutes of Health Grant CA34469 and assisted by the services of the Cell Culture Facility Core and the Imaging and Cytometry Core, which are supported by National Institutes of Environmental Health Sciences Grant P30-ES06639.
Abbreviations
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- DRE
- dioxin responsive element
- EGF
- epidermal growth factor
- EMSA
- electrophoretic mobility shift assay
- ERK
- extracellular signal-regulated kinase
- GST
- glutathione-S-transferase
- MAPK
- mitogen-activated protein kinase
- MEK
- mitogen-activated protein kinase kinase
- MBP
- myelin basic protein
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- AHR
- aryl hydrocarbon receptor
- AHRT aryl hydrocarbon receptor nuclear translocator
- DMSO, dimethylsulfoxide
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}