


Abstract
We examined the interrelationships of internalization and down-regulation of the β2-adrenergic receptor in response to treatment of the BEAS-2B human epithelial cell line with both a series of agonists at high occupancy and with various concentrations of fenoterol that gave occupancies from 0.93 to 0.001. We found that the extent of internalization measured after a 30-min treatment increased as a function of coupling efficiency, with ephedrine, dobutamine, albuterol, fenoterol, and epinephrine giving 0, 7, 17, 48, and 55% internalization, respectively. With the exception of dobutamine, the rates of down-regulation (k deg) also showed a dependence on agonist coupling efficiency, giving (in terms of fraction of receptors lost/h) 0.082 with ephedrine, 0.250 with dobutamine, 0.148 with albuterol, 0.194 with fenoterol, and 0.212 with epinephrine. Comparison of down-regulation to internalization showed that weak agonists caused down-regulation in the absence of significant internalization. The extent of internalization caused by fenoterol over a 1000-fold range of occupancy was proportional to agonist occupancy. However, although no internalization was observed with the low concentrations (0.2 and 2 nM fenoterol), these concentrations did cause significant down-regulation. Thus, as with partial agonists, it was clear that down-regulation occurred in the absence of measurable internalization. The kinetics of agonist-induced down-regulation are consistent with a scheme in which down-regulation proceeds by two pathways; a high-affinity, low-capacity component (EC50 = 0.5 nM) clearly dissociated from internalization and a low-affinity, high-capacity component (EC50 = 160 nM) closely correlated with internalization.
Important therapy for a variety of diseases such as asthma and premature contractions of the uterus involves chronic treatment with β2-adrenergic agonists such as albuterol, fenoterol, and many others. In spite of this, we know very little about how these agonists alter receptor levels and in particular how their effect on receptor levels may be influenced by agonist strength. In previous studies, we examined the effect of agonist strength on the rapid phase of desensitization that included measurement of internalization and phosphorylation of the receptor (January et al., 1997). Good proportionality was observed between the strength of an agonist (coupling efficiency) and its capacity at high occupancy to induce the rapid phase of desensitization, internalization, and phosphorylation. Studies of several families of receptors, including the muscarinic, opiate, and other receptors, indicate that this is a general phenomenon with few exceptions (as recently reviewed in Clark et al., 1999).
In this study, we have examined the effect of coupling efficiency on down-regulation of the β2-adrenergic receptor (β-AR) and the relationship between the extent of internalization and down-regulation. Surprisingly few studies of the effect of agonist strength on down-regulation of the β-AR have been published, although there have been several studies of the mechanism of down-regulation, in particular with a focus on mRNA stability and transcriptional regulation (Bouvier et al., 1989; Perkins et al., 1991; Collins et al., 1992). One notable previous study indicated that there was strong down-regulation in response to weak agonists; however, no attempt was made to quantitatively define agonist strength or to examine its relationship to internalization (Pittman et al., 1984). A more recent study (Gagnon et al., 1998) examined the role of clathrin-mediated endocytosis in down-regulation using a variety of transfected cell types and demonstrated that down-regulation of the β-AR in these systems is due at least in part to trafficking through a clathrin-coated pit pathway. It has been suggested (Koenig and Edwardson, 1997) that the extent of down-regulation may be proportional to the level of internalized receptor and that down-regulation may proceed at a rate independent of the level of internalization, indicating that it does not saturate. This model, however, has not been examined thoroughly to the best of our knowledge with any seven-transmembrane receptor either as a function of agonist coupling efficiency or with varied concentrations of a strong agonist.
The β-AR is a seven-transmembrane-spanning G-protein-coupled receptor that has been studied extensively, and many of the effects of its stimulation by hormone binding have been well characterized. The activated β-AR is a substrate for protein kinase A (PKA)- and G-protein-coupled receptor kinase-mediated phosphorylation at serine and threonine residues on the third intracellular loop and the carboxyl terminus. The G-protein-coupled receptor kinase-phosphorylated β-AR is a binding substrate for β-arrestin, and the receptor/arrestin complex internalizes into early endosomes by a clathrin-mediated process (Pitcher et al., 1998; Krupnick and Benovic, 1998; Clark et al., 1999). This internalization occurs rapidly (t 1/2 ≈3 min) on agonist stimulation and is followed by recycling of receptors from endosomes back to the plasma membrane (t 1/2 ≈11 min) such that a steady-state level of internalized receptor is attained within 30 min of initial agonist exposure (Morrison et al., 1996). The pool of internalized β-AR is thought to be the substrate for receptor down-regulation that occurs as a fraction of the receptor-rich early endosomes are sorted into a late endosome/lysosome pathway, resulting in proteolytic degradation of the receptors (Moore et al., 1999). For the muscarinic acetylcholine receptor, this process is thought to occur in a directly linear relationship with endocytosed receptor; i.e., as the internalized receptor pool increases, the rate of lysosomal degradation of receptor increases (Koenig and Edwardson, 1994).
In this study, we measured the capacity of five β-AR agonists of differing coupling efficiencies to induce internalization and down-regulation in the BEAS-2B human lung epithelial cell line that we and others have shown to be an excellent model system for the study of β-AR regulation (Kelsen et al., 1997; January et al., 1998). Coupling efficiencies, a quantitative measure of how well the β-AR activates adenylyl cyclase, were determined as previously described (Whaley et al., 1994) and recently reviewed (Clark et al., 1999). This has allowed us to determine the relationship of agonist coupling efficiency both to the endocytosed receptor pools and to the process of down-regulation; that is, is there a direct proportionality between the relatively rapidly formed internalized receptor pool generated by a specific agonist and the rate of down-regulation derived from extended treatment with the same agonist. In addition, we examined the effect of a 10,000-fold concentration range of the strong agonist fenoterol on internalization and down-regulation. Based on the simplified two-state model of receptor activation, reduced concentrations of a strong agonist that match the coupling efficiency of saturating concentrations of a weaker agonist should cause similar effects on internalization and down-regulation (Clark et al., 1999).
Our studies demonstrate that down-regulation occurs in the absence of measurable internalization with the weak agonists or with very low occupancy of the receptor with a strong agonist. We also show that down-regulation saturates as either a function of agonist coupling efficiency or over the high range of concentrations of the strong agonist fenoterol. Furthermore, it appears that the initial rate of dobutamine-induced down-regulation deviates from the predicted order based on its coupling efficiency. A model for the down-regulation process is proposed based on analysis of the kinetics of fenoterol-induced down-regulation that shows saturation and amplification relative to internalization and is therefore not a linear function of internalized receptor. Most notably, the model predicts that there are two pathways of down-regulation of fenoterol; one driven by very low occupancy of the β-AR by agonist and another requiring high occupancy that has an EC50 (163 nM) that is nearly identical with that for internalization (100 nM).
Experimental Procedures
Materials.
Agonists, vehicles, antagonists, and buffering agents were purchased from Sigma (St. Louis, MO) with the exception of thiourea, which was purchased from Aldrich Chemicals (Milwaukee, WI). [125I]Iodocyanopindolol ([125I]CYP) was prepared using cyanopindolol acquired from Sandoz (East Hanover, NJ) and 125I from New England Nuclear (Boston, MA) by the method previously described (Whaley et al., 1994). The radiolabeled antagonist [3H]CGP-12177 was purchased from New England Nuclear, and digitonin was purchased from Gallard/Schlesinger (New York, NY).
Cell Culture.
BEAS-2B cells were acquired from American Type Culture Collection (Rockville, MD) and maintained in Ham's F-12 medium (Sigma) plus sodium bicarbonate and supplemented with 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA) and 100 U/ml penicillin and 100 μg/ml streptomycin (Mediatech). BEAS-2B cells were subcultured using 0.25% trypsin/EDTA and were seeded into 12-well tissue culture dishes and allowed to grow into a confluent monolayer. Experiments were performed 24 to 48 h after monolayers were attained, and only subcultures numbering between 10 and 30 were used.
Determination of β-AR Density.
Receptor density was determined by first homogenizing cells scraped from 150-mm dishes of confluent BEAS-2B cells in 20 mM HEPES, pH 8; 1 mM EDTA, pH 7 buffer. The resulting lysate was then layered over an ice-cold sucrose gradient (23/43%) and centrifuged at 45,000g in a Beckman SW28.1 ultracentrifuge rotor. Membrane fractions were isolated and frozen at −80°C until assayed. Assays were conducted at 30°C using [125I]CYP as the labeled β-specific ligand with or without 2 μM alprenolol to assess nonspecific binding. After 50 min of incubation, the reaction was stopped by addition of 2.5 ml of cold 50 mM Tris, pH 7.5, 10 mM MgCl2 buffer. The contents of each tube were poured over Whatman GF/C glass fiber filters and washed through with 3 more volumes of Tris/Mg2+ buffer. Filters were dried and counted in a Beckman 4000 gamma counter. Using a saturating concentration of [125I]CYP and the calculation that counts per minute of signal from the radioligand is proportional to binding at the estimated ratio of 1 cpm ≈ 3760 fmol of ligand, receptor number was calculated and standardized to protein concentration. β-AR density was found to be 28.00 (±2.0) fmol of receptor/mg of membrane protein in BEAS-2B cells.
To control for the possibility that BEAS-2B cells expressed sufficient levels of β1-adrenergic receptors to interfere with the measurement of β2-receptors, we contrasted the ability of several receptor-specific drugs to displace [125I]CYP binding to cells. The binding of 200 pM [125I]CYP was measured alone and in the presence of the β2-selective drugs, 100 nM salmeterol, 100 nM ICI 118–551, 1.0 μM alprenolol, or the β1-selective drug, 100 nM atenolol. The incubations were carried out in the presence or absence of 0.2% digitonin to permeabilize cells. After homogenization, aliquots of each sample were washed through Millipore 96-well glass fiber filtration plates in triplicate to separate free [125I]CYP from that bound to receptors, and the dried filters were counted in a Beckman 4000 gamma counter. Counts obtained in the presence of β2-specific competitors (salmeterol, ICI 118–551) were not significantly different from counts in the presence of alprenolol, the nonspecific β-AR antagonist, whereas the [125I]CYP signal was not significantly diminished by atenolol. These data demonstrated that all the [125I]CYP binding was occurring at the β2-receptor subtype.
Determination of Agonist Kd, EC50, and Vmax for Adenylyl Cyclase Activation.
The K d for agonist binding to the β-AR in membrane preparations was determined by displacement of ≈60 pM [125I]CYP by increasing concentrations of agonist in the presence of 10 μM GTP. Nonspecific binding, determined by addition of 2 μM alprenolol, was subtracted, and data analysis was performed using GraphPad software (GraphPad, San Diego, CA). K d values were calculated using the Cheng-Prussoff correction. EC50 and V max values for activation of adenylyl cyclase were determined as previously described (Salomon et al., 1974) with the following modifications. Briefly, cell membranes were prepared as above and incubated in buffer containing 20 mM HEPES, pH 7.7, 1 mM EDTA, 5 mM free Mg2+, 8 mM creatine phosphate, 16 U/ml creatine kinase, 1 μM GTP, 100 μM ATP, 0.1 mM 1-methyl-3-isobutylxanthine, various concentrations of agonist, and 2 μCi [α32P]ATP for 10 min at 30°C. The reaction was stopped by addition of 0.5 ml of 5% TCA containing 1 mM cAMP (including ≈10,000 cpm [3H]cAMP/assay tube), and the [32P]cAMP was isolated by Dowex and alumina column chromatography and counted in a Beckman LS-7500 scintillation counter. Adjustments for recovery were made based on recovered [3H]cAMP counts, and dose response curves, EC50 values, andV max values were obtained using GraphPad software.
Determination of Receptor Internalization.
BEAS-2B cells grown to confluency in 12-well dishes were treated with near-saturating concentrations of the β-AR agonists epinephrine, fenoterol, albuterol, dobutamine, ephedrine, or the vehicle (referred to elsewhere in this paper as AT) containing only the antioxidants ascorbic acid (0.1 mM) and thiourea (1 mM). Saturating concentrations were derived from prior competitive binding assays that determined approximateK d values for the agonists and using the formula θ = [A]/([A] +K d) where θ = fractional occupancy, [A] = agonist concentration, andK d is the dissociation constant for the agonist. Concentrations of agonists that yielded 93 to 98% receptor occupancy were used. The same solutions were used for both the internalization and down-regulation studies.
After treatment for 30 min, cells were washed twice with warm (22°C) PBS, then placed on ice, and washed an additional four times with ice-cold PBS to remove agonist. After the last wash, 1 ml of serum-free F-12 medium containing 5 nM [3H]CGP-12177, a hydrophilic β-AR antagonist that binds only surface receptors, was added to each well, either with or without 2 μM alprenolol. Dishes were then incubated for 1 h on ice, washed three times with ice-cold PBS to remove unbound [3H]CGP-12177, and cells were removed from wells using 0.25% trypsin/EDTA and transferred to scintillation vials containing 5 ml of Universol scintillation fluid (ICN, Costa Mesa, CA). Wells were washed again with 0.25% trypsin/EDTA, which was added to the first batch, and the vials were sealed, shaken, and counted for 5 min each in a Beckman LS-7500 scintillation counter. Counts obtained in the presence of alprenolol were subtracted from total counts to adjust for nonspecific binding of [3H]CGP-12177, and receptor density was computed as proportion of receptor remaining on surface of cells relative to AT control. Each assay point was done in triplicate, and data shown are from one representative experiment or combined experiments as explained in the figure legends.
Determination of Receptor Down-Regulation.
BEAS-2B cells were grown to confluent monolayers in 12-well dishes and treated in triplicate with near-saturating concentrations of the respective agonists or vehicle control for various times. After treatment, the cells were washed four times with warm (22°C) PBS, then incubated for 50 min in 1 ml/well radiolabeled incubation solution comprised of 20 mM HEPES, pH 8; 1 mM EDTA, pH 7; 100 μM phentolamine (an α-adrenergic receptor antagonist), AT, ≈200 pM [125I]CYP, and 0.2% digitonin at 37°C either with or without 2 μM alprenolol. Digitonin was added because of its ability to permeabilize cell membranes (Seibold et al., 1998), insuring that the radioligand enters the cells to bind receptors in intracellular compartments. Dishes were removed from incubation, 0.75 ml of ice-cold 50 mM Tris, 10 mM MgCl2 buffer was added, and the resulting lysate was passed through a 1000-μl pipette tip three times. Triplicate aliquots of 200 μl each were taken from each well and filtered through a Millipore 96-well glass filter tray, followed by three washes with cold Tris/Mg2+ to wash through free [125I]CYP. [125I]CYP bound to β-ARs remained on the filter. The filters were dried and counted on a Beckman 4000 gamma counter. Nonspecific binding of [125I]CYP, as ascertained by binding of [125I]CYP in the presence of the β-AR antagonist alprenolol (2 μM), was subtracted from total binding to yield net β-receptor binding. Data are presented as the proportion of receptors remaining relative to the AT-treated control. The accuracy of this technique was verified by comparison to [3H]CGP-12177 binding in the presence of 0.1% digitonin. CGP-12177 can only enter cells on permeabilization, in this case using digitonin, to bind all receptors. Results obtained using [3H]CGP-12177 after a 6-h 10 μM epinephrine treatment of BEAS-2B cells did not differ significantly from those using the [125I]CYP method described above (data not shown).
Analysis of Down-Regulation Data.
Down-regulation is measured over a period of hours. Phosphorylation, interaction with arrestin, and the internalization/resurfacing cycle attain their steady states in much less than an hour. It is therefore reasonable in down-regulation studies to assume that these related processes are at steady states for the duration of the measurements. This being the case, the rate constants for down-regulation should be invariant with time for the duration of the experiment.
The rate of change in the concentration of receptor is given by:
Integration followed by insertion of the boundary condition that [R] = [R
o] whent = 0 gives:
Dependence of the Rate of Down-Regulation on Agonist Concentration.
The rate constant for down-regulation (k
deg) has an agonist-independent component (k
basal) and terms that depend on agonist concentration. The simplest model with a saturable dependence on agonist would be given by:
The Use of Experimental Data to Determine the Rate Constants for Down-Regulation.
Inspection of eq. 3 shows that whent = 0 (before addition of agonist), the level of receptors is given byv
s/k
basal. After long times of treatment when a new steady state is reached, eq. 3shows that receptor level isv
s/k
deg. Therefore, we have the relationship:
Results
Determination of Coupling Efficiencies for Agonist Stimulation of Adenylyl Cyclase.
Dose-response curves for stimulation of adenylyl cyclase were generated using the five β2-adrenergic agonists, epinephrine, fenoterol, albuterol, dobutamine, and ephedrine, as shown in Fig.1. The EC50 for adenylyl cyclase activation by epinephrine, fenoterol, and albuterol was determined from these curves by GraphPad analysis. Because of the low levels of receptor in the BEAS-2B cells, accurate EC50 values could not be determined for dobutamine and ephedrine. The K d for each agonist, determined by the method described under Experimental Procedures, are presented in Table1. Coupling efficiencies based on the EC50 and K d values for epinephrine, fenoterol, and albuterol were calculated as previously described (Whaley et al., 1994; Clark et al., 1999) and are given in Table 1 as well. Coupling efficiencies for dobutamine and ephedrine were obtained from our previous measurements in HEK293 cells (January et al., 1997). In addition, relative rankings of coupling efficiencies are given as percentage of coupling efficiency of epinephrine. These data serve to rank the strength of each agonist (in terms of activation of adenylyl cyclase) in the following relative order: epinephrine > fenoterol > albuterol > dobutamine > ephedrine. The relative order of these agonists has been invariant in studies of HEK293 cells, l-cells, S49 lymphoma cells, and DDT1 MF-2 cells (data not shown).
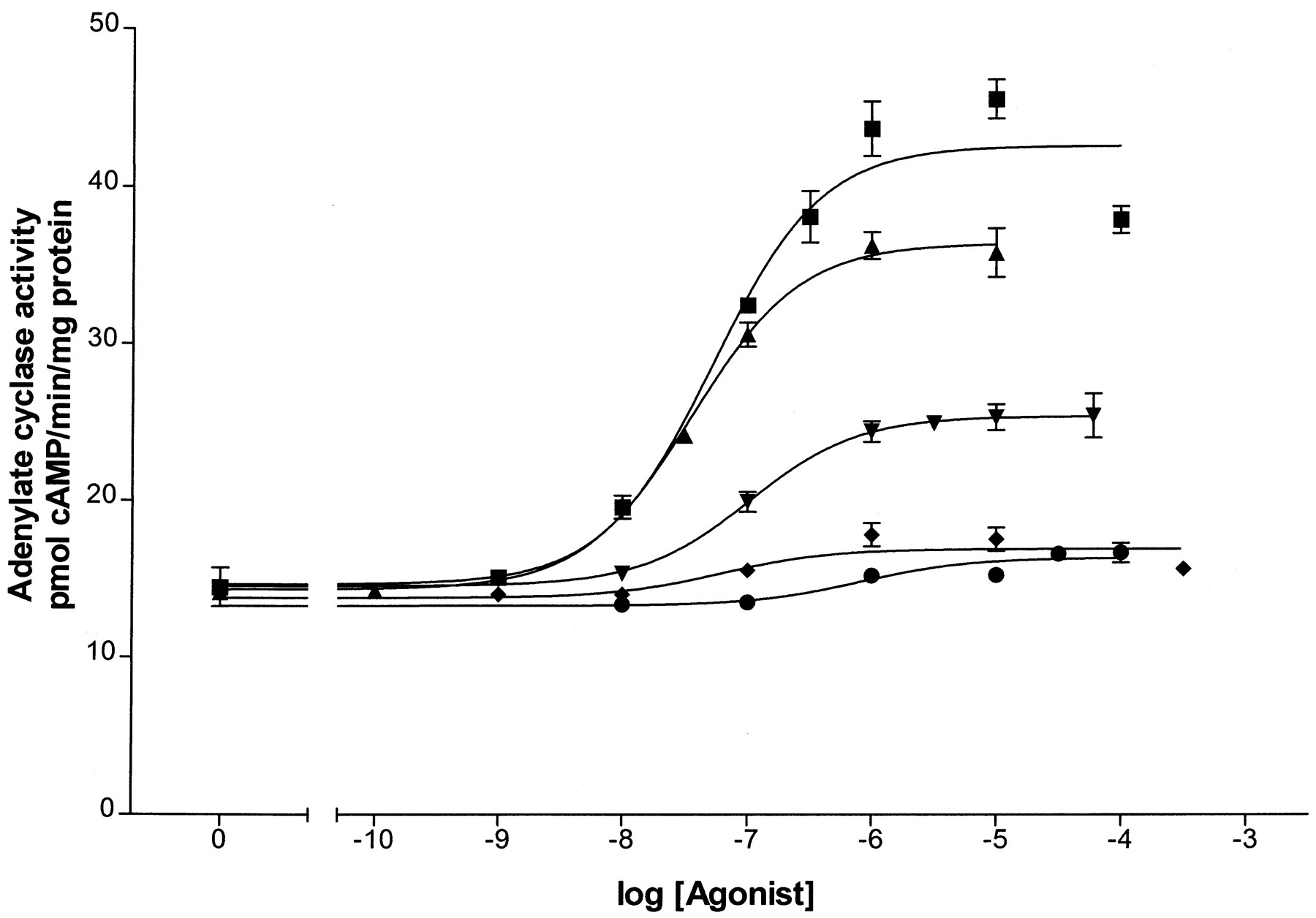
Dose-response curves of adenylyl cyclase activity generated by five β-AR agonists of various strengths. The production of cAMP is measured as described under Experimental Procedures and plotted against agonist concentration used in the assay. Sigmoidal dose-response curves were generated by GraphPad software. Stimulation of β-AR was provided by epinephrine (▪), fenoterol (▴), albuterol (▾), dobutamine (♦), or ephedrine (●). Assays were performed in triplicate using pooled membrane fractions from three 150-mm dishes of BEAS-2B cells.
Values and coupling efficiencies of agonists in BEAS-2B cells
Internalization of the β-AR on Treatment with Various Agonists.
Internalized receptor was measured after 30-min treatments with near-saturating concentrations of each of the five agonists and assayed by the method described under Experimental Procedures. Treatment for 30 min was sufficient to reach quasi-steady-state values. Longer incubations could not be used because of incipient down-regulation. Figure 2A shows the fraction of β-AR internalized, as percentage of control levels, plotted in order of decreasing agonist strength. Epinephrine, the strongest agonist in terms of coupling efficiency, induced the largest endosomal pool of receptors, and the degree to which this pool was formed was proportional to the coupling efficiency of each partial agonist in excellent agreement with our previous studies of HEK293 cells (Morrison et al., 1996; January et al., 1997). The weakest agonist in terms of coupling efficiency, ephedrine did not induce measurable endocytosis of receptor.

Internalization (loss of surface receptor) of β-AR on 30-min treatment with agonist. A, BEAS-2B cells treated with near-saturating concentrations of each of the five agonists were assayed for loss of surface receptor as described underExperimental Procedures. Agonists used were 10 μM epinephrine (Epi), 2 μM fenoterol (Fen), 6 μM albuterol (Alb), 15 μM dobutamine (Dob), and 100 μM ephedrine (Eph). Data shown are from three independent experiments (±S.E.M.), each performed in triplicate. B, loss of surface receptor due to treatment for 30 min with various concentrations of fenoterol. Concentrations ranging from 0.2 to 2000 nM were used to induce internalization of β-AR. Data shown are the results of three independent experiments (±S.E.M.) each performed in triplicate.
Internalization of β-AR after Treatment with Various Concentrations of Fenoterol.
Cells were treated with various concentrations of the strong agonist fenoterol to determine internalization of receptor. The concentrations used corresponded to a fraction of β-AR occupied, as calculated using the formula θ = [A]/([A] + K d), that varied from 0.0014 to 0.93. Thus, a broad range of receptor occupancy levels were represented. Figure 2B shows that the development of internalized receptor pools was occupancy-dependent through this range of concentrations. No significant internalization was observed with either 2 or 0.2 nM fenoterol.
Rates of Down-Regulation of β-AR on Exposure to Various Agonists.
To determine rates of receptor down-regulation, BEAS-2B cells were treated with near-saturating concentrations of each of the five agonists for 2 to 24 h, after which receptor levels were measured as described under Experimental Procedures. Results are shown in Fig. 3A, and the rate constants for down-regulation (k deg) are given in Table 2.k deg was estimated as described underExperimental Procedures using eqs. 3 and 6. The solid lines in Fig. 3A represent the best fit of the data using these formulations. It should be noted that k deg in this context is a complex function of the basal and high- and low-affinity rates of down-regulation (see eq. 5) as will be discussed below. The rates of down-regulation were not greatly different between treatments with epinephrine, fenoterol, albuterol, and dobutamine. Surprisingly, dobutamine, with a coupling efficiency just 2% of the strongest agonists, consistently showed the most extensive down-regulation at the earlier times in paired experiments with the stronger agonists, although the extent of down-regulation after 16- and 24-h treatment was identical with the strong agonists. Ephedrine, the weakest agonist in terms of coupling efficiency, was the only agonist that caused markedly less down-regulation relative to the strongest agonists; nonetheless, it still reduced β-AR levels to 32% of the control level after 24 h.

Time courses of down-regulation (loss of total receptor) of β-AR after treatments with agonists. A, cells were treated with near-saturating concentrations of each of five agonists, epinephrine (▪), fenoterol (▴), albuterol (▾), dobutamine (♦), and ephedrine (●) for indicated times, then assayed for total β-AR remaining as described under Experimental Procedures.For 24-h treatments, cells had supplemental agonist added once, at ≈9 h. Data are the results (±S.E.M.) from at least three separate experiments, each performed in triplicate. Curves represent best fits as generated by GraphPad using eqs. 3 and 6 as described underExperimental Procedures. B, cells were treated with denoted concentrations of fenoterol, for indicated times, then assayed for total β-AR. No supplemental agonist was provided. Concentrations used were 0.2 (●), 2 (♦), 20 (▾), 200 (▴), and 2000 nM (▪). Results shown are from three independent experiments, each performed in triplicate; error bars indicate S.E.M. Curves represent best fits as generated by GraphPad using eqs. 3 and 6 as described underExperimental Procedures.
Internalization and rates of down-regulation induced by various agonist treatments
Rates of Down-Regulation of β-AR on Exposure to Various Concentrations of Fenoterol.
BEAS-2B cells were treated for 2 to 24 h with different concentrations of fenoterol as indicated, then receptor number was measured as described under Experimental Procedures (Fig. 3B). The rate constants for down-regulation estimated from eqs. 3 and 6 are given in Table3. The two highest concentrations of fenoterol, which resulted in receptor occupancy of greater than 50%, yielded rates of down-regulation that were similar. As occupancy levels were reduced, the rates of down-regulation were reduced as well; however, even with occupancy of only 0.1%, the levels of β-AR at 16 and 24 h were reduced to 76 and 67% of control levels, respectively.
Internalization and rates of down-regulation induced by various concentrations of fenoterol, a strong agonist
Discussion
To quantitatively express the relationships between either agonist coupling efficiency, or agonist concentration (for fenoterol), and internalization and down-regulation as well as the relationship of internalization to down-regulation, we have plotted the data from Figs.2 and 3 and Tables 1 through 3 as shown in Figs.4 and 5. Using coupling efficiency as a measure of agonist strength, it can be seen that internalization of β-AR increases as a function of agonist strength and approaches a maximum with epinephrine, as shown in Fig. 4A. The k deg values for down-regulation, however, when plotted against coupling efficiencies of the various agonists, display characteristics that were unexpected. First, the rates differ very little over the 50-fold range of agonist strength. The weakest agonist, ephedrine, had a somewhat lowerk deg of down-regulation relative to the other more powerful agonists, and the extent of down-regulation at 24 h was much reduced relative to the other agonists (Figs. 3A and4C). Second, dobutamine, a very weak agonist in terms of coupling efficiency, showed a slightly faster initial rate of down-regulation than that observed with the strongest agonists, as shown in Fig. 4B. Third, there is a clear dissociation of the process of internalization from that of down-regulation, both at 6 h, approximately thet 1/2 for down-regulation, and at 24 h, at which time the down-regulation process is complete, as is shown in Fig. 4C.
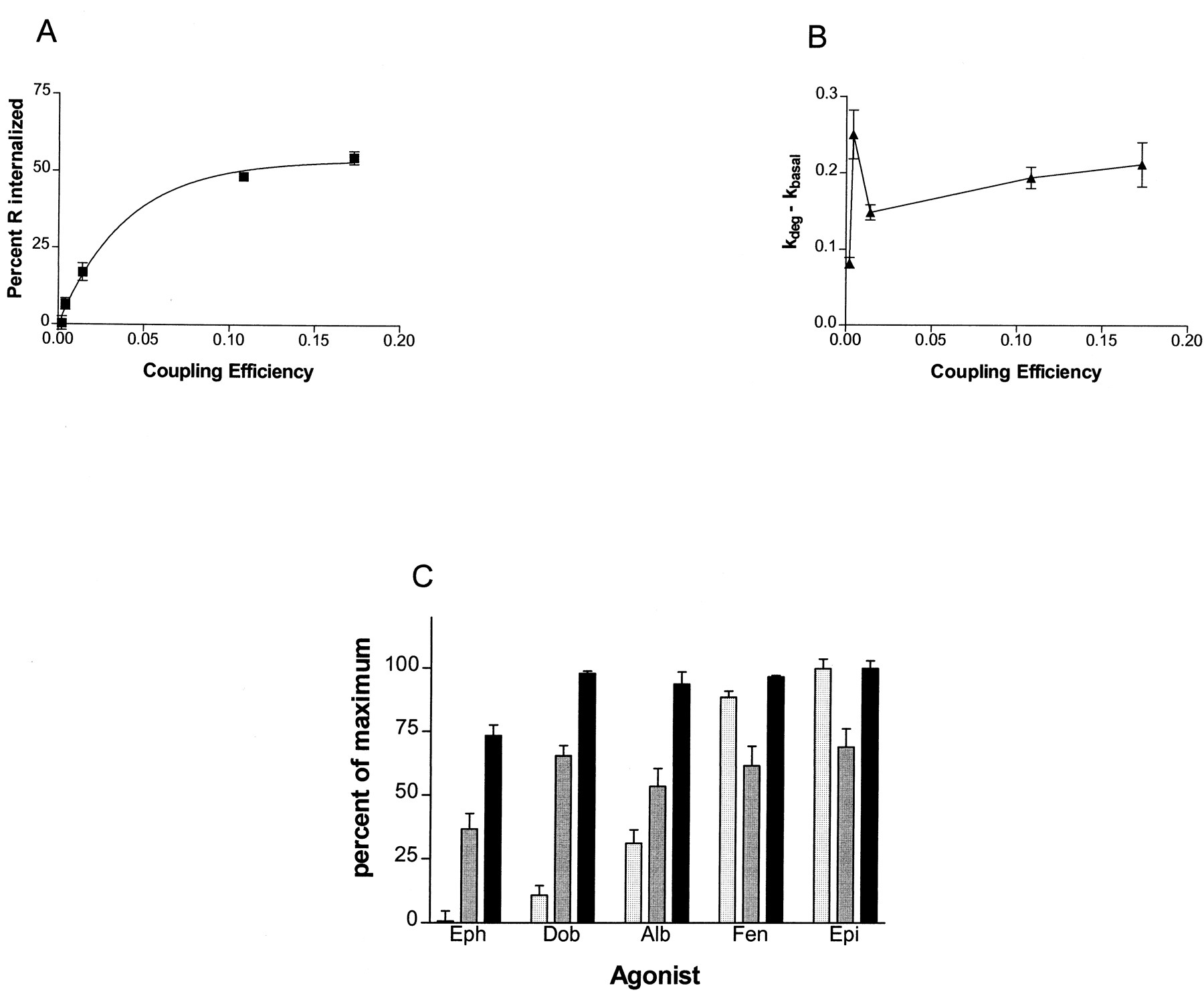
Relationships between coupling efficiencies of the five agonists and internalization and down-regulation of β-AR. A, internalization of receptor as a function of coupling efficiency of agonist (see Table 1 for the derivation of coupling efficiency for each agonist). From left to right, the points on the graph correspond to ephedrine, dobutamine, albuterol, fenoterol, and epinephrine. The curve on the graph is a hyperbolic association generated by GraphPad software, r 2 = 0.99. B, rate of down-regulation of β-AR as a function of coupling efficiency of agonist. Rates of down-regulation (as the fraction of receptors remaining h−1) were obtained as described underExperimental Procedures using GraphPad software (see Table 2 for rates and standard errors) and plotted against coupling efficiencies of the five agonists. From left to right, the points correspond to ephedrine, dobutamine, albuterol, fenoterol, and epinephrine. Note that dobutamine causes down-regulation at a rate that is much greater than its coupling efficiency would predict. C, comparison among extent of internalization of receptor at steady state (speckled columns; attained by 30 min), extent of down-regulation of receptor at 6 h (gray columns), and extent of down-regulation of receptor at 24 h (black columns), plotted as percentage of maximum of each parameter measured, which is in fact that generated by treatment with epinephrine.

Relationships between occupancy of β-AR by fenoterol and receptor internalization and down-regulation. A, internalized receptor (▪; at steady state) and rate of down-regulation of receptor (▴; k deg− k basal) as a function of fraction of receptor occupied by various concentrations of fenoterol (see Table 3for concentrations of fenoterol that generated these occupancy levels). The internalization curve on the graph is a hyperbolic association generated by GraphPad software, r 2 = 0.99. The down-regulation rate curve was generated by GraphPad from data obtained as in Fig. 4 and given in Table 3;r 2 = 0.998. B, comparison between extent of internalization of receptor at steady state (speckled columns; attained by 30 min) and extent of down-regulation of receptor at 24 h (gray columns), plotted as percentage of maximum of each parameter measured.
The amplification of down-regulation versus internalization that is shown here demonstrates that there is not a simple relationship of the two processes. Similar plots comparing internalization and down-regulation over the range of fenoterol concentrations (Fig. 5, A and B) point to the same phenomenon. That is, there is 1) a nearly linear proportionality between fraction occupied and internalization (Fig. 5A), 2) the rate of down-regulation saturates as a function of fenoterol concentration (Fig. 5A), and 3) there is a clear dissociation of internalization from down-regulation at low concentrations (Fig.5B).
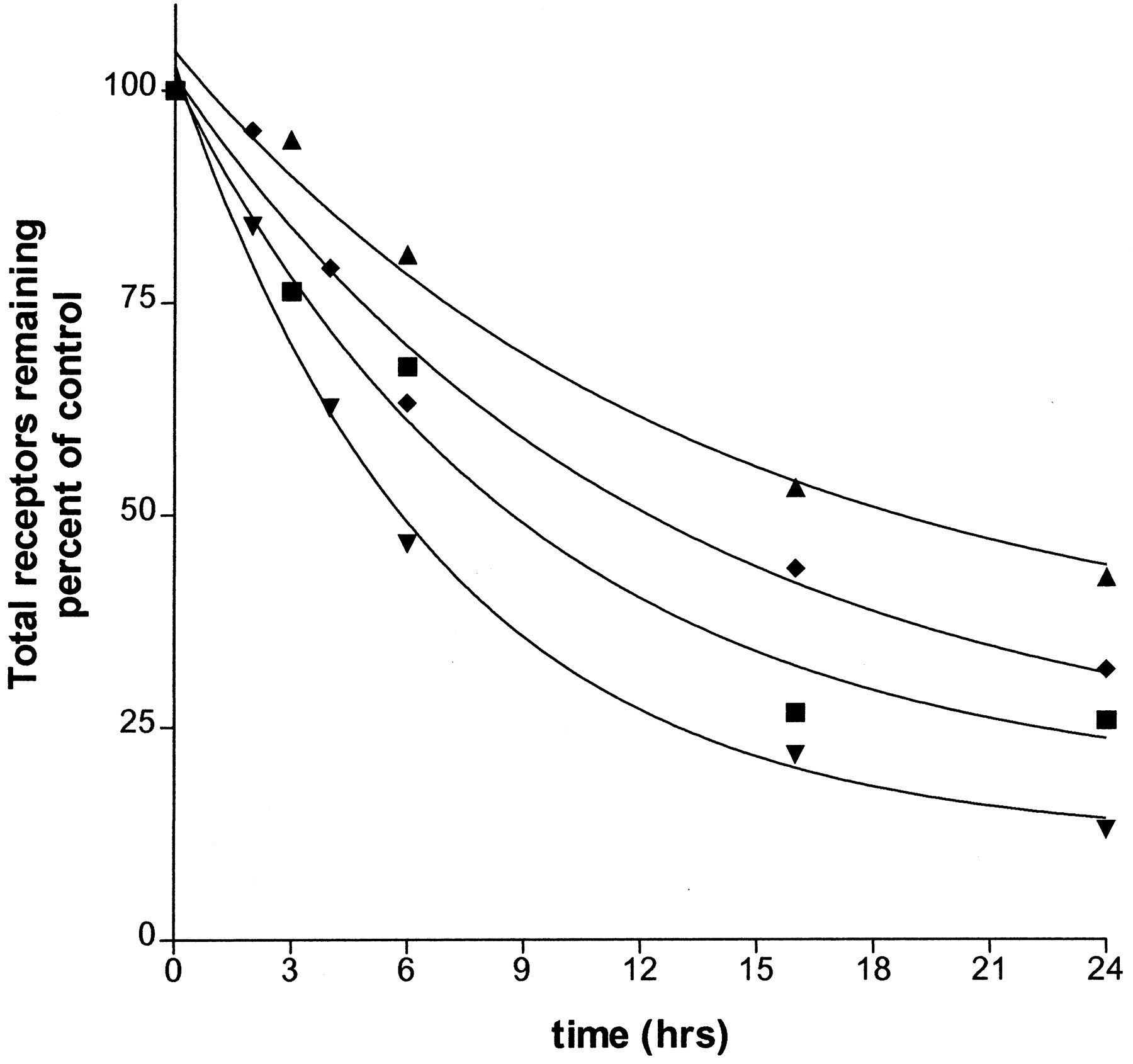
Parallels can be drawn between down-regulation induced by weak agonists and that induced by low concentrations of a strong agonist. To the extent that the two-state model of receptor activation holds with regard to partial agonists, that is, that partial agonists stabilize less activated receptor than strong agonists, then a comparison of the data with a range of concentrations of a strong agonist is possible, considering that low concentrations of a strong agonist yield proportionately low levels of receptor occupancy and hence less activated receptor. As shown in Fig. 6, the down-regulation induced by ephedrine, an agonist with a coupling efficiency that is ≈1% that of epinephrine, is similar in profile to that induced by 2 nM fenoterol, a concentration that yields receptor occupancy of ≈1%. Treatment with albuterol, an agonist with ≈8% the coupling efficiency of epinephrine, yields a down-regulation profile which resembles that of 20 nM fenoterol.

Parallels between down-regulation due to weak agonist treatment and low concentrations of a strong agonist. Time course data from treatment with 100 μM ephedrine (♦) and 6 μM albuterol (▾) are shown together with data from time course experiments using 2 nM fenoterol (▴) and 20 nM fenoterol (▪). Profiles generated from low concentrations of fenoterol (which yield low receptor occupancy) approximate the down-regulation profiles from treatments with weak agonists; i.e., ephedrine ≈2 nM fenoterol, albuterol ≈20 nM fenoterol. Curves were generated by GraphPad using the scheme presented in the modeling discussion under Experimental Procedures.
Taken together, our data point to the following conclusion: there is a good correlation between β-AR internalization and either the coupling efficiency of the agonist used to induce internalization or the occupancy level of receptor in the case of low concentrations of a strong agonist, with the caveat that the range of partial agonists (Fig. 4A) shows some saturation with the two strongest agonists similar to what we previously reported (January et al., 1997). There is considerable down-regulation even when levels of internalized receptor are imperceptibly small, as in the case of dobutamine or ephedrine treatment and treatments with low concentrations of fenoterol. Generally, the rate of down-regulation correlates with coupling efficiency. Finally, as Fig. 6 illustrates, the similarity of the behavior of the weak agonists ephedrine and albuterol with low concentrations of fenoterol are consistent with predictions of the two-state model. However, a notable exception to the correlation between agonist strength and rate of down-regulation is evidenced by the data from dobutamine treatments that show a slightly greater rate of down-regulation of β-ARs than the coupling efficiency would predict. Consistent with our results, dobutamine has been shown to be a potent down-regulator of β-ARs in leukocytes when administered for treatment of congestive heart failure (Teng et al., 1993). Furthermore,Blevins et al. (2000) have shown that dobutamine displays unusual properties with regard to a quantitative analysis of GTP shifts relative to the other agonists. In preliminary studies of DDT1-MF2 cells, we found that the down-regulation induced by dobutamine was proportionate to its coupling efficiency, relative to other agonists, indicating that some feature of dobutamine's unique properties may be cell-type specific (data not shown). Clearly, more work is needed to determine what makes this compound occasionally yet reproducibly behave disproportionately relative to other β-AR agonists.
Measurement of the down-regulation at a range of fenoterol concentrations provided the data needed for a quantitative analysis of the kinetics of down-regulation for a strong agonist in a cell line expressing only endogenous receptor. Fenoterol was chosen over epinephrine because of its greater stability over the long term. The increased conformational changes in receptor with increasing fenoterol concentration are changes only in amount and not in type as would be the case with the partial agonists. This enabled us to test and eliminate several hypothetical pathways that have been proposed as routes to down-regulation.
Current schemes for the down-regulation pathway feature internalization as the determining event as diagrammed in Fig.7A, model I. However, our data summarized in Fig. 5 are not quantitatively consistent with this simple model. The rate of down-regulation at 20 nM fenoterol is at least 50% that at 2 μM even though the steady-state amount internalized is only approximately 20% of that at the greater concentration. Furthermore, extensive down-regulation is observed with 0.2 and 2 nM fenoterol, and no internalization is observed. The scheme shown in model I is not saved by supposing that the down-regulation process is saturable because down-regulation as a whole has first order kinetics. That is, when the amount of receptor in the endocytosed compartments is reduced by prior desensitization, the rate of subsequent down-regulation follows strict proportionality with the amount of receptor remaining in the cell. This is not consistent with the pathway shown in model I being saturable over the higher range of concentrations in the experiment, and therefore the hypothesis fails.

A, the upper diagram (model I) shows a single mode of down-regulation with endocytosis as a necessary prerequisite. The current data are not consistent with this straightforward model in which the total rate of agonist-promoted down-regulation is simply proportional to the steady-state concentration of internalized receptor. The lower diagram (model II) indicates a model in which there are two independent pathways of agonist-dependent down-regulation. This model gives an adequate description of the experimental data for fenoterol when both processes are Michaelian in nature. The low-affinity process represented by a single arrow in the lower diagram corresponds to the entire endocytosis/down-regulation process of the upper diagram. EC50.2 in the lower diagram therefore corresponds to thek r/k e ratio of the upper figure. B, graphic representation of fenoterol data shows the effect of the high-affinity down-regulation pathway (solid line) on the otherwise simple dose-response curve of the low-affinity component (dashed line). Note that the low affinity component alone corresponds well to the dose-response data for internalization (▪). ▪, internalization (steady state); ▴, down-regulation (rate constant); —, biphasic fit to down-regulation data; - - -, the low-affinity component of down-regulation.
Conversely, if one supposes that internalized receptor is immune to down-regulation and that down-regulation occurs proportionately to occupancy of receptor by agonist in the membrane, then again, down-regulation is greater at low fenoterol concentrations than the model predicts. As before this proposal cannot be saved by supposing a saturable process because the down-regulation is a first order process.
Kinetic models that are consistent with the data can be constructed if a pool of receptor in addition to the endocytosed pool can be invoked. Here we propose a scheme (Fig. 7A, model II), the key features of which are that down-regulation can occur by either of two pathways; one pathway involving a high agonist affinity component with a low capacity that is independent of internalization and a second pathway using a low-affinity, high-capacity event that is closely correlated with internalization. This scheme provides an explanation for the dissociation of internalization from down-regulation either with the low concentrations of fenoterol or for the weakest partial agonists. That is, at low concentrations of fenoterol or with very weak agonists, down-regulation is proposed to occur almost exclusively by the high-affinity process. At high concentrations of fenoterol or with the range of relatively strong agonists, down-regulation proceeds predominantly via the low-affinity pathway. Using eq. 5 underExperimental Procedures and the fenoterol data in Fig. 5B (as summarized in Table 3), we were able to evaluate the EC50 values and k degvalues for the two-component scheme using GraphPad. The EC50 values calculated for the low- and high-affinity components were 163 nM and 0.53 nM, respectively. The corresponding rate constants for the low and high components were 0.138 and 0.065 h−1, respectively (also see Fig. 7B).
Although the mechanistic basis of the two proposed pathways for down-regulation are unknown, we speculate that the high-affinity component may involve PKA because its activation typically shows a huge amplification relative to agonist occupancy of the β-AR and with imperceptible increases in cAMP (Clark et al., 1999). Preliminary work with forskolin lends some support to this view, in that pretreatment with 100 μM forskolin, but not 10 μM, induces significant down-regulation in these cells (24-h pretreatment with 100 μM forskolin resulted in receptor levels that were 61.7% those of control; n = 7, S.E.M. = 0.087; whereas 10 μM forskolin had no significant effect, at least in the one trial performed). Penn et al. (1994) also did not find down-regulation after chronic 10 μM forskolin or 1 μM PGE2treatments, indicating that PKA may not be involved or that only very high forskolin concentrations trigger the response.
The strong correlation of the low-affinity component of down-regulation with the EC50 for internalization by fenoterol (≅100 nM) suggests that endocytosed β-AR is the precursor for down-regulation of this pathway. Gagnon et al. (1998) clearly demonstrated a dependence on internalization of down-regulation in three transfected systems, although in two of those systems blockage of internalization resulted in only partial blockage of down-regulation, opening questions about other mechanisms and cell or system specificity. Consistent with our model II, preliminary studies we have performed indicate that hypertonic sucrose may only partially block down-regulation. However, these speculations must be viewed in light of the fact that agonist occupancy of the β-AR causes phosphorylation at several sites and at high occupancy also causes interactions with other cellular proteins, such as binding of β-arrestin. Any one of these modifications may make the receptor more (or less) susceptible to down-regulation by the occupancy. It is easy to imagine that a particular phosphorylation renders the receptor vulnerable to down-regulation. The down-regulation then occurs itself without further involvement of the agonist. Of interest is a recent study of the relationship of internalization to down-regulation in several cell lines using overexpressed β-ARs that supports the proposal that significant down-regulation occurs under conditions such as hypertonic sucrose where internalization is blocked (Jockers et al., 1999). The methods used to block internalization in that study have been the subject of some debate, although the results point to the same conclusion, i.e., that a down-regulation pathway separate from internalization may exist. Early work in our laboratory, using low concentrations of epinephrine, gave preliminary indications that a low-occupancy pathway of down-regulation was plausible (Proll et al., 1992).
To summarize, our data demonstrate that even extremely weak β-AR agonists drive a disproportionately large down-regulation and that, consistent with this, very low levels of strong agonists show similar effects. Agonist-induced internalization of the β-AR per se then is a poor predictor of down-regulation. It should be noted that a myriad of processes might be involved during the hours-long course of agonist treatment that is necessary to witness receptor down-regulation. These may include differential trafficking, binding protein interactions, or even transcription of new proteins such as proteases or chaperones. However, we show that a relatively simple two-component model does provide an excellent description of the kinetics of down-regulation. Another conclusion from our studies is that the measurement of down-regulation can be a very sensitive measure of coupling efficiency for the very weak agonists, with the caveat that exceptions are to be expected given the oversimplification of the two-state model (Clark et al., 1999). Nonetheless, as a general rule, very weak agonists that give no indication of being able either to activate adenylyl cyclase or to cause internalization (as is the case in the BEAS-2B cells used in these studies) may be more accurately classified as partial agonists based on the measurement of down-regulation. Further tests of these phenomena in animal models and other cultured cell lines expressing endogenous β-AR should reveal whether our results will aid in developing better rationale for the use of partial agonists in the treatment of various disease states such as asthma and cardiovascular disease.
Footnotes
- Received December 2, 1999.
- Accepted May 24, 2000.
-
Send reprint requests to: Dr. R. B. Clark, Department of Integrative Biology and Pharmacology, The University of Texas Medical School, 6431 Fannin, Houston, TX 77030. E-mail:dclark{at}farmr1.med.uth.tmc.edu
-
This work was supported by National Institutes of Health Grants GM 31208 (to R.B.C.) and RR07710 (to R.B.).
Abbreviations
- β-AR
- β2-adrenergic receptor
- [125I]CYP
- [125I]iodocyanopindolol
- PKA
- protein kinase A
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}