


Abstract
The aryl hydrocarbon receptor (AHR) is believed to mediate many of the toxic, carcinogenic, and teratogenic effects of environmental contaminants such as dioxins, polycyclic aromatic hydrocarbons, and polyhalogenated biphenyls. Ligands for the AHR have been shown to influence cell proliferation, differentiation, and apoptosis, but the mechanism by which the AHR affects the cell cycle is not known. Increased levels of mature transforming growth factor-β (TGFβ) has been correlated with reduced cell proliferation and increased rates of apoptosis and fibrosis. Based on the increase in portal fibrosis and small liver size observed in AHR-null (Ahr−/− ) mice, the relationship between TGFβ expression and apoptosis in this mouse line was analyzed. Livers from Ahr−/− mice had marked increase in active TGFβ1 and TGFβ3 proteins and elevated numbers of hepatocytes undergoing apoptosis compared with wild-type mice. Furthermore, increases in TGFβ and apoptotic cells were found in the portal areas of the liver, where fibrosis is found in theAhr−/− mice. In vitro, primary hepatocyte cultures from Ahr−/− mice exhibited a high number of cells in later stages of apoptosis and an elevated secretion of active TGFβ into the media compared with cultures from wild-type mice, which have previously been shown to secrete only latent forms of the molecule. Conditioned media fromAhr−/− hepatocytes stimulated apoptosis in cultured hepatocytes from wild-type mice. Taken together, these findings suggest that the phenotypic abnormalities inAhr−/− mice could be mediated in part by abnormal levels of active TGFβ and altered cell cycle control.
The AHR is a transcription factor that mediates the toxic and carcinogenic effects of a group of chemicals including polycyclic aromatic hydrocarbons, polyhalogenated biphenyls and dioxins (TCDD) (Swanson and Bradfield, 1993; Fernandez-Salgueroet al., 1996). TCDD a widespread environmental contaminant that causes marked acute toxicity and is a nongenotoxic carcinogen in rodents (Safe et al., 1989). The AHR belongs to the periodicity/ARNT/simple-minded basic-helix-loop-helix superfamily of transcription factors that mediate the activation of target genes (Whitlock, 1993). Dioxin binding to the AHR results in its translocation to the nucleus where it dimerizes with a second periodicity/ARNT/simple-minded basic-helix-loop-helix protein called the ARNT (Reyes et al., 1992; Swanson and Bradfield, 1993). The functional AHR-ARNT heterodimer can interact with regulatory sequences called AHR response elements located upstream of target genes such as CYP1A1, CYP1A2,UDP-glucuronosyltransferase 1*06, and others.
The administration of TCDD to rodents causes an increase in the transcription of genes encoding xenobiotic metabolizing enzymes and induces a wide range of alterations in the immune system, hormonal, and endocrine imbalances and changes in cell proliferation and differentiation (Pohjanvirta et al., 1994). The mechanisms by which TCDD is able to induce alterations related to endogenous processes are under study. Among the possibilities is that TCDD disrupts the function of an unidentified endogenous ligand for the AHR, resulting in alteration of AHR-dependent signal transduction pathways. Direct evidence for a role for AHR in cell cycle control was obtained by comparing the Hepa 1c1c7 cell line with a clonal derivative lacking the receptor (Ma and Whitlock, 1996). In addition, it was shown that during the growth of NIH 3T3 fibroblasts, AHR expression was modulated by serum through a process dependent on tyrosine kinase activity and independent of dioxin (Vaziri et al., 1996). Other results indicated that the AHR can be found in the nucleus of HeLa cells in the absence of exogenous ligand (Singh et al., 1996). A critical endogenous role for the AHR also is supported by its constitutive pattern of expression early in embryonic development (Peters and Wiley, 1995) and by the fact that both AHR and ARNT are expressed during long periods of the developmental program of the mouse embryo (Abbottet al., 1994). Indeed, the AHR-null mouse presented phenotypic changes consistent with this hypothesis (Fernandez-Salgueroet al., 1995, 1997; Schmidt et al., 1996). Among the phenotypes reported for Ahr −/− mice are a small liver size and elevated pockets of hepatic fibrosis (Fernandez-Salguero et al., 1995; Schmidt et al., 1996).
TGFβ has been associated with the induction of increased amounts of fibrosis (Flier and Underhill, 1993; Border and Noble, 1994; Sandersonet al., 1995; Bernasconi et al., 1995) and decreased rates of cell proliferation in rodent liver (Oberhammeret al., 1992). In addition, different transgenic mouse lines expressing high levels of mature TGFβ1 protein in liver exhibited accelerated apoptosis in vivo, which was more pronounced in the lines with the higher level of expression of the transgene (Sanderson et al., 1995). To address the mechanism of the observed liver phenotype in Ahr −/− mice, TGFβ expression and apoptosis were analyzed. The following results suggest that in the absence of dioxin, the AHR is involved in cell cycle control in a signal transduction pathway that involves retinoic acid and TGFβ.
Materials and Methods
Mice.
AHR-deficient mice (C57BL6/CNrX 129/Sv) were produced as described previously (Fernandez-Salguero et al., 1995). Control male 5–6-month-old (Ahr+/+ ,Ahr+/− ) and AHR-null (Ahr−/− ) mice were housed in a specific pathogen-free facility using air-filtered controlled-environment racks and autoclaved water, cages, and bedding and were fed autoclaved Purina rodent chow. All manipulations of mice were done in sterile conditions and in accordance with the National Institutes of Health guidelines recommended and enforced by the National Cancer Institute Animal Care and Use Committee. Mice were genotyped by restriction fragment length polymorphism analysis of tail genomic DNA as described previously (Fernandez-Salguero et al., 1995).
Immunohistochemistry.
Mice were killed by CO2 narcosis, and the organs were quickly removed and fixed in formalin (10% formaldehyde in PBS). Tissues were embedded in paraffin and sectioned at 4–6 μm. TGFβ1 expression was analyzed by immunohistochemistry using antibodies designated CC and LC, both of which were raised against amino acids 1–30 (Flanders et al., 1989, 1991; Barcellos-Hoff et al., 1995). The sameAhr+/+ andAhr −/− mice were used for immunostaining with both anti-TGFβ1 and anti-TGFβ3 antibodies (Flanderset al., 1991). Immunostaining was performed according to a method described previously (Flanders et al., 1989, 1991;Barcellos-Hoff et al., 1995).
Isolation and culture of primary hepatocytes.
Mice were anesthetized by an intraperitoneal injection of 2.5% avertin (tribromoethyl alcohol and tertiary amyl alcohol), and the hepatocytes isolated by in situ collagenase perfusion as described previously (Maslanski and Williams, 1982). Briefly, the portal vein was cannulated, and the liver was perfused with 0.5 mm EGTA in Ca2+/Mg2+-free Hanks’ balanced salt solution and 0.05% collagenase (Waco Bioproducts, Richmond, VA) in Waymouth MB752/1 media (GIBCO BRL, Grand Island, NY) containing 10,000 units/ml penicillin, 100 μg/ml streptomycin, and insulin at 37°. The perfused liver was removed, placed in Waymouth MB752/1 media supplemented with 10% fetal bovine serum (Hyclone Labs, Logan, UT), trimmed of excess tissues, minced, and filtered through nylon into a 50-ml sterile tube. The resulting cell suspension was centrifuged for 5 min at 50 × g, and viable hepatocytes were selected by centrifugation in a Percoll isodensity gradient as described previously (Kreamer et al., 1986). Viable hepatocytes were counted by trypan blue exclusion using an hematocytometer and seeded at 7 × 104cells/cm2 onto plastic dishes. Four hours later, the medium was aspirated, and the cells received fresh media containing insulin as the sole hormone. The media was changed once daily thereafter.
Cell proliferation analysis.
DNA replication was determined at each time point by measuring the level of [3H]methylthymidine incorporation. Hepatocytes were seeded at 2.5 × 105 cells/ml (final volume, 3 ml) and incubated for 4 hr with 1 ml of [3H]methylthymidine (6.7 Ci/mmol, 1 μCi/ml; DuPont, Wilmington, DE) at a final concentration of 1 μCi/ml at different time points. The medium was aspirated, and the cells were washed in PBS; 10% cold trichloroacetic acid was added to the plates. Culture dishes were kept on ice for 30 min, and DNA was isolated as described previously (Laird et al., 1991). DNA synthesis was determined as counts of [3H] incorporated/min/mg of protein by liquid scintillation counting. Protein concentration was determined from an aliquot of scraped hepatocytes by using the BCA method (Pierce Chemical, Rockford, IL).
Preparation of conditioned media for quantification of TGFβ.
Liver perfusion and isolation of hepatocytes were performed as described with the following modification. Hepatocytes (1.5 × 106 cells/60-mm dish) were cultured in MB752/1 media supplemented as listed above (1.5 ml/plate). The media was replaced 4 hr after plating the cells, and fresh media contained insulin as the sole hormone. Forty-eight hours later, the supernatant from three or four plates was centrifuged at 2000 rpm for 15 min at 4°. Bovine serum albumin and phenylmethylsulfonyl fluoride (Sigma Chemical, St. Louis, MO) were added to the supernatant at final concentrations of 100 μg/ml, and the conditioned medium was separated into aliquots, placed into different Eppendorf tubes, and saved at −80° until measurement of TGFβ.
Bioassay for TGFβ.
Mink lung epithelial cells (CCL64) were plated onto 24-well plates at a density of 2.5 × 104 cells/well in 1 ml of Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (GIBCO BRL) and allowed to adhere overnight. The next day, the medium was aspirated, and wells were washed with fresh medium containing 1% serum and replaced with medium conditioned by either t mice were use orAhr −/− hepatocytes diluted in Dulbecco’s modified Eagle’s medium with 1% fetal bovine serum with or without the TGFβ-blocking antibody 1D11 (25 μg/ml; Genzyme, Cambridge, MA) or control IgG; additional wells were treated with medium plus TGFβ1 standard concentrations (each with total volume, 0.3 ml/well). CCL64 cells then were grown for an additional 24 hr with 10 μCi of [3H]thymidine (Amersham, Arlington heights, IL) added for the final 2 hr of incubation. The medium was aspirated, 0.5 ml of trypsin-EDTA (GIBCO BRL) was added, and the plates were incubated for 30 min at 37° before harvesting onto a 24-well filter plate and counting on a Top Count according to manufacturer’s instructions (Packard Instrument Company, Meriden, CT).
Analysis of apoptosis in vitro and in vivo.
Primary hepatocytes were analyzed for apoptotic features in vitro as described previously (Sheik et al., 1995). Primary cultures of hepatocytes were prepared and harvested at the indicated times. The cells were washed in PBS and fixed in ice-cold absolute methanol for 3–5 min, rehydrated in PBS, and incubated with DAPI solution for 30 min in the dark. The cells were washed, mounted with coverslips using 10% polyvinyl alcohol, and analyzed using a Zeiss fluorescence microscope at 420 nm. Apoptotic cells exhibiting crescent-shaped areas of condensed chromatin located near the periphery of the nucleus or fragmented nuclei were scored as positive. Apoptotic nuclei were counted in five to seven randomly selected fields using a ×40 Neofluar objective. At least 500-1000 nuclei were counted for each genotype and time point. The results are expressed as number of apoptotic nuclei divided by the total number of nuclei counted. To analyze apoptosis in vivo, liver sections were fixed, embedded in paraffin, and sectioned at 4–6 μm as indicated above. DAPI immunofluorescence was performed as described previously.
Northern blot analysis.
Total liver RNA fromAhr+/− , Ahr−/− , TGFβ1 homozygous null-mice (TGFβ1−/−; (Kulkarni et al., 1993), and mice with different genetic background and mouse kidney were isolated by tissue homogenization in guanidinium thiocyanate solution followed by centrifugation in a gradient of cesium trifluoroacetate (CsTFA). Poly(A+) RNA was purified from total RNA by oligo(dT) cellulose chromatography, and 3 μg of poly(A+) RNA was subjected to electrophoresis on a 1% agarose containing 2.2 m formaldehyde gel and transferred to Gene Screen Plus membranes in 20× standard saline citrate buffer (3 m NaCl, 30 mm sodium citrate, pH 7.0). The RNA was fixed to the membranes by baking at 80° for 2 hr, and the membranes were blocked by prehybridization for 3 hr at 65° in 0.5 mNaH2PO4, pH 7.0, 1% bovine serum albumin, 7% sodium dodecyl sulfate, and 1 mm EDTA. Mouse TGFβ1 and TGFβ3 cDNAs were labeled by random priming with the Klenow fragment of the DNA Polymerase I using [α-32P]dCTP (Pharmacia, Piscataway, NJ). The probes were added to the membranes at 1.5 × 106 cpm/ml in the same solution indicated above and hybridized overnight at 65°. The filters were washed in 0.1× standard saline citrate and 0.5% sodium dodecyl sulfate, and the membranes were exposed to X-ray film.
DNA fragmentation analysis.
Genomic DNA was isolated fromAhr+/+ , Ahr+/− , andAhr−/− primary hepatocytes cultures by proteinase K digestion and 2-propanol precipitation (Laird et al., 1991). Briefly, the cells were washed in PBS and incubated for 4 hr in lysis buffer at 37°. The DNA was precipitated with one volume of 2-propanol and centrifuged, and the pellet washed in 80% ethanol, dried, and resuspended in 10 mm Tris·HCl, pH 7.5, and 1 mm EDTA. Fifteen micrograms of DNA was electrophoresed in 0.8% agarose gels and visualized by staining with ethidium bromide.
Treatment of control hepatocytes with conditioned media fromAhr−/− hepatocytes.
Liver perfusion was performed as described previously under isolation and culture of primary hepatocytes. Hepatocytes from Ahr+/+ mice were plated at a cell density of 1 × 106 cells/plate (3 cm diameter, Corning Glassworks, Corning, NY) in MB752/1 medium supplemented with 1% fetal bovine serum (1 ml/plate) and left for 3–4 hr to attach. Monolayers were washed with PBS to remove dead or loosely attached cells and exposed to different treatments as described in the legend of Fig. 7. Hepatocytes were harvested 48 hr after treatments, and DNA was extracted and electrophoresed in 0.8% agarose gel as described.
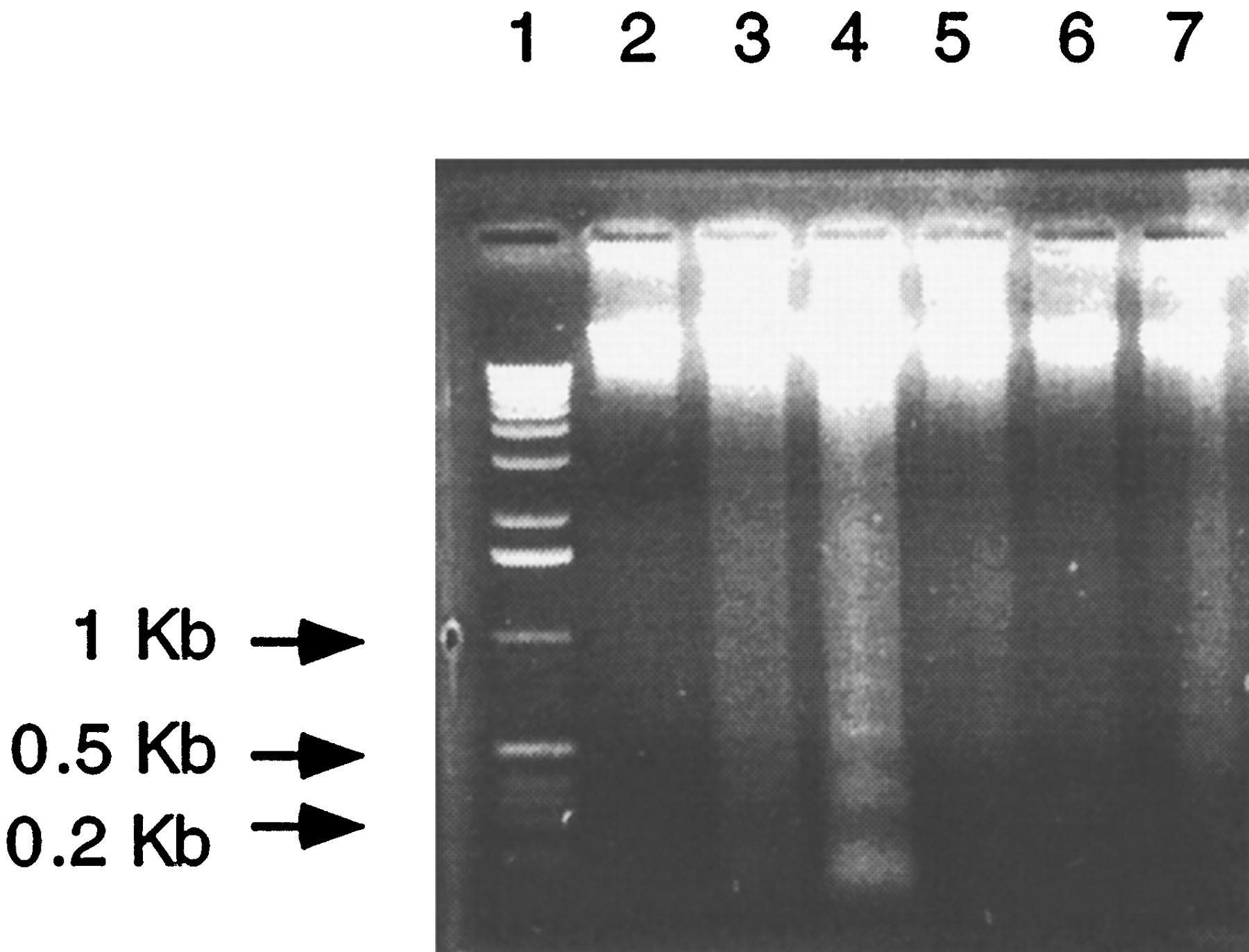
Treatment of control hepatocytes with conditioned media from Ahr −/− hepatocytes. Hepatocytes from wild-type mice, conditioned media, and DNA extraction were prepared as described in the text. Twenty micrograms of DNA was loaded in each lane. Lane 1, 1-kb ladder marker. Lane 2, day 0. Lane 3, control hepatocytes; 48 hr.Lane 4, hepatocytes treated with conditioned media for 48 hr; conditioned media diluted 1:4. Lane 5, hepatocytes treated with conditioned media plus antibody 1D11; condition media and antibody were preincubated for 1 hr at 4°; final concentration of the antibody in the media was 30–40 μg/ml.Lane 6, hepatocytes treated with MB752/1 media plus phenylmethylsulfonyl fluoride and bovine serum albumin. Lane 7, hepatocytes treated with MB752/1 media plus antibody 1D11). Three or four culture plates were used per treatment, and two separate cultures were analyzed, and the same results were obtained.
Results
TGFβ1 and TGFβ3 expression in Ahr−/− mice.
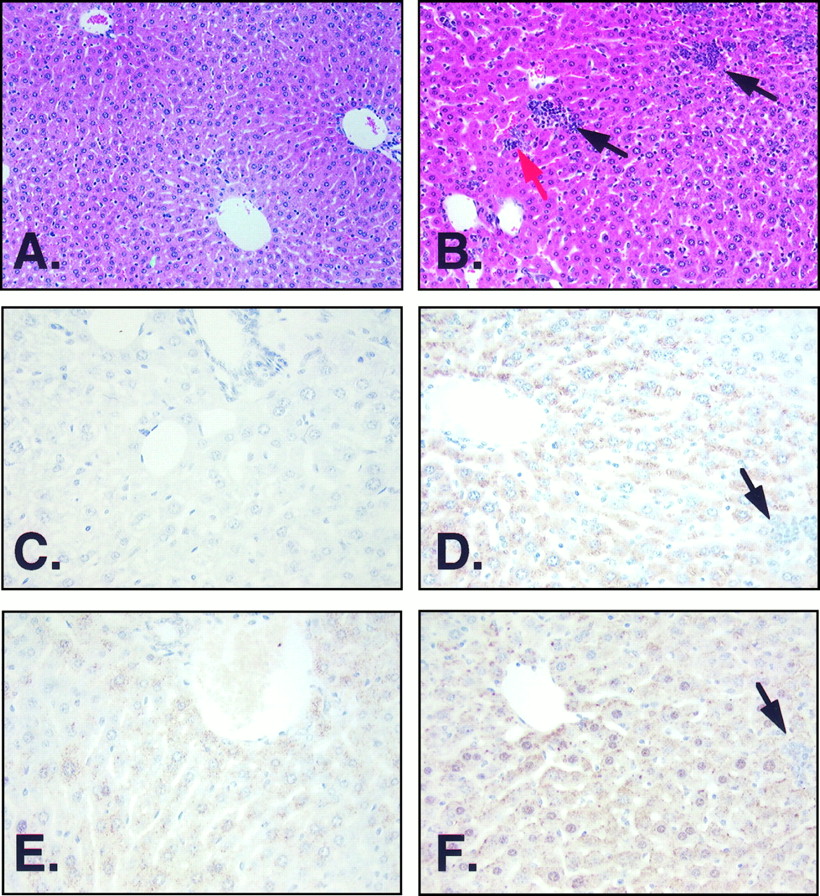
To establish a link between TGFβ expression and the increased fibrosis and smaller liver size reported inAhr−/− mice, the expression of TGFβ1 and TGFβ3 in the livers of control and Ahr−/− mice was examined by immunohistochemistry. TGFβ1 immunostaining was markedly increased in the portal areas of the liver ofAhr −/− (Fig.1D) compared with wild-type mice (Fig.1C). The level of TGFβ1 in the liver of wild-type mice essentially was undetectable by this technique as reported previously (Nagyet al., 1991). Immunodetectable levels of TGFβ3 protein also were elevated in Ahr −/− (Fig. 1F) compared with wild-type mice (Fig. 1E); this protein was, however, expressed in the wild-type mice in contrast to the lack of expression of TGFβ1. It should be noted that with the antibodies used, both proteins were localized in the cytosol of the hepatocytes, with an undetectable level of staining in the extracellular matrix. TGFβ1 and TGFβ3 expression also was analyzed by immunohistochemistry in heart (data not shown), and similar to the results found in liver,Ahr −/− mice exhibited higher levels of expression for both isoforms. This elevated expression of TGFβ1 and TGFβ3 may contribute to the presence of fibrosis in the heart ofAhr −/− mice (Fernandez-Salguero et al., 1997).

TGFβ1 and TGFβ3 immunohistochemistry. Liver sections were stained for TGFβ1 (C and D) and TGFβ3 (E and F). A and B correspond to hematoxylin-eosin-stained sections. A, C, and E, Obtained from liver sections from wild-type mice. B, D, and F, Obtained from Ahr −/− mice. Note the difference in expression between wild-type and Ahr −/−mice in both cytokines, particularly in TGFβ1. The expression of both proteins were exclusively intracellular, and no signal was obtained in the extracellular matrix. Black arrows (B, D, and F), Presence of granulocytes, mainly neutrophils, together with some foci of erythroid precursors scattered through the parenchyma. Red arrow, Existence of extramedullary hematopoiesis represented by erythroid precursors, probably coming from the bone marrow. Wild-type mice did not show any foci in the parenchyma (A). Hepatocytes present in Ahr −/− livers (B) were more heterogeneous than those in wild-type mice (A).
To determine whether Ahr −/− hepatocytes secreted active TGFβ, unlike normal hepatocytes, which have previously been shown to secrete only latent forms of the molecule (Lawrence, 1991; Gressner et al., 1997), a TGFβ bioassay was performed. A 4-fold dilution of conditioned media collected fromAhr −/− hepatocytes (48 hr after culturing) produced an inhibition of CCL64 cell growth equivalent to 0.090 ng/ml TGFβ1, which could be completely reversed by the addition of the anti-TGFβ blocking antibody 1D11 but not by control IgG (Fig.2). Activation of latent TGFβ in this sample by heating the conditioned media at 80° for 8 min revealed activity of ≥0.5 ng/ml, indicating the secretion of both active and latent forms by null hepatocytes. The 20 μg/ml of 1D11 antibody is insufficient to completely reverse TGFβ at concentrations of >0.5 ng/ml and thus only partially reversed inhibition by conditioned medium after heat activation (Fig. 2). The same results were obtained from three separate experiments. Time course determination revealed that there was no active TGFβ secreted in the media ofAhr −/− hepatocytes in the 4-hr collection and that the 24-hr time point was intermediate between the 4- and 48-hr collections (data not shown).

Concentrations of TGFβ in medium conditioned byAhr −/− hepatocytes 48 hr after seeding the cells. Hepatocytes were cultured in MB752/1 media without fetal bovine supplementation for 48 hr. After the time indicated, the medium was collected and TGFβ was quantified as described in the test. Experiments were performed in triplicate. No active TGFβ was detected in the medium of Ahr −/− hepatocytes in the 4-hr collection, and the 24-hr time point was intermediate between the 4- and 48-hr collections (data not shown).
To determine whether the genes encoding TGFβ1 and TGFβ3 were more actively expressed in the Ahr −/− mouse liver, Northern blot analysis was performed. Livers fromAhr+/+, Ahr+/− , andAhr−/− mice revealed no significant difference in mRNA levels (Fig. 3), even thoughAhr −/− mice had increased levels of intracellular TGFβ1 and TGFβ3 proteins as determined by immunohistochemistry. These results suggest that the increased immunolocalization of TGFβ in situ in AHR-null mice does not result from transcriptional up-regulation; rather, the increased detection may be the result of post-translational regulatory mechanisms, including activation and release of mature TGFβ from latency-associated proteins. Previous studies have demonstrated that under certain conditions, TGFβ immunoreactivity may reflect changes in activation status rather than merely protein abundance (Barcellos-Hoff, 1996). This may be the case inAhr −/− mice, particularly in light of our demonstration of active TGFβ by culturedAhr −/− hepatocytes.

Northern blot analysis of TGFβ1 and TGFβ3 expression in Ahr +/+,Ahr +/−, andAhr −/− mouse livers. Poly(A+) RNAs (3 μg/lane) were electrophoresed in agarose/formaldehyde gels, blotted to nylon membranes, and hybridized with the mouse cDNAs for the TGFβ1 and TGFβ3 genes as described in the text. Lanes +/+, +/−, and −/−, wild-type (Ahr +/+), heterozygous (Ahr +/−), and homozygous-null (Ahr −/−) mice, respectively. Mouse actin cDNA was used as a probe to control for gel loading and RNA integrity.Last three lanes, 10 μg of total RNA from TGFβ1 homozygous-null mice (TGFβ1−/−) as negative control, wild-type mice with a different genetic background, and wild-type mouse kidney as positive control for TGFβ1, respectively.
Apoptosis in livers of Ahr−/− mice.
To determine whether hepatocytes from livers ofAhr−/− mice had increased apoptosis, DAPI immunofluorescence was performed. As noted above, the morphology of hepatocytes in the portal areas of liver sections fromAhr +/+ mice differed from those present in the portal areas of Ahr −/− mice of the same age and sex (Fig. 4). Liver sections fromAhr −/− mice exhibited increased numbers of hepatocytes presenting peripheral accumulation of chromatin in their nuclei (Fig. 4, arrows), a feature indicative of apoptosis. TGFβ expression and apoptosis analyses were performed on tissue sections obtained from the same mice, and as observed, TGFβ1 and TGFβ3 expression and the apoptotic cells inAhr −/− mice were mainly localized in the portal areas of the liver. These results suggest the existence of a spatial correlation in the portal areas of the liver ofAhr −/− mice where overexpression of TGFβ1 and TGFβ3 and apoptosis were coincident. It should be noted that the portal areas in the liver ofAhr −/− mice were reported to exhibit increased levels of fibrosis (Fernandez-Salguero et al., 1995; Schmidt et al., 1996).

Morphological analysis for apoptosis in the liver. Liver sections from Ahr +/+ andAhr −/− mice were prepared, and DAPI immunofluorescence was carried out as indicated in the text.Arrows, Ahr −/− hepatocytes showing chromatin condensation. All sections presented corresponded to portal areas of the liver. Liver sections from one wild-type and two AHR-null mice are shown.
Analysis of apoptosis in primary cultures of hepatocytes fromAhr−/− mice.
Primary cultures of hepatocytes were produced from wild-type andAhr−/− mice by collagenase perfusion. Although the total recovery of hepatocytes from wild-type mice was 24–26 × 106 cells/g of liver, it was only 11–13 × 106 cells/g of the liver fromAhr−/− mice. Cell viability, as determined by trypan blue exclusion, was 85–90% and 80–85% for hepatocyte cultures from control and Ahr −/− mice, respectively. In addition, hepatocytes obtained fromAhr −/− mice represented a more heterogeneous population with respect to cell size than control cells. This is consistent with the histochemical analysis presented in Fig. 1. Pretreatment of the plate surface with an extracellular matrix such as Matrigel did not improve the plating efficiency ofAhr −/− hepatocytes compared with aliquots from the same preparation plated directly onto plastic dishes. These results indicate that under similar conditions of hepatocyte isolation from wild-type mice, Ahr −/− cells had lower plating efficiency after dissociation from the liver and culturing. No difference was observed betweenAh r+/+ andAhr+/− hepatocyte cultures, indicating that AHR gene dosage is not relevant for these processes. To date, characterization studies of Ahr+/− mice revealed that they are indistinguishable (Fernandez-Salguero et al., 1995, 1996; Schmidt et al., 1996).
Because cultured rodent primary hepatocytes are able to enter into the S phase of the cell cycle (Loyer et al., 1996), the replication status of Ahr+/− andAhr−/− hepatocytes was determined. The rate of [3H]thymidine incorporation inAhr+/− and Ahr−/− hepatocyte cultures is shown in Fig. 5A.Ahr+/− hepatocytes exhibited a [3H]thymidine incorporation profile that increased with time up to 72 hr in culture and was similar to results reported by others (Loyer et al., 1996). In contrast,Ahr −/− hepatocytes exhibited no increase DNA synthesis with time, suggesting that cell replication is blocked. To determine whether the cell death found inAhr −/− cultures was due to apoptosis or necrosis, the DNA from these cultures was analyzed. Fig. 5B shows that DNA from Ahr −/− hepatocytes, isolated at 44 hr after plating, had DNA fragmentation patterns typical of apoptosis; this was not observed in cultures fromAhr+/− mice. Lower degrees of DNA fragmentation also were found in Ahr −/− cells after 4 hr of culturing (data not shown).

DNA synthesis and apoptosis detection by DNA fragmentation in Ahr −/− mice. A, Rate of incorporation of [3H]methylthymidine in primary cultures of hepatocytes isolated by two-step collagenase perfusion fromAhr +/− (▪) andAhr −/− (•) mice as a function of time in culture. The level of incorporation was normalized by the total amount of protein in the cultures. Dead cells were removed by aspiration, and the attached cells were used in the labeling experiment. Results obtained from one Ahr +/− and two different Ahr−/− mice are shown. This experiment was repeated three times, and similar results were obtained. B, DNA fragmentation analysis in primary cultures of hepatocytes 44 hr after plating. Lanes 1 and 2, 20 and 5 μg of genomic DNA fromAhr +/− cells, respectively.Lane 3, 20 μg of DNA fromAhr −/− primary hepatocytes. A significant degree of DNA fragmentation, visualized as DNA laddering, was clearly visible in the DNA from Ahr −/− cells. The 1-kb DNA ladder (GIBCO BRL, Gaithersburg, MD) was used as size marker.
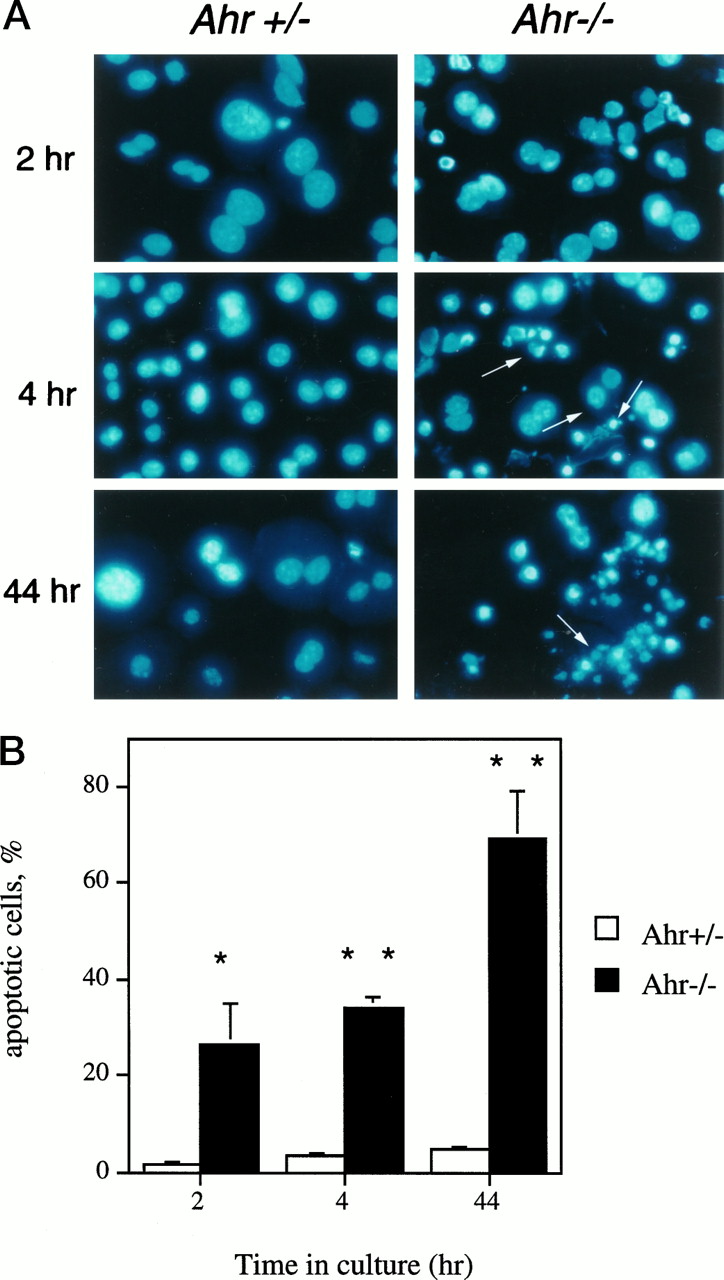
To further confirm the presence of apoptotic cells in primary cultures of hepatocytes from the Ahr −/− mice, nuclei were stained with DAPI to detect the morphological features of apoptosis. As shown in Fig. 6A, a significant increase in the number of cells exhibiting chromatin condensation and nuclear fragmentation (Fig. 6A, arrows) was observed in the nuclei of Ahr −/−hepatocytes with increased time in culture.Ahr+/− cells, on the contrary, did not exhibit this phenotype. Although chromatin condensation at the periphery was the predominant apoptotic feature in liver sections fromAhr −/− mice (Fig. 4), primary hepatocyte cultures from Ahr −/− mice presented a significant increase in the number of cells at the later stages of apoptosis with marked evidence of chromatin condensation and nuclear fragmentation (Fig. 6A). These results suggest that on dissociation from the tissue and disruption of cell/cell interactions, the lack of AHR expression seems to accelerate the process of cell death in vitro. Apoptosis developed rapidly inAhr −/− cultures: after 4 hr, >30% of the cells had chromatin condensation and nuclear fragmentation, and by 44 hr, ∼70% of the cells were apoptotic with significant nuclear fragmentation (Fig. 6B). Cultures with a majority of cells with chromatin condensation are indicated by one asterisk, whereas cultures exhibiting nuclear fragmentation are indicated by two asterisks. The increased rates of apoptosis in cultures fromAhr −/− mice was not due to the perfusion method used because, as shown in Fig. 6, A and B,Ahr+/− mice perfused at the same time and maintained under the same culturing conditions did not show a significant degree of apoptosis (<2–4%). These results are in agreement with the observed lower efficiency ofAhr −/− hepatocytes to attach to the culture plates, with the low level of DNA synthesis, and with the presence of DNA fragmentation. Therefore, the presence of apoptosisin vivo and in vitro, together with increased levels of TGFβ1 and TGFβ3 in the portal areas, strongly supports a role for this cytokine in the genesis of portal fibrosis in the liver of Ahr −/− mice.

Morphological analysis for apoptosis in primary cultures of hepatocytes. A DAPI immunofluorescence was performed on primary hepatocyte cultures from Ahr +/−and Ahr −/− mice as a function of time in culture. Nuclear morphology of the control hepatocytes showed a homogeneous distribution of chromatin with no obvious indication of condensation or fragmentation. In contrast,Ahr −/− cultures had a significant number of cells, with crescent-shaped areas showing condensation of chromatin and fragmented nuclei (arrows) that could be observed 4 hr after plating. These numbers increased with time; by 44 hr, a major population of Ahr −/− hepatocytes was at later stages of apoptosis as evident by nuclear fragmentation. B, Quantification of apoptotic cells in Ahr +/−and Ahr −/− hepatocytes: between 500 and 1000 nuclei were counted for each genotype and time point, and the results referred to the total number of nuclei considered (100%). Apoptosis proceeded rapidly inAhr −/− hepatocytes such that by 4 hr, 20–30% of the cells presented both chromatin condensation and nuclear fragmentation. Forty-four hours after culturing, this fraction increased to close to 70% of the hepatocytes. ∗, Chromatin condensation in the nuclei. ∗∗, Presence of nuclear fragmentation.Error bars, mean ± standard deviation values.
Treatment of control hepatocytes with conditioned media fromAhr−/− hepatocytes.
To determine whether apoptosis seen in Ahr−/− hepatocytes was directly due to TGFβ, hepatocytes from wild-type mice were treated with conditioned media collected from Ahr−/−hepatocytes alone or conditioned media plus monoclonal antibody 1D11. The DNA, extracted from hepatocytes treated with conditioned media for 48 hr and electrophoresed through 0.8% agarose gel, exhibited a ladder pattern characteristic of digestion into oligonucleosomal fragments (Fig. 7). On the other hand, no DNA digestion was evident in hepatocytes treated with conditioned media pretreated with the antibody 1D11 (Fig. 7). These results indicate that TGFβ and probably other proteins secreted fromAhr −/− hepatocytes stimulated apoptosis.
Discussion
To further understand the development of fibrosis and the small liver size observed in AHR-null mice in the absence of treatment with dioxin (Fernandez-Salguero et al., 1995, Schmidt et al., 1996), changes in the expression of TGFβ1 and TGFβ3 were analyzed. These cytokines represent relevant candidates to explain such alterations in Ahr −/− mice because they have been associated with increased fibrosis, diminished cell proliferation and elevated apoptosis (Lin and Chou, 1992; Border and Noble, 1994; Jurgensmeier et al., 1994; Bernasconi et al., 1995; Ohno et al., 1995; Sanderson et al., 1995). The portal areas ofAhr −/− mice exhibited increased immunodetectable levels of intracellular TGFβ1 and TGFβ3 proteins as determined by immunohistochemistry. Primary cultures of hepatocytes from AHR-null mice secreted higher levels of active and latent TGFβ into the conditioned media as determined by its ability to inhibit proliferation of mink lung epithelial cells and to produce apoptosis in wild-type hepatocytes. TGFβ1 and TGFβ3 overexpression and apoptotic features in vivo were mostly limited to the portal areas of the liver where increased fibrosis was reported previously (Fernandez-Salguero et al., 1995; Schmidt et al., 1996). Apoptosis in Ahr −/− mice was more pronounced in primary cultures of hepatocytes, which exhibited early onset of chromatin condensation, nuclear fragmentation, and DNA laddering together with very low levels of DNA synthesis.
Several studies have shown that overexpression of TGFβ1 can result in increased levels of apoptosis in vitro. In support of this observation, TGFβ1-treated normal fibroblasts are able to induce apoptotic elimination of UV-transformed fibroblasts, indicating that transformed cells have an increased sensitivity to this cytokine (Jurgensmeier et al., 1994). TGFβ1 also is able to induce apoptosis not only in human hepatoma cell lines (Lin and Chou, 1992) but also in primary cultures of rat hepatocytes (Ohno et al., 1995; Cain et al., 1996). These studies support the possibility that elevated TGFβ causes the increased apoptosis in AHR-null mice. The number of apoptotic cells was shown to be highest in the portal zone of the liver. Indeed, TGFβ expression is not homogeneous in different areas of the liver (Ohno et al., 1995), with TGFβ1 and TGFβ3 expression mainly localized to the portal areas. This is the same region of the liver that shows spontaneous fibrosis in the AHR-null mice (Fernandez-Salguero et al., 1995; Schmidt et al., 1996). Thus, the results reported herein further strengthen the possibility of a direct relationship between TGFβ and apoptosis in livers. Earlier studies also indicated that TGFβ1 expression was associated with an increased rate of apoptosis in vivo because different transgenic mouse lines overexpressing the mature form of TGFβ1 in liver exhibited apoptosis that was more pronounced in the lines with the higher level of expression of the transgene (Sanderson et al., 1995). Overexpression of this cytokine was related to increased extracellular matrix deposition and fibrosis, not only in mouse models (Sandersonet al., 1995) but also in humans exhibiting different tissue injuries and diseases (Flier and Underhill, 1993). In addition, it was reported that the intravenous injection of mature TGFβ1 in rats induces apoptosis in liver and that the apoptotic activity of TGFβ1 is related to effects on cell proliferation and differentiation (Oberhammer et al., 1992). TGFβ1 also was associated with lower levels of DNA synthesis (Nakamura et al., 1985), with organ growth regulation (Braun et al., 1988), and with time-dependent DNA fragmentation and chromatin condensation in hepatocyte cultures (Cain et al., 1996). Furthermore, high levels of TGFβ1 were shown to elicit a significant increase in apoptosis in fetal mouse hepatocyte cultures (Fabregat et al., 1996), in rat primary hepatocyte cultures, and in human hepatoma cell lines (Fan et al., 1996). Interestingly, primary cultures of hepatocytes fromAhr −/− mice secreted high levels of active TGFβ1 into the conditioned media, suggesting that the antiproliferative activity of TGFβ could account for the observed lack of DNA replication and for the increased rate of apoptosis in these cultures and in vivo. In support of this observation is the presence of apoptosis detected as DNA ladder by conventional agarose gel electrophoresis (180–200-oligonucleosome integer fragments), in hepatocytes from wild-type mice after treatment with medium from Ahr −/− hepatocytes, and the ability of the antibody to antagonize this effect. Therefore, our results suggest that in the absence of AHR expression, high levels of active TGFβ1 and TGFβ3 could be responsible not only for a higher rate of apoptosis but also for a lower rate of cell proliferation in mouse liver. The observation that TGFβ1 is able to control apoptosis in fetal hepatocytes (Fabregat et al., 1996) indicates that during embryonic development, a process involving the AHR, TGFβ1, and TGFβ3 could participate in regulating the extent of cellular proliferation in the liver, thus contributing to the control of the size of this organ in the adult.
The pattern of expression of TGFβ1 and TGFβ3 inAhr −/− mouse liver and its correlation to apoptosis and fibrosis could be used to determine whether the mode of action of these cytokines is autocrine versus paracrine. Two hypothesis have been developed to explain signaling through TGFβ at a distance. One postulates that TGFβ would have a paracrine mode of action by generating a concentration-dependent gradient acting at a distance from the cell of origin (Lecuit et al., 1996; Nellen et al., 1996). A second hypothesis suggests that TGFβ would induce autocrine signaling only between adjacent cells and that this will trigger, by a relay mechanism, a cascade of various inducing signals throughout the intercellular matrix (Bissell et al., 1995;Reilly and Melton, 1996), thus limiting TGFβ mobility to only few layers of neighboring cells. Our results indicate that inAhr −/− mouse liver, TGFβ1 and TGFβ3 proteins were mainly localized in the hepatocytes surrounding the portal areas, and suggest that an autocrine mode of action would account for the phenotype observed in these areas of the liver. In agreement with this hypothesis, it was shown that although the level of TGFβ1 in normal or undamaged liver essentially is undetectable by immunostaining, after induction of fibrosis, there is a localized increase in the immunoreactivity for TGFβ within fibrotic tissue but not within the normal adjacent cells (Nagy et al., 1991).
The mechanism by which AHR controls TGFβ expression could involve retinoic acid. Ahr −/− mice have a significant alteration in the extent of metabolism of retinoic acid (Andreola et al., 1997). Thus, the lack of AHR causes a lower expression of an enzyme that catalyzes retinoic acid catabolism, resulting in accumulation in the liver of retinyl esters and retinoic acid. The latter can cause an increase in retinoic acid-responsive genes through activation of retinoic acid receptor-α. Indeed, the type II transglutaminase, which is capable of converting latent TGFβ1 and TGFβ3 to the active form (Nunes et al., 1997;Gleizes et al., 1997), has a retinoic acid receptor response element (Nagy et al., 1996) and is elevated in livers ofAhr −/− mice (Andreola et al., 1997). The role of this enzyme in activation of latent TGFβ in theAhr −/− mice is supported by the fact that TGFβ mRNAs were not increased in livers of Ahr −/− mice compared with wild-type mice, again suggesting that the increased immunodetection of TGFβ may reflect post-translational modifications, including activation of latent TGFβ complex (Barcellos-Hoff, 1996). The increase in immunohistochemically stained TGFβ in the absence of new protein synthesis could be the result of preferential staining of the active forms of the cytokine as demonstrated previously (Barcellos-Hoff et al., 1995). An association between retinoic acid levels and TGFβ expression has been demonstrated in keratinocyte cultures in which retinoic acid increases the level of secretion of active TGFβ and decreases DNA synthesis (Flanders et al., 1989). Retinoic acid also is able to activate TGFβ in cocultures of endothelial smooth muscle cells by a transglutaminase-mediated mechanism (Glick et al., 1989). The results reported herein support the possibility of an endogenous role for the AHR in liver development through interaction with the retinoic acid/TGFβ signal transduction pathways.
Footnotes
- Received February 17, 1998.
- Accepted April 23, 1998.
-
Send reprint requests to: Dr. Frank J. Gonzalez, NCI/NIH, Bldg. 37, Rm. 3E-24, Bethesda, MD 20892. E-mail: fjgonz{at}helix.nih.gov
-
↵1 Current affiliation: Laboratorio de Bioquı́mica y Biologı́a Molecular, Facultad de Ciencias, Universidad de Extremadura, 06080-Badajoz, Spain.
Abbreviations
- AHR
- aryl hydrocarbon receptor
- DAPI
- 4′-diamino-2-phenylindole
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- PBS
- phosphate-buffered saline
- TGFβ
- transforming growth factor-β
- ARNT
- aryl-hydrocarbon receptor nuclear translocator
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}