


Abstract
We compared the phosphorylation and internalization properties of constitutively active alpha-1b adrenergic receptor (AR) mutants carrying mutations in two distant receptor domains, i.e., at A293 in the distal part of the third intracellular loop and at D142 of the DRY motif lying at the end of the third transmembrane domain. For the A293E and A293I mutants the levels of agonist-independent phosphorylation were 150% and 50% higher than those of the wild-type alpha-1b AR, respectively. On the other hand, for the constitutively active D142A and D142T mutants, the basal levels of phosphorylation were similar to those of the wild-typealpha-1b AR and did not appear to be further stimulated by epinephrine. Overexpression of the guanyl nucleotide binding regulatory protein-coupled receptor kinase GRK2 further increases the basal phosphorylation of the A293E mutant, but not that of D142A mutant. Both the wild-type alpha-1b AR and the A293E mutant could undergo β-arrestin-mediated internalization. The epinephrine-induced internalization of the constitutively active A293E mutant was significantly higher than that of the wild-typealpha-1b AR. In contrast, the D142A mutant was impaired in its ability to interact with β-arrestin and to undergo agonist-induced internalization. Interestingly, a double mutant A293E/D142A retained very high constitutive activity and regulatory properties of both the A293E and D142A receptors. These findings demonstrate that two constitutively activating mutations occurring in distant receptor domains of the alpha-1b AR have divergent effects on the regulatory properties of the receptor.
Thealpha-1b adrenergic receptor (AR) belongs to the superfamily of guanyl nucleotide binding regulatory protein (G protein)-coupled receptors (GPCR). The seven transmembrane domains (TMD) common to all GPCRs contribute to the formation of the ligand binding pocket, whereas amino acid sequences of the intracellular domains appear to mediate receptor–G protein coupling (Savarese and Fraser, 1992). Agonist binding to a GPCR is believed to induce a conformational change of the receptor that results in its productive coupling to heterotrimeric G proteins, thus leading to intracellular signaling events.
After receptor activation, exposure to the agonist can induce a series of biochemical events, resulting in desensitization of various GPCRs. A prominent role in agonist-induced desensitization of several GPCRs is played by GPCR kinases (GRK) whose activation does not require the production of second messengers (Premont et al., 1995). Once the receptor is occupied by the agonist, it is recognized by the kinase and becomes phosphorylated. The subsequent uncoupling of the receptor and G protein is then mediated by arrestin proteins that specifically bind to the phosphorylated receptor (Wilden et al., 1986; Benovic et al., 1987). GRKs have the unique ability to recognize and phosphorylate their GPCR substrates only in their active (i.e., agonist-occupied) conformation.
Exposure to the agonist can also induce receptor internalization that results in the decrease of cell surface receptors and their translocation toward an intracellular compartment (Fonseca et al., 1995; Koenig and Edwardson, 1997). Several lines of evidence indicate that internalization does not mediate receptor desensitization because it occurs when receptors are already uncoupled from the G protein. On the other hand, internalization seems to play an important role in the resensitization of the receptor as has been shown for thebeta-2 AR (Pippig et al., 1995; Krueger et al., 1997). Recently, elegant studies provided evidence that binding of β-arrestin to the phosphorylated receptor represents an early step in the agonist-induced internalization of beta-2 AR (Ferguson et al., 1996; Goodman et al., 1996).
We recently described that mutations of Asp-142 belonging to the highly conserved DRY sequence lying at the end of TMDIII and of Ala-293 in the distal part of third intracellular (i3) loop could constitutively activate the alpha-1b AR (Kjelsberg et al., 1992; Scheer et al., 1996). A combined approach with experimental and computer-simulated mutagenesis of the receptor suggested that these two amino acids play a key role in the activation process of thealpha-1b AR, i.e., its transition from the inactive to active state (Scheer et al., 1996, 1997).
Because constitutively active receptor mutants are thought to mimic, at least in part, the agonist-occupied form of wild-type GPCRs, it has been postulated that they can be substrates for GRK-mediated phosphorylation in the absence of the agonist. This hypothesis is supported by previous findings that indicated that constitutively active beta-2 AR and alpha-2 AR carrying mutations in the distal portion of i3 loop can be phosphorylated by GRK2 in the absence of the agonist (Ren et al., 1993; Pei et al., 1994). These results suggested that activating mutations in the C-terminal portion of the i3 loop of the ARs induce receptor conformations with docking complementarity with both the G protein heterotrimers and regulatory proteins, including GRKs. However, activating mutations have been recently described in different structural domains of GPCRs (Scheer and Cotecchia, 1997). Thus, an important question is whether constitutively active receptors carrying mutations in different receptor domains share similar regulatory properties.
To investigate the relationship between constitutive activity and the regulatory properties of receptor mutants, in this study we compared the phosphorylation and internalization properties of constitutively active alpha-1b ARs carrying mutations in two distant receptor domains, i.e., at A293 in the distal part of i3 loop and at D142 lying at the end of TMDIII. Our results indicate that the constitutively active alpha-1b AR mutated at either one of the two positions are strikingly different with respect to their regulatory properties. This study provides information that might have several implications for the elucidation of the mechanisms underlying the activation and regulation of the alpha-1b AR both at a biochemical and structural level.
Experimental Procedures
COS-7 Cell Culture and Transfections.
COS-7 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and gentamicin (100 μg/ml) and transfected by the diethylaminoethyl-dextran method. The cDNA encoding the hamsteralpha-1b AR (Cotecchia et al., 1988) and its mutants were subcloned in pRK5 (Didsbury et al., 1991), that of GRK2 and its K220R mutant (Kong et al., 1994) in pCMV5 (Andersson et al., 1989) and that of the β-arrestin-1 mutant V53D (Ferguson et al., 1996) in pCDNA1. For phosphorylation assays, cells were plated in 55-mm dishes (1 × 106 cells) for most experiments and in 100-mm dishes (3 × 106 cells) for experiments involving the D142A and D142T mutants. The transfected DNA encoding the receptors was 2.5 μg per 1 × 106 cells. For inositol phosphate (IP) determination, COS-7 cells (0.15 × 106) were plated in 12-well plates and the transfected DNA encoding the receptors ranged from 0.2–3 μg per 1 × 106 cells as indicated in the figures. For internalization experiments, cells (0.3 × 106) were grown in six-well plates and the transfected DNA was 0.2–3 μg per 1 × 106 cells for the receptors. The amount of transfected DNA encoding GRK2, its K220R mutant and the β-arrestin-1 mutant V53D was 3-fold higher than that of the receptors. Assays were performed 48 h after transfection.
Mutagenesis of the alpha-1b AR.
The cDNA of the hamster alpha-1b AR was mutated by polymerase chain reaction-mediated mutagenesis with Taq DNA polymerase. The mutated DNA fragments were sequenced by automated DNA sequencing (Microsynth, Balgach, Switzerland) and subcloned in the pRK5 expression vector containing the wild-type alpha-1b AR.
IP Measurement.
Transfected cells were labeled for 12 h with myo-[3H]inositol at 4 μCi/ml in inositol-free DMEM supplemented with 1% FBS. Cells were then preincubated for 10 min in phosphate-buffered saline containing 20 mM LiCl, and then stimulated for 45 min with epinephrine. Total IPs were extracted and separated as described previously (Cotecchia et al., 1988).
32P-Labeling and Immunoprecipitation of the Receptors.
Transfected COS-7 cells were equilibrated in phosphate-free DMEM for 2 h and then incubated in the same buffer containing 32Pi (0.2 mCi/ml) for 2 h at 37°C. The incubation was then continued in the absence or presence of epinephrine as indicated. A separate set of dishes was incubated under similar conditions, but in the absence of 32Pito measure receptor binding. Immunoprecipitation of the phosphorylated receptors was performed as described previously (Lattion et al., 1994). For most experiments, antibodies raised against the last 24 amino acids (residues 492–515) of the alpha-1b AR were used at 1:50 dilution. Similar results were obtained with antibodies raised against the first 22 amino acids of the receptor (data not shown). After autoradiography, the amount of radioactivity associated with the phosphorylated receptor was quantified by liquid scintillation spectroscopy. To assess receptor phosphorylation, the receptors were expressed at similar levels and the 32P content (counts per minute) of the immunoprecipitated receptors was directly compared for statistical analysis.
Western Blot Analysis of GRK2 and β-Arrestin.
Overexpression of GRK2, of its mutant K220R and of β-arrestin-1 mutant V53D was assessed on the cytosolic fractions of transfected cells as described previously (Diviani et al., 1996). For immunodetection of GRK2 and its mutants K220R, the antiserum (used at 1:1000 dilution) was raised against a glutathioneS-transferase fusion protein encoding residues 467 to 688 of rat GRK3, as described previously (Diviani et al., 1996). For immunodetection of the β-arrestin-1 mutant V53D, the antiserum (used at 1:1000 dilution) was raised against a glutathioneS-transferase fusion protein encoding residues 172–268 of bovine β-arrestin.
Ligand Binding.
Membrane preparations derived from cells expressing the alpha-1b AR or its mutants and ligand binding assays with {β-(4-hydroxy-[125I]iodophenyl)ethylaminomethyl}tetralone ([125I]HEAT) were performed as described previously (Cotecchia et al., 1992). Prazosin (10-6 m) was used to determine nonspecific binding. [125I]HEAT concentration was 250 pM for measuring receptor expression at a single saturating concentration and 80 pM for competition binding analysis of epinephrine. Intact cell receptor binding assays were performed as described (Lattion et al., 1994) by incubating cell monolayers grown in 35-mm dishes with [3H]prazosin (2 nM) in 2.5 ml of DMEM at 4°C for 7–10 h. After binding, cells were washed three times with ice-cold phosphate-buffered saline containing 0.1% bovine serum albumin, scraped in water, and counted. Phentolamine (10-4 M) was used to determine nonspecific binding that was 50% of total binding. Data were analyzed by nonlinear least-square regression analysis (DeLean et al., 1982).
β-Arrestin-Green Fluorescent Protein (GFP) Distribution.
The construction of the plasmid encoding a β-arrestin-GFP conjugate, transfection of HEK-293 cells and fluorescence microscopy were as described previously (Barak et al., 1997). The transfected DNA per 106 cells was 0.025–1.0 μg for thealpha-1b AR and A293E mutant, 3 μg for the D142A receptor, 0.5 μg for the β-arrestin-GFP, and 1 μg for GRK2.
Statistical Analyses.
Results are expressed as mean ± S.E. Statistical significance was assessed by paired Student’st test.
Materials.
COS-7 cells were purchased from American Tissue Culture Collection (Manassas, VA); DMEM, gentamicin, FBS, and restriction enzymes were purchased from Life Technologies, Inc. (Gaithersburg, MD); Taq polymerase was purchased from Boehringer Mannheim (Indianapolis, IN); [125I]HEAT and [3H]inositol were purchased from DuPont/NEN (Wilmington, DE); Dowex AG1-X8 from Bio-Rad (Richmond, CA); epinephrine was purchased from Sigma Chemical Co. (St. Louis, MO); and prazosin was purchased from Research Biochemical International, Inc. (Natick, MA).
Results
Phosphorylation of Constitutively Active Mutants Carrying Mutations at Position A293 and D142.
We previously reported two series of constitutively active mutants resulting from all-amino acid scanning mutagenesis at two positions, i.e., at A293 in the distal part of the i3 loop and at D142 of the DRY motif lying at the end of TMDIII (Kjelsberg et al., 1992; Scheer et al., 1996). To compare the phosphorylation properties of the constitutively active receptors mutated at A293 or D142, we have chosen a mutant with high (D142T and A293E) and a mutant with intermediate (D142A and A293I) levels of constitutive activity for each series of receptors. We previously reported that the maximal expression levels of the D142A and D142T mutants in COS-7 cells was 3- to 4-fold lower than that of the receptors mutated at position 293. Thus, to directly compare their properties the receptor mutants were expressed at similar expression levels of 0.3–0.4 pmol/mg of protein.
As shown in Fig. 1A, when expressed at similar levels the receptor mutants displayed varying levels of constitutive activity measured as an increase in agonist-independent IP production above that of the wild-type receptor. As previously reported (Kjelsberg et al., 1992; Scheer et al., 1996), the IP response mediated by the constitutively active mutants could be stimulated by epinephrine with the exception of the A293E mutant for which the agonist-induced response was observed in previous studies only at its maximal expression levels.

IP response and phosphorylation of thealpha-1b AR and its constitutively active mutants. A, COS-7 cells were transfected with the cDNA encoding the wild-typealpha-1b AR (WT) or its mutants D142A, D142T, A293E, A293I and D142A/A293E. Cells were incubated in the absence (Bas) or presence of 10-4 M epinephrine (Epi) for 45 min. Total IP were measured as described in Experimental Procedures. Receptor expression measured in membrane preparations was in the range of 0.3 to 0.4 pmol/mg of proteins for all receptors. The results are the mean ± S.E. from three independent experiments. a, P < .05 as compared with wild-typealpha-1b AR in the absence of epinephrine. B, COS-7 cells were transfected with the cDNA encoding the wild-typealpha-1b AR (WT) or its mutants D142A, D142T, A293E, A293I, D142A/A293E. After labeling with32Pi, cells were incubated in the absence (Bas) or presence of 10-4 Mepinephrine (Epi) for 15 min. Immunoprecipitation of the phosphorylated receptors and quantification of its 32P content were performed as described in Experimental Procedures. Receptor expression was in the range of 0.4 to 1.0 pmol/mg of proteins. Receptor phosphorylation is expressed as percent of the control that indicates the 32P content (cpm) of the wild-typealpha-1b AR in the absence of epinephrine. The results are the mean ± S.E. from three to five independent experiments. a, P < .05 as compared with the basal phosphorylation of the wild-type alpha-1b AR; b,P < .05 as compared with the basal phosphorylation of each respective receptor.
Receptor phosphorylation was measured in COS-7 cells transiently expressing the wild-type or mutated receptors in the absence (basal) or presence of epinephrine. In cells expressing the wild-typealpha-1b AR exposure to 10-4 M epinephrine for 15 min increased receptor phosphorylation by 68% above basal (Figs. 1B and2). Interestingly, the levels of basal phosphorylation of the A293I and A293E mutants were 50% and 140% greater than those of the wild-type receptor, respectively. Stimulation of the A293I and A293E mutants with epinephrine could also significantly increase receptor phosphorylation above basal (Figs. 1B and 2).

Phosphorylation of the alpha-1b AR and its constitutively active mutants. COS-7 cells were transfected with the cDNA encoding the wild-type alpha-1b AR (WT) (lanes 1 and 2) or its mutants A293E (lanes 3 and 4) and D142A (lanes 5 and 6). The experimental conditions are as in Fig. 1B. The amount of receptors loaded in each lane was 50 fmol. Positions of prestained molecular mass markers are indicated in kDa.
Surprisingly, the constitutively active D142A and D142T mutants displayed basal levels of phosphorylation similar to those of the wild-type alpha-1b AR and did not appear to undergo any significant epinephrine-induced increase in phosphorylation (Figs. 1B and 2). Thus, constitutively active receptors mutated at position D142 displayed clearly different phosphorylation features than those mutated at position A293. Whereas for the A293E and A293I receptors, the level of constitutive activity was correlated with their increased values of basal phosphorylation, the D142T and D142A mutants did not exhibit a significant increase in basal or agonist-induced phosphorylation despite their high levels of constitutive activity.
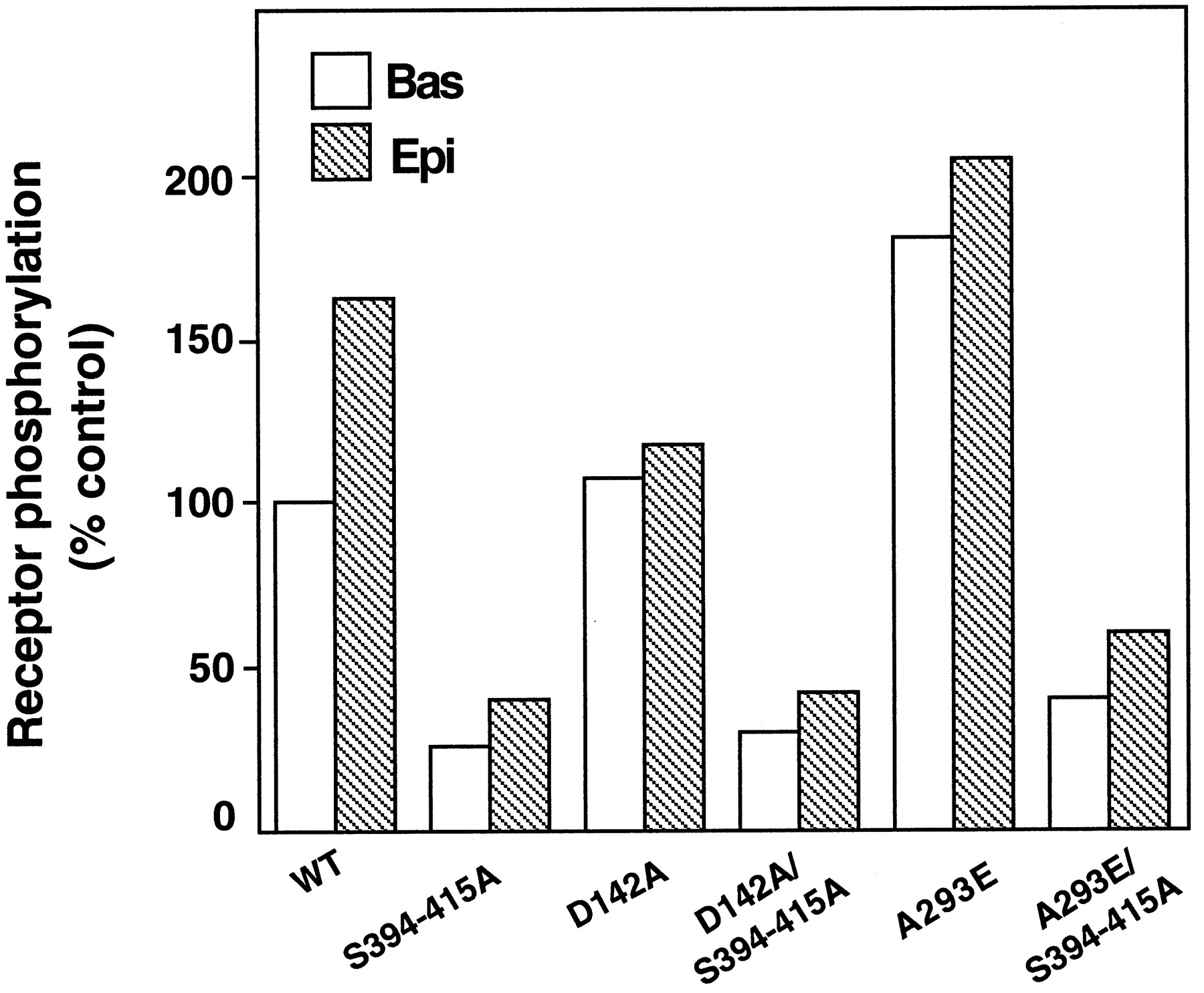
Our recent findings indicated that a cluster of serines in the intermediate portion of the C-tail of the alpha-1b AR contains the main sites for GRK- and protein kinase C (PKC)-mediated phosphorylation of the receptor (Diviani et al., 1997). This cluster of serines was mutated in the wild-type alpha-1b AR as well as in the A293E and D142A mutants. The cluster of serines containing the main sites of phosphorylation for GRK (Ser-404, Ser-408, and Ser-410) and PKC (Ser-394 and Ser-400) was mutated in the wild-type, A293E, and D142A, so as to construct receptors the S394 to 415A, A293E/S394 to 415A, and D142A/S394 to 415A, respectively. TheK i of epinephrine for the A293E/S394 to 415A and D142A/S394 to 415A was not significantly different than that of the A293E and D142A, respectively (data not shown).
In agreement with previous findings (Diviani et al., 1997), all the receptors carrying mutations of the phosphorylation sites displayed low levels of basal as well as agonist-induced receptor phosphorylation (Fig. 3). These results strongly suggest that the increased basal phosphorylation of the A293E mutant mainly occurs at the same sites involved in GRK- and PKC-mediated phosphorylation of the wild-type alpha-1b AR.

Phosphorylation of the wild-typealpha-1b AR and its constitutively active mutants carrying substitutions of the phosphorylation sites. COS-7 cells were transfected with the cDNA encoding the alpha-1b AR (WT), the constitutively active receptors A293E and D142A or the receptor mutants S394 to 415A, A293E/S394 to 415A and D142A/S394 to 415A in which eight serines were mutated into alanine. Phosphorylation was measured as described in Experimental Procedures and Fig. 1B. Receptor expression was in the range of 0.2 to 0.5 pmol/mg of proteins. Receptor phosphorylation is expressed as percent of the control that indicates the 32P content (cpm) of the WT receptor in the absence of epinephrine. The results are the mean of two independent experiments.
Mutation of the phosphorylation sites did not have any effect on the basal or agonist-induced IP response of the wild-typealpha-1b AR. On the other hand, for both the A293E/S394 to 415A and D142A/S394 to 415A receptors, the agonist-independent IP response was 69% and 73% higher than that of the constitutively active A293E and D142 mutants, respectively (data not shown). Our interpretation of these findings is that removal of the phosphorylation sites can have a small enhancing effect on the receptor-mediated activity. However, such enhancement is too small to be appreciated at the wild-type alpha-1b AR, whereas it can be observed for the A293E and D142A mutants characterized by higher efficacy as compared with the wild-type receptor (Lee et al., 1996; Perez et al., 1996).
Effects of GRK2 on Phosphorylation of A293E and D142A Mutants.
Previous findings indicated that constitutively activebeta-2 AR and alpha-2 AR mutants were substrates for GRK2-mediated phosphorylation in the absence of agonist (Ren et al., 1993; Pei et al., 1994). Thus, we wanted to investigate the effect of GRK2 on the phosphorylation of the constitutively active receptors mutated at position 293 or 142. For these studies, the A293E and D142A mutants were chosen because they displayed similar constitutive activity when expressed at similar levels (Fig. 1A).
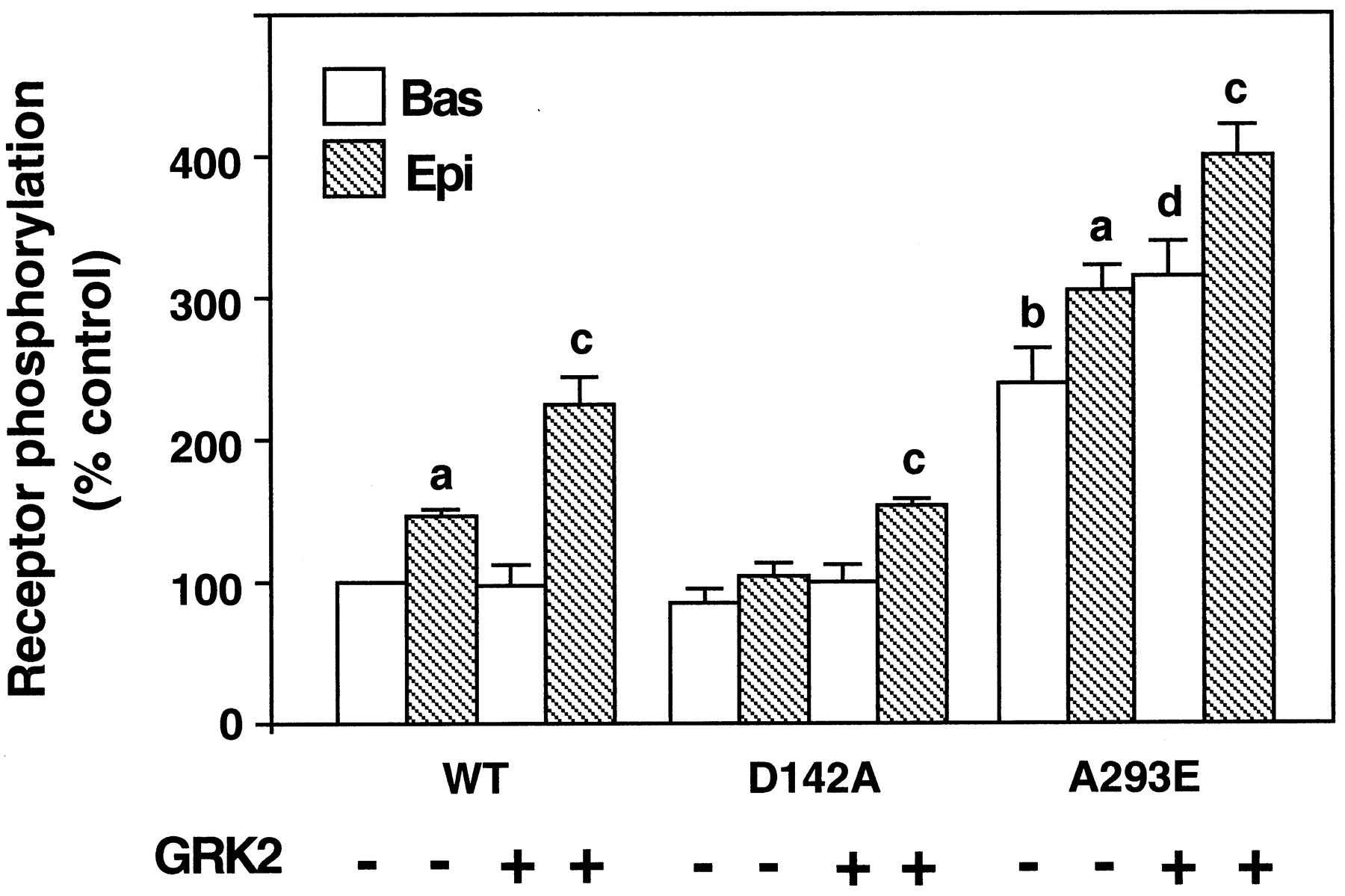
GRK2 or its dominant negative mutant K220R (Kong et al., 1994) were transiently coexpressed with either the wild-type or mutated receptors in COS-7 cells (Fig. 4). Western blot analysis indicated that for both GRK2 and its dominant negative mutant the expression level was 5- to 10-fold higher than that of the endogenous kinase, as reported previously (Diviani et al., 1996), In agreement with previous findings (Diviani et al., 1996), in cells overexpressing GRK2 epinephrine-induced phosphorylation of thealpha-1b AR was significantly increased above that of the receptor expressed alone. Overexpression of GRK2 resulted also in a significant increase of the basal phosphorylation of the A293E that was 31% above that of the receptor expressed alone. This result strongly suggests that the constitutively active A293E mutant is a substrate for GRK2-mediated phosphorylation in the absence of the agonist. This hypothesis is further supported by the observation that coexpression of the dominant negative GRK2 mutant resulted in ∼50% decrease in the agonist-independent phosphorylation of the A293E mutant (data not shown). Overexpression of the dominant negative GRK2 mutant partially impaired epinephrine-induced phosphorylation of the wild-typealpha-1b AR in agreement with previous findings (Diviani et al., 1996), but did not have any effect on that of the D142A mutant.

Effect of bovine GRK2 on the phosphorylation of the wild-type alpha-1b AR and its constitutively active mutants. COS-7 cells were transfected with the cDNA encoding thealpha-1b AR (WT) or its mutants D142A and A293E alone or in combination with the cDNA encoding the bovine GRK2. Phosphorylation was measured as described in Experimental Procedures and Fig. 2B. Receptor expression was in the range of 0.4 to 1.0 pmol/mg of proteins. Receptor phosphorylation is expressed as percent of the control that indicates the 32P content (cpm) of the wild-type alpha-1b AR in the absence of epinephrine. The results are the mean ± S.E. from three independent experiments. a, P < .05 as compared with the basal phosphorylation of each respective receptor in the absence of GRK2; b,P < .05 as compared with the basal phosphorylation of the wild-type alpha-1b AR in the absence of GRK2; c,P < .05 as compared with the epinephrine-induced phosphorylation of each respective receptor in the absence of GRK2; d,P < .05 as compared with the basal phosphorylation of the A293E in the absence of GRK2.
In contrast to what was observed for the A293E receptor, the basal phosphorylation of the D142A mutant was not significantly increased in the presence of GRK2. As already observed in Fig. 1B, stimulation of the D142A receptor with epinephrine did not significantly increase receptor phosphorylation. However, a significant 50% increase of agonist-induced phosphorylation of the D142A above basal could be observed in cells overexpressing GRK2 (Fig. 4).
Together these findings reveal a striking difference between the A293E and D142A constitutively active mutants with respect to their interaction with GRK2 and their ability to undergo agonist-induced phosphorylation. The A293E receptor seems to be a substrate of GRK2-mediated phosphorylation both in the absence and in the presence of the agonist. On the other hand, the D142A mutant might be impaired in its interaction with GRK2.
Effects of Combined Mutations at A293 and D142 Positions.
Our findings indicate that mutations at position A293 and D142 of thealpha-1b AR share similar effects in triggering receptor activation, but induce different effects on receptor phosphorylation. To further investigate the relative role of the structural domains of the alpha-1b AR including A293 and D142 we constructed a receptor mutant A293E/D142A carrying the double mutation.
As shown in Fig. 1A, the D142A/A293E mutant displayed a constitutive activity that was 71% and 93% higher as compared with that of the A293E or D142A receptors, respectively. On the other hand, the levels of agonist-independent phosphorylation of the A293E/D142A mutant was significantly decreased by 40% as compared with that of the A293E receptor (Fig. 1B). The K i of epinephrine for the A293E/D142A mutant was not significantly different than that of the D142A or A293E receptors. (data not shown).
These findings have two main implications. First, they provide additional evidence that the conservation of D142 is important for receptor phosphorylation. Second, they suggest that the effects of mutations at A293 and D142 are additive on the constitutive activity whereas they have opposite effects on the phosphorylation of thealpha-1b AR.
Agonist-Induced Internalization of A293E and D142A Mutants.
Recent evidences suggest that receptor phosphorylation is required to facilitate the internalization of the beta-2 AR and muscarinic receptors (Tsuga et al., 1994; Ferguson et al., 1995). In particular, it has been proposed that binding of β-arrestin to the phosphorylated receptor represents an early step in the agonist-induced internalization of beta-2 AR (Ferguson et al., 1996;Goodman et al., 1996). This was supported by the finding that a dominant negative mutant of β-arrestin-1 (V53D) could partially inhibit receptor internalization (Ferguson et al., 1996). Thus, the internalization of the A293E and D142A mutants was compared with that of the wild-type alpha-1b AR in the absence or presence of a dominant negative mutant of β-arrestin-1 (V53D).
As shown in Fig. 5, treatment of COS-7 cells transiently expressing the wild-type alpha-1b AR with epinephrine for 45 min induced a 32% decrease of cell surface receptors as compared with untreated cells. The extent of agonist-induced internalization of the alpha-1b AR was similar when the receptor was expressed in the range of 75 to 300 fmol per 1 × 106 cells (data not shown). Overexpression of the V53D β-arrestin-1 mutant reduced receptor internalization to 18%. Western blot analysis indicated that the expression of the β-arrestin-1 mutant was a ∼5-fold higher than that of the endogenous arrestin (data not shown). A more profound inhibition of receptor internalization could not be achieved in our experiments probably because the amount of β-arrestin-1 mutant expressed was not maximal despite the large amount of transfected DNA. Alternatively, β-arrestin might be only partially involved in the internalization process of the alpha-1b AR.
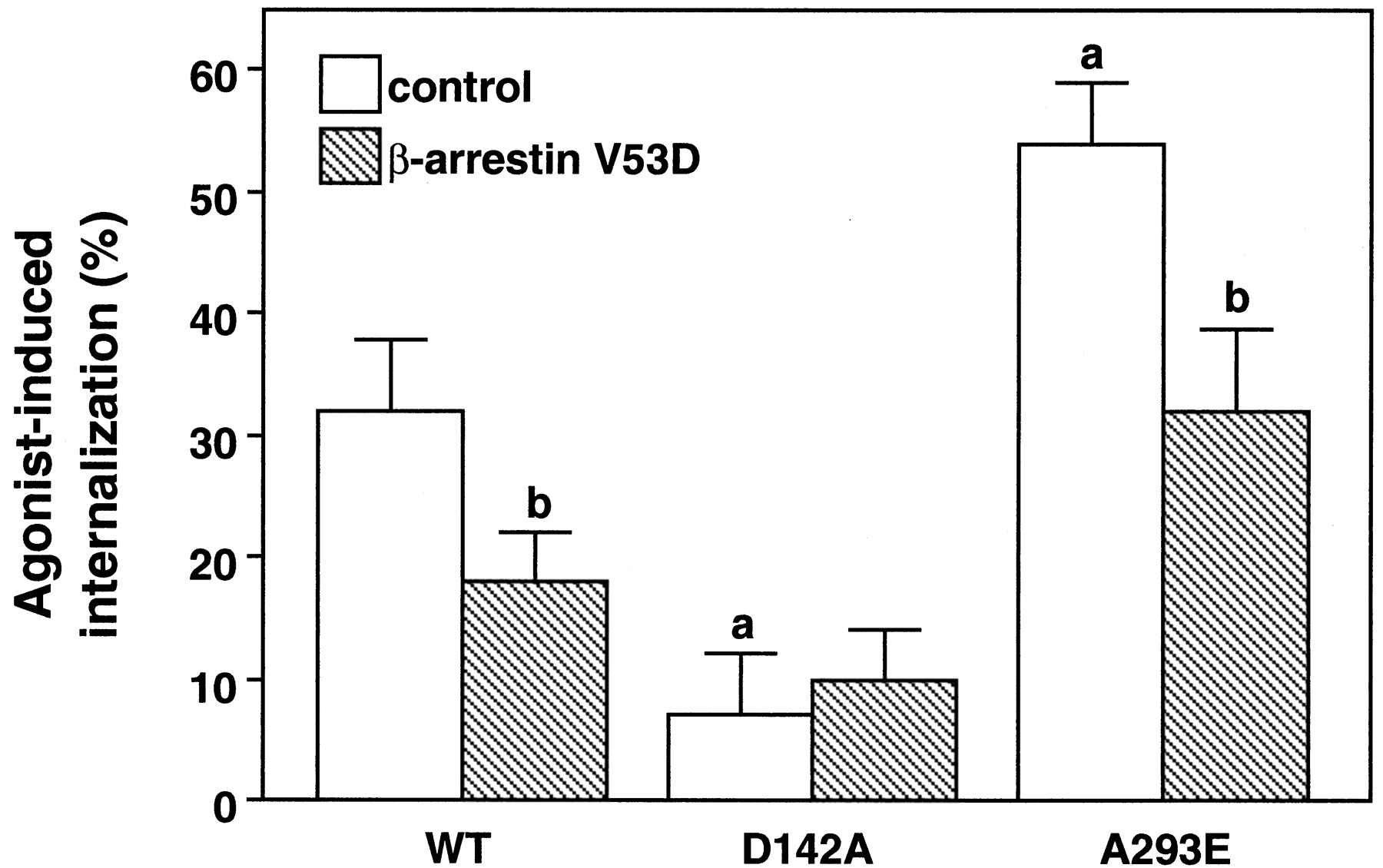
Epinephrine-induced internalization of thealpha-1b AR and its constitutively active mutants. COS-7 cells were transfected with the cDNA encoding thealpha-1b AR (WT) or its constitutively active D142A and A293E mutants alone (control) or in combination with the β-arrestin mutant (V53D). The transfected DNA per 1 × 106 cells was 1 μg for the wild-type alpha-1b AR and A293E receptors, and 3 μg for the D142A. Cell surface receptors were measured as described in Experimental Procedures with [3H]prazosin. Receptor expression ranged from 180 to 270 fmol per 1 × 106 cells for the wild-typealpha-1b AR and the A293E receptors and 60–90 fmol per 1 × 106 cells for the D142A. Percent (%) internalization indicates the decrease of cell surface receptors induced by exposure to 10-4 M epinephrine for 45 min. The results are the mean ± S.E. from three to five experiments performed in triplicate. a, P < .05 as compared with the wild-type alpha-1b AR in the absence of β-arrestin (V53D); b, P < .05 as compared with the same receptor in the absence of β-arrestin (V53D).
Interestingly, epinephrine-induced internalization of the constitutively active A293E mutant was significantly higher than that of the wild-type alpha-1b AR and this could be partially inhibited by overexpression of the V53D β-arrestin-1 mutant (Fig. 5). On the other hand, the D142A receptor mutant was impaired in its ability to undergo epinephrine-induced internalization that was never >6%.
As previously reported for the beta-2 AR (Ferguson et al., 1996), coexpression of the dominant negative β-arrestin-1 mutant resulted in a significant 52% increase of epinephrine-induced phosphorylation of the wild-type alpha-1b AR as compared with that of the receptor expressed alone (data not shown). However, overexpression of the V53D β-arrestin-1 mutant did not have any significant effect on the phosphorylation of the D142A receptor (data not shown).
It could be expected that the A293E mutant displaying increased basal phosphorylation could also be constitutively internalized in the absence of the agonist. Overexpression of the V53D β-arrestin-1 mutant did not significantly increase the number of cell surface receptors for any of the receptors tested as compared with receptors expressed alone (data not shown). However, these observations cannot exclude a tonic internalization of the A293E receptor mutant that would be hardly measurable in our experimental approach measuring ligand binding to intact cells.
Interaction of Receptors with β-Arrestin-GFP Conjugate.
Previous studies demonstrated that β-arrestin-GFP conjugate was a valuable tool to investigate the role of β-arrestin in the internalization process of the beta-2 AR (Barak et al., 1997). Thus, to better characterize the interaction between β-arrestin and the constitutively active mutants, we examined how a β-arrestin-GFP responded to agonist-induced activation of the receptor by fluorescence microscopy. For these studies, HEK cells were used because, as compared with COS-7 cells, their smaller size results in a better contrast produced by the redistribution of β-arrestin-GFP. The use of HEK cells is supported by previous evidences indicating that several regulatory properties of GPCRs are comparable in HEK and COS-7 cells. 1) The relationship between receptor phosphorylation and arrestin translocation was demonstrated both in HEK and COS-7 cells (Barak et al., 1997; Menard et al., 1997). 2) Our previous findings indicated that the regulatory properties of thealpha-1b AR coexpressed with various GRKs and arrestins were virtually identical in COS-7 and HEK cells (Diviani et al., 1996).
Figure 6 shows that epinephrine-induced activation of the alpha-1b AR coexpressed with β-arrestin-GFP and GRK2 results in a clear translocation of β-arrestin toward the cell membrane. This is demonstrated by an enhancement of the cell membrane fluorescence and a concomitant loss of cytosolic fluorescence in Fig. 6B as compared with Fig. 6A.

Confocal microscopy of β-arrestin-GFP distribution in cells expressing the alpha-1b AR and its constitutively active mutants. HEK cells were transfected with the cDNA encoding the alpha-1b AR (A and B) or its constitutively active D142A (C and D) and A293E mutants (E and F) in combination with the cDNAs coding for β-arrestin-GFP and GRK2. The transfected DNA encoding the receptors was 1 μg per 1 × 106 cells. The distribution of β-arrestin-GFP is shown before (A, C, and E) or after 10-min stimulation with 2 × 10-5 Mepinephrine (B, D, and F). Receptor expression was 3 pmol/mg of protein for the alpha-1b AR and A293E, and 0.7 pmol/mg of protein for the D142A.
Interestingly, in cells expressing the constitutively active A293E mutant the cell membrane fluorescence in the absence of epinephrine was clearly more pronounced than in cells expressing similar levels of the wild-type alpha-1b AR (Fig. 6E). Stimulation with epinephrine, could further enhance the translocation of β-arrestin-GFP to the cell membrane resulting in a decrease of cytosolic fluorescence (Fig. 6F). These findings strongly suggest that in cells expressing the constitutively active A293E receptor mutant β-arrestin are, at least in part, constitutively translocated to the cell membrane. The translocation of β-arrestin-GFP in cells expressing either the alpha-1b AR or the A293E mutant was smaller when GRK2 was not overexpressed (data not shown).
On the other hand, agonist-induced stimulation of the constitutively active D142A receptor mutant did not induce any apparent translocation of the β-arrestin-GFP that remained diffused throughout the cytosol both in the absence and presence of epinephrine (Fig. 6, C and D).
To rule out that the reduced signal in the β-arrestin-GFP translocation assay in cells expressing the D142A receptor derived from the lower expression of this mutant, arrestin translocation was compared in HEK cells expressing similar levels of D142A and wild-typealpha-1b AR. Fluorescence microscopy was used to visually look for positives in a large cohort of cells (∼50,000 cells per dish). As shown in Fig. 7, in cells expressing the alpha-1b AR agonist-induced increase of fluorescence at the cell margin was proportional to receptor expression (compare Fig. 7, D and F). However, when the alpha-1b AR was expressed at levels similar to those of the D142A agonist-induced translocation of arrestin was observed in cells expressing the wild type, but not in those expressing the mutated receptor (Fig. 7, B and D).

Fluorescence microscopy of β-arrestin-GFP distribution in cells expressing the alpha-1b AR and the D142A mutant. HEK cells were transfected with the cDNA encoding the constitutively active D142A mutant (A and B) and thealpha-1b AR (C–F) in combination with the cDNAs coding for β-arrestin-GFP and GRK2. The transfected DNA per 1 × 106 cells was 3 μg for the D142A, 0.025 μg for thealpha-1b AR in C and D, and 0.1 μg for thealpha-1b AR in E and F. The distribution of β-arrestin-GFP is shown before (A, C, and E) or after 10-min stimulation with 2 × 10-5 M epinephrine (B, D, and F). Receptor expression was 2 pmol/mg of protein for the D142A and for the alpha-1b AR in C and D, and 4 pmol/mg of protein for the alpha-1b AR in E and F.
Altogether these findings suggest that the enhanced basal phosphorylation of the constitutively active A293E mutant might result in the constitutive translocation of β-arrestin toward the receptor that could lead to increased agonist-induced internalization of the receptor mutant. On the other hand, the decreased ability of the D142A to undergo agonist-induced phosphorylation might result in its apparent lack of interaction with β-arrestin. as well as of epinephrine-induced receptor internalization.
Discussion
The goal of this study was to investigate the relationship between the constitutive activity and the regulatory properties of different constitutively active alpha-1b AR mutants. Our findings demonstrate that two constitutively activating mutations occurring in distant receptor domains of the alpha-1b AR, i.e., at A293 in the distal portion of the i3 loop and at D142 lying at the end of TMDIII, have opposite effects on the regulatory properties of the receptor.
Active Conformation of Alpha-1b AR Induced by Mutations at A293 is a Substrate for GRK2.
The analysis of the A293E and A293I mutants suggests a correlation between the constitutive activity induced by the mutations of Ala-293 and agonist-independent phosphorylation of the receptor (Fig. 1). Three lines of evidence suggest that the increased basal phosphorylation of the A293E mutant is, at least in part, mediated by GRK2. 1) Coexpression with GRK2 significantly increased both the basal and agonist-dependent phosphorylation of the A293E receptor (Fig. 4). 2) Mutations of the phosphorylation sites on the receptor, including those for GRK2, dramatically decreased both basal and agonist-induced phosphorylation of the A293E mutant (Fig. 3). 3) Overexpression of a kinase-deficient GRK2 mutant could partially reduce the basal phosphorylation of the A293E receptor (data not shown). These results are in striking agreement with previous findings that showed that for thebeta-2 AR and alpha-2 AR, mutations of the amino acid homologous to A293 in the alpha-1b AR, resulted in both increased constitutive activity and agonist-independent phosphorylation mediated by GRK2 (Ren et al., 1993;Pei et al., 1994).
The observation that constitutively active ARs carrying mutations at the distal portion of the i3 loop are substrates for GRK2-mediated phosphorylation in the absence of the agonist support the hypothesis that they mimic the agonist-occupied form of the wild-type receptors.
A293E Mutant Displays Increased β-Arrestin-Mediated Internalization.
In this study we report for the first time, that agonist-induced internalization of the alpha-1b AR might be, at least in part, mediated by β-arrestin as recently shown for the beta-2 AR (Ferguson et al., 1996; Goodman et al., 1996). This was mainly demonstrated by the fact that coexpression of the alpha-1b AR with the dominant negative β-arrestin-1 mutant V53D inhibited receptor internalization by ∼40% (Fig. 5).
Interestingly, the agonist-induced internalization of the constitutively active A293E mutant was significantly increased by ∼68% above that of the wild-type alpha-1b AR (Fig. 5). Several lines of evidence might explain this finding. On one hand, the efficacy of a number of full or partial agonists at the A293E mutant is greater than at the wild-type alpha-1b AR, as reported previously (Perez et al., 1996). This is also in agreement with a recent study that showed that the down-regulation of the Gqα subunit induced by a constitutively active alpha-1b AR mutant carrying a triple mutation in the distal portion of the i3 loop was greater than that mediated by the wild-type receptor (Lee et al., 1996).
On the other hand, our findings strongly suggest that the affinity of the A293E mutant for β-arrestin is increased as compared with that of the wild-type receptor. This was mainly demonstrated by the fact that in cells expressing the constitutively active A293E mutant the β-arrestin-GFP conjugate was, at least in part, constitutively translocated to the cell membrane as compared with cells expressing the wild-type alpha-1b AR (Fig. 6).
The constitutive translocation of β-arrestin toward the receptor as well as the enhanced agonist-induced receptor internalization are consistent with the increased basal phosphorylation of the constitutively active A293E mutant. Our findings further support the notion suggested for different GPCRs that GRK-mediated phosphorylation of the receptor can facilitate its interaction with β-arrestin that promotes receptor internalization (Tsuga et al., 1994; Ferguson et al., 1995).
D142 Plays a Role in Interaction of Alpha-1b AR with Regulatory Proteins.
A striking finding of our work is that mutations of D142 can impair receptor phosphorylation and internalization (Figs. 1 and 5). Our results, however, indicate that mutation of D142 into alanine did not entirely abolish receptor phosphorylation. In fact, overexpression of GRK2 could partially rescue the phosphorylation of the D142A mutant resulting in a significant increase of epinephrine-induced phosphorylation of the receptor (Fig. 4). In addition, the basal phosphorylation of the double mutant D142A/A293E was still significantly higher than that of the wild-type alpha-1b AR (Fig. 1B). Altogether, these findings suggest that the mutation of D142 into alanine reduces the affinity of the receptor for GRK2.
The expression of the D142A mutant was 3- to 5-fold lower than that of the wild-type alpha-1b AR. This is consistent with what has been reported for other GPCRs in which mutations of the homologous aspartate could dramatically reduce receptor expression (Lu et al., 1997). One might speculate that constitutive internalization might be responsible for the apparent lack of agonist-induced internalization of the constitutively active D142A mutant. This hypothesis seems to be ruled out by several observations. 1) Overexpression of the dominant negative mutant of β-arrestin-1 did not increase the expression of the D142A mutant at the cell surface or agonist-induced phosphorylation of the receptor (data not shown). 2) in HEK cells that express high amounts of β-arrestin agonist-induced stimulation of the D142A receptor did not induce any apparent translocation of the β-arrestin-GFP conjugate to the cell membrane (Figs. 6 and 7). We conclude that the mutation of D142 into alanine might reduce the affinity of the alpha-1b AR for GRK2 and/or β-arrestin or for both.
These findings suggest that the aspartate of the DRY sequence plays a role in the interaction of the alpha-1b AR with regulatory proteins, including GRK2 and β-arrestin. This observation is in agreement with recent findings that suggest that sequences from both the i2 and i3 loops are involved in the regulatory mechanisms of other GPCRs (Moro et al., 1994; Bourne, 1997). It is now well established that the interaction of rhodopsin kinase (GRK1) with the intracellular loops of rhodopsin underlies the activation process of the kinase that can subsequently phosphorylate residues located in the C-tail of the receptor (Palczewski et al., 1991; Shi et al., 1995; Thurmond et al., 1997). However, despite these observations, the structural determinants of GPCRs that interact with different GRKs and/or arrestin proteins, as well as the molecular mechanisms underlying the activation of these regulatory proteins, remain largely unknown. One interpretation of our findings could be that the negative charge of D142 is directly interacting with members of the GRK and/or arrestin family. Alternatively, mutations of D142 in the alpha-1b AR can perturb the conformation of the i2 or i3 loops that might directly bind and activate GRK2, leading to the interaction of the phosphorylated receptor with β-arrestin.
An important question is to what extent the apparent constitutive activity of the D142A mutant reflects enhanced activation versus reduced desensitization. The definition of receptors carrying mutations at position D142 as “constitutively active” is supported by their peculiar pharmacological properties (extremely high affinity for agonist binding) as well as by their features described by the molecular dynamics analysis of the theoretical receptor structures (Scheer et al., 1996; Scheer et al., 1997). In addition, several lines of evidence seem to exclude that reduced desensitization is the main mechanism underlying the high degree of constitutive activity of the D142A mutant. 1) Removal of the phosphorylation sites does not significantly enhance the constitutive activity of the wild-typealpha-1b AR (as reported in Results section). Thus, the attenuation of desensitization could hardly explain the high spontaneous activity of the constitutively active receptor mutants. 2) A marked degree of constitutive activity is detectable for both the A293E and D142A mutants also in a membrane assay (in which desensitization should not occur) measuring receptor-mediated stimulation of GTPγ35S binding to Gαq (our unpublished results).
However, an intriguing hypothesis is that the output of both the D142A and A293E receptor mutants results from the balance between two opposite processes—activation and desensitization. On one hand, the constitutive activity of the D142A mutant in intact cells might be enhanced because of its reduced desensitization. On the other hand, the constitutive activity of the A293E mutant might be underestimated because of its increased desensitization. This hypothesis might also apply to the interpretation of the properties of other constitutively active GPCRs.
Conformations of Alpha-1b AR Underlying Its Activation and Regulatory Processes.
A challenging task for most GPCRs is the identification of the structural motifs and conformations of the receptor that can selectively interact with a large number of signaling proteins that regulate the balance between receptor activation and desensitization.
In the rhodopsin system, it has been shown that, first, the receptor domains interacting with transducin and rhodopsin kinase are in part overlapping, and second, the light-activated meta II state can both activate transducin and be phosphorylated by rhodopsin kinase (Palczewski et al., 1991; Shi et al., 1995; Thurmond et al., 1997). Altogether, these findings suggest that the receptor conformations underlying the activation and desensitization processes of rhodopsin are similar. This hypothesis is also supported by studies that show that constitutively active rhodopsin mutants carrying mutations in different transmembrane domains, can display light-independent phosphorylation (Rim and Oprian, 1995).
Our findings indicate that the constitutively activealpha-1b AR mutants carrying mutations of A293 might display receptor conformations with docking complementarity for both the G proteins, on one hand, and GRK2 and β-arrestin, on the other. Thus, by analogy with rhodopsin, it can be suggested that also in thealpha-1b AR the structural domains involved in the interaction of the receptor with the G proteins and the regulatory proteins GRKs and arrestins can overlap.
On the other hand, the finding that the D142A mutant was constitutively active, but impaired in the processes of receptor phosphorylation and internalization suggests that, among the multiple conformations underlying receptor activation, not all favor the interaction of the receptor with GRK2 and/or β-arrestin.
Conclusions.
Our study demonstrates that the agonist-independent activity of different alpha-1b AR constitutively active mutants does not necessarily correlate with their ability to undergo enhanced phosphorylation and internalization. The results of this study have also other implications. 1) The DRY sequence is identified as an important structural determinant for the phosphorylation and internalization of the alpha-1b AR. 2) Additional evidence is provided about the role of β-arrestin in the regulation of the alpha-1b AR. 3) The divergent regulatory properties of the constitutively active mutants might help to interpret the structural differences observed in the molecular dynamics analysis of their putative structures (Scheer et al., 1996,1997). This might improve our theoretical model describing the receptor conformers associated with the active and inactive states of thealpha-1b AR.
Acknowledgments
We are sincerely grateful to Dr. R. J. Lefkowitz for the antiserum against GRK2; Dr. J. F. Benovic for the K220R mutant of GRK2; Dr. M. G. Caron for the dominant negative mutant of β-arrestin1 (V53D); and Dr. F. Mayor, Jr., for the antiserum against β-arrestin. We acknowledge the excellent technical help of M. Munoz and M. Nenniger in cell culture. We thank Dr. Peter Greasley for critical revision of the manuscript.
Footnotes
- Received May 29, 1998.
- Accepted October 30, 1998.
-
Send reprint requests to: Dr. Susanna Cotecchia, Institut de Pharmacologie et de Toxicologie. 27, Rue du Bugnon, Faculté de Médecine, 1005 Lausanne, Switzerland. E-mail:susanna.cotecchia{at}ipharm.unil.ch
-
This work was supported by Fonds National Suisse de la Recherche Scientifique Grant 31–51043.97 and by European Community Grant BMH4-CT97–2152.
Abbreviations
- AR
- adrenergic receptor(s)
- G protein
- guanyl nucleotide-binding regulatory protein
- GRK
- G protein-coupled receptor kinase
- GPCR
- G protein-coupled receptors
- i2 and i3
- second and third intracellular (loops), respectively
- IP
- inositol phosphate
- DMEM
- Dulbecco’s modified Eagle’s medium
- TMD
- transmembrane domain
- FBS
- fetal bovine serum
- PKC
- protein kinase C
- [125I]HEAT
- {β-(4-hydroxy-[125I]iodophenyl)ethylaminomethyl}tetralone
- GFP
- green fluorescent protein
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}