


Abstract
Cardiovascular regulation is tightly controlled by signaling through G protein-coupled receptors (GPCRs). β-Adrenergic receptors (ARs) are GPCRs that regulate inotropy and chronotropy in the heart and mediate vasodilation, which critically influences systemic vascular resistance. GPCR kinases (GRKs), including GRK2 (or βARK1), phosphorylate and desensitize agonist-activated βARs. Myocardial GRK2 levels are increased in heart failure and data suggest that vascular levels may also be elevated in hypertension. Therefore, we generated transgenic mice with vascular smooth muscle (VSM) targeted overexpression of GRK2, using a portion of the SM22α promoter, to determine its impact on vascular βAR regulation. VSM βAR signaling, as determined by adenylyl cyclase and mitogen-activated protein (MAP) kinase activation assays, was attenuated when GRK2 was overexpressed 2- to 3-fold. In vivo vasodilation in response to βAR stimulation using isoproterenol was attenuated and conscious resting mean arterial blood pressure was elevated from 96 ± 2 mm Hg in nontransgenic littermate control (NLC) mice (n = 9) to 112 ± 3 mm Hg and 117 ± 2 mm Hg in two different lines of SM22α-GRK2 transgenic mice (n = 7 and n = 5, respectively; p < 0.05). Interestingly, medial VSM thickness was increased 30% from 29.8 ± 1.6 μm in NLC mice (n = 6) to 39.4 ± 1.6 μm in SM22α-GRK2 mice (n = 7) (p < 0.05) and vascular GRK2 overexpression was sufficient to cause cardiac hypertrophy. These data indicate that we have developed a unique mouse model of hypertension, providing insight into the contribution that vascular βAR signaling makes toward resting blood pressure and overall cardiovascular regulation. Moreover, they suggest that GRK2 plays an important role in vascular control and may represent a novel therapeutic target for hypertension.
β-Adrenergic receptors (ARs) are critical G protein-coupled receptors (GPCRs) in the regulation of the cardiovascular system. βARs (β1, β2 and β3 subtypes) are involved in the regulation of myocardial function as well as taking part in control of vascular tone (Brodde 1991; Chruscinski et al., 1999; Rohrer et al., 1999). β1 and β2ARs primarily couple to the heterotrimeric G protein Gs, which then dissociates and activates adenylyl cyclase via the α-subunit and can also trigger signaling events mediated by the βγ-subunits (Gβγ), such as the activation of mitogen-activated protein kinases (MAPKs). Studies so far suggest that stimulation of the β1AR primarily mediates inotropy and chronotropy in the heart (Brodde and Michel, 1992;Chruscinski et al., 1999; Rohrer et al., 1999). In contrast, studies from β1AR, β2AR, and β1/β2 knockout mice imply that vascular smooth muscle (VSM) relaxation seems to be controlled by all three βAR subtypes (Chruscinski et al., 1999;Rohrer et al., 1999).
Importantly, several studies have shown that βAR levels and signaling can be significantly altered by cardiovascular disease. Among the most well characterized βAR derangements are those that take place in chronic heart failure, where β1ARs are selectively down-regulated both at the level of mRNA and protein while both β1- and β2ARs are functionally uncoupled (Brodde 1991). On the vascular side, impairment in βAR-mediated vasodilation caused by an alteration in receptor/G-protein coupling increases systemic vascular resistance and has been described in both human and animal models of hypertension (Brodde and Michel 1992; Feldman 1990). In heart failure, defective βAR coupling is probably the result of increased cardiac expression and activity of one member of the GPCR kinase (GRK) family, GRK2 (βAR kinase or βARK1) (Ungerer et al., 1993). GRK2, like other GRKs, phosphorylates and desensitizes only agonist-occupied GPCRs and its substrates include numerous GPCRs, including βARs, resulting in decreased sensitivity to further catecholamine stimulation (Lefkowitz 1993). Elevations in cardiac GRK2 activity have been documented to precede symptoms of heart failure in well-defined animal models, including the spontaneously hypertensive heart failure rat (Anderson et al., 1999). Interestingly, elevations in GRK2 levels have been found in the lymphocytes of hypertensive patients (Gros et el., 1997, 1999) and in the vasculature of hypertensive rats (Gros et al., 2000). The 55% increase in GRK2 levels in the lymphocytes of hypertension patients also translates into a reduction in β-adrenergic stimulated adenylate cyclase activity (Gros et al., 1999). This strongly suggests that GRK2 plays a critical role in vascular βAR signaling alterations shifting the balance between vasoconstrictor and vasodilator mechanisms resulting in this cardiovascular disorder.
Studies completed using transgenic mice illustrate that one of the most important mechanisms for rapidly regulating in vivo cardiac βAR function is the activity of GRK2 (Koch et al., 1995). Transgenic overexpression of GRK2 in the heart, to levels seen in human heart failure, leads to attenuated inotropic responses to catecholamines (Koch et al., 1995; Akhter et al., 1999) and detrimental consequences after cardiac ischemia (Chen et al., 1998). Parallel studies with myocardial-targeted transgenic expression of a peptide inhibitor of GRK2 (βARKct) have shown that decreased activity of this kinase has positive effects on in vivo heart function (Koch et al., 1995; Akhter et al., 1999). The βARKct inhibits the actions of GRK2 by competing for Gβγ-mediated membrane translocation, a process required for GRK2 activation (Pitcher et al., 1992; Koch et al., 1993). Recent studies have even documented that inhibition of cardiac GRK2 via the βARKct can rescue or prevent the development of heart failure (Rockman et al., 1998; White et al., 2000), indicating that GRK2 is a critical regulator of myocardial βAR signaling and function
Given the important role of GRK2 in the regulation of cardiac βARs and function as well as the importance of βARs in the vascular system, we were interested in altering the expression and activity of vascular GRK2 to determine its impact on regulation of VSM signaling and blood vessel tone. Herein, we describe the biochemical and physiological implications of transgenic vascular-targeted overexpression of GRK2 on the βAR system both in vitro and in vivo.
Materials and Methods
Transgene Construction and Development of Transgenic Mice.
A 481-base pair portion of the SM22α promoter (−441 to +41 relative to transcription start) was amplified using PCR. This portion of the promoter was ligated into a described previously plasmid containing the SV40 intron poly(A+) signal (Koch et al., 1995) along with a 2070-base pair fragment containing the coding sequence for bovine GRK2. The SM22α-GRK2 transgene underwent pronuclear injection done by the Duke Comprehensive Cancer Center Transgenic Facility. Offspring are screened by slot blot analysis of genomic DNA using a probe to the SV40 sequence. Second generation adult animals (2–12 months of age) were used for all studies. Institutional review board approval for all mouse experiments was obtained from Duke University.
Transgene Expression.
Total RNA was extracted with RNAzol (Biotecx Laboratories, Houston, TX). Reverse transcription was performed using ProSTAR First Strand RT-PCR Kit (Stratagene, La Jolla, CA). The SV40 portion of the transgene was amplified using primers 5′-TGAATGGGAGCAGTGGT-3′ and 5′-TATGCCTGTGTGGAGTAAGAA-3′ at a concentration of 300 nM, 1.5 mM MgCl2, 200 μM dNTPs, and 1 unit of Tfl polymerase (Promega, Madison, WI). Reaction conditions were: 94°C, 5 min; 94°C, 30s; 65°C, 30s; 72°C, 45s; and 72°C, 5 min. PCR products were run on a 1.2% agarose-TAE gel and visualized with ethidium bromide staining. Protein expression of transgene was determined using protein immunoblotting as described, from cell extracts using polyclonal GRK2 antibodies (Koch et al., 1995).
Determination of Protein Expression.
Ten aortas were pooled and the VSM layers of the aortas were digested enzymatically (Iaccarino et al., 1999). Frozen samples were pulverized using a tissue smasher and homogenized as described previously (Koch et al., 1995). To determine GRK2 protein expression, 40 μg of protein was resolved on a 12% SDS-PAGE gel and transferred to nitrocellulose. The membrane was immunoblotted for GRK2 using the appropriate primary (Santa Cruz Biotechnologies, Santa Cruz, CA) and secondary antibodies and standard chemiluminescent detection (ECL kit; Amersham Biosciences, Piscataway, NJ).
Cell Culture.
Primary cultures of VSM from thoracic aortas were obtained and cultured as described previously (Iaccarino et al., 1999).
GRK Activity Assays.
Cytosolic protein (100 μg) are incubated in a volume of 100 μl of radioimmunoprecipitation assay buffer supplemented with 0.1 mM ATP (containing γ-[32P]ATP), 10 mM MgCl2, and rhodopsin-enriched rod outer segments as described previously (Koch et al., 1995).
Adenylyl Cyclase Activity.
To determine adenylyl cyclase activity, cultured cells were grown until 4 days after confluence. Cells were then labeled overnight in 3.0 μCi/ml [3H]adenine (PerkinElmer Life Sciences, Boston, MA) in medium 199 and then preincubated in medium 199 with 10 mM HEPES and 1 mM 3-isobutyl-1-methylxanthine (IBMX) for 30 min. Cells were then stimulated with the appropriate concentration of isoproterenol or 10 mM forskolin for 15 min. After incubation, cAMP was determined by anion exchange chromatography and a percentage incorporation of the total 3H uptake was calculated.
MAPK Activity.
To study MAPK activity, cells were stimulated with isoproterenol at the described concentration for 5 min. Cells were then harvested and homogenized in ice-cold radioimmunoprecipitation assay buffer with 1 mM sodium orthovanadate (Iaccarino et al., 1999). MAPK activity as assessed by via kinase activity assays was performed as described previously (Iaccarino et al., 1999). Protein immunoblotting for the activated phosphorylated forms of ERK1/2 and JNK1/3 (New England Biolabs, Beverly, MA) was normalized to total MAPK levels using antibodies specific to total ERK1/2 and JNK1 (Santa Cruz Biotechnologies).
In Vitro Physiology.
Aortas were dissected and cut into 2.5-mm rings. Rings were then placed in a 37°C chamber (Kent Scientific, Redmond, WA) containing Krebs-Henseleit buffer and bubbled with 95% O2/5% CO2. Two stainless steel wires were placed through each ring and one wire is attached to a fixed end. The other was attached to a force transducer and contraction is measured by force displacement (PowerLab, ADInstruments, Mountainview, CA). Responses to various agonists were tested in the presence and absence (mechanically scraped using a thin wire or chemical inhibition of nitric-oxide synthase using 100 μMl-NAME) of endothelial cells. For isoproterenol responses, pretension was established at 60% of the maximum phenylephrine response (3 × 10−7 M phenylephrine).
In Vivo Physiology.
Mice were anesthetized with ketamine (100 mg/kg body weight) and xylazine (5 mg/kg body weight). Subsequently, a flexible plastic catheter (flame-stretched PE50 tubing) was placed in the left carotid artery to monitor arterial pressure and tunneled subcutaneously to exit at the nape of the neck. The catheter was then flushed with 100 μl of heparinized phosphate-buffered saline (30 U/ml), sealed, and attached to the skin between the scapulae. Twenty-four hours later, blood pressure measurements were recorded from awake, unrestrained mice. Intra-arterial blood pressure was recorded continuously through the carotid catheter using PowerLab (ADInstruments, Mountain View, CA) data acquisition and software. In both anesthetized and awake animals, systolic (SBP) and diastolic blood pressure (DBP), and heart rate were measured. Mean arterial pressure (MAP) was calculated as DBP + 1/3 (SBP − DBP). The recordings from animals in each experimental group were then integrated and averaged. To analyze acute blood pressure responses, a second catheter was placed in the jugular vein to infuse agonists. Immediately after an equilibration period, mice received a bolus injection at 2- to 5-min intervals.
Histology and Morphology.
First, mice were sacrificed and aorta wall thickness was determined by perfusing for 10 min at 100 mm Hg with phosphate-buffered saline. Subsequently, mice were whole-body perfusion-fixed at 100 mm Hg using 10% neutral-buffered formalin. The aorta were then removed and fixed for another 4 h in 10% neutral-buffered formalin at 4°C, rinsed and stored in 70% ethanol. Aortas were embedded in paraffin and sectioned on a cryostat. The resulting sections were stained with a modified Verhoeff VanGieson/Masson's trichrome stain. Heart-to-body weight ratios (mg/g) were calculated from weighing mice before sacrifice and then weighing the blotted dry hearts after dissection.
Statistical Analysis.
Data are expressed as mean ± S.E.M. Data were analyzed using two-way analysis of variance (ANOVA) or unpaired Student's t test as indicated.
Results
In Vivo VSM-Specific GRK2 Targeting.
To generate mice with targeted VSM expression of the GRK2 transgene, we amplified a 481-base pair portion of the SM22α promoter from mouse genomic DNA that spans from −441 to +41 relative to the transcription start site. This portion of the promoter is necessary and sufficient to direct robust transcription in VSM but not in nonvascular tissues as shown in both in vitro and in vivo marker gene studies (Solway et al., 1995; Kim et al., 1997). The complete reading frame of bovine GRK2 was ligated to this SM22α promoter and transgenic mice were generated as described previously (Koch et al., 1995). Two independent lines of SM22α-GRK2 transgenic mice were established: SM22α-GRK2-10 and SM22α-GRK2-25. No gross phenotypic changes or unusual neonatal mortality were observed in these transgenic mice compared with nontransgenic littermate control (NLC) mice. Second-generation adult animals 2 to 4 months of age were used for most studies.
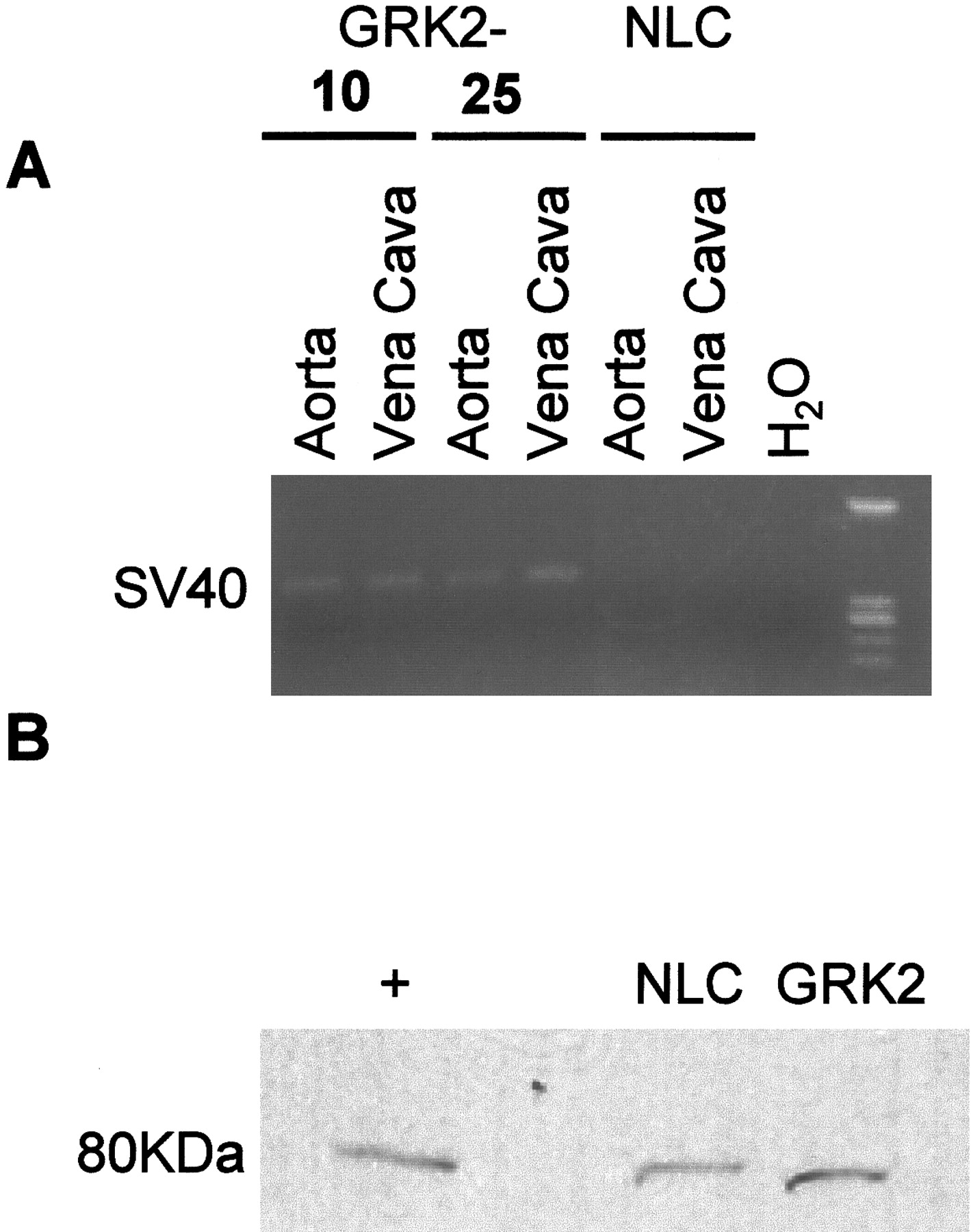
To assess mRNA transgene expression in the two lines of SM22α-GRK2 mice, RT-PCR was used. Positive expression was seen in only the transgenic GRK2 lines in both aorta and vena cava but not in samples from NLC mice (Fig. 1A). To assess GRK2 protein expression, we harvested aortas from 10 mice each for NLC and GRK2-10 mice. These aortas were pooled and enzymatically digested to isolate the smooth muscle layer. We then extracted protein, resolved 40 μg of protein on a 12% SDS-PAGE gel, transferred to nitrocellulose, and immunoblotted for GRK2. As shown in the representative autoradiograph (Fig. 1B), there was an increase of approximately 80% in GRK2 expression in GRK2-10 mice compared with NLC mice. Furthermore, RT-PCR analysis revealed the SM22α-GRK2 mRNA in several mouse organs from transgenic mice; however, the level of GRK2 protein expression in heart, liver, kidney, and brain was no different between NLC mice and the vascular transgenic mice (data not shown). Thus, although these tissues are highly vascularized, the RT-PCR positive GRK2 transgene expression, no doubt found in VSM cells, cannot be detected via protein analysis when sampling from cell heterogeneous tissue samples. Therefore, the level of in vivo overexpression in vascular tissue is subtle but similar to the increase seen in hypertensive patients (Gros et al., 1999).

Expression of the GRK2 transgene. A, expression of the GRK2 transgene was determined by RT-PCR for the SV40 portion of the construct from various tissues isolated from NLC and 2 different lines of SM22α transgenic mice. B, in vivo protein levels of GRK2 were determined using immunoblotting techniques. The medial smooth muscle layer from 10 aortas were pooled, and protein was extracted and resolved on a 12% SDS-PAGE gel. The + lane is protein isolated from the heart of transgenic mice with cardiac overexpression of GRK2.
β-AR Uncoupling in VSM.
To study the signaling implications of vascular GRK2 overexpression, we generated primary smooth muscle cells cultured from aorta of SM22α-GRK2 mice. Aorta VSM cells from SM22α-GRK2 mice had an overexpression of GRK2 of approximately 3-fold (Fig. 2A). These levels of overexpression seen in culture are consistent with in vivo expression results described above, although we find slightly higher overexpression (∼300% versus 80%). However, these cell extracts may represent a more accurate measure of GRK2 levels because cultures of aorta VSM cells are pure, whereas pooled aorta have a heterogeneous cell mixture. As shown in Fig. 2A, cell extracts from both lines of SM22α-GRK2 transgenic mice (GRK2-10 and GRK2-25) had almost identical transgene expression. Consistent with the Western blot results, VSM cellular GRK activity, as demonstrated by the ability of extracts from these cultured cells to phosphorylate the in vitro substrate rhodopsin, was also increased in transgenic extracts (Fig. 2B).

In vitro expression and activity of vascular GRK2 overexpression in transgenic mice. A, primary cell cultures were established from thoracic aorta of NLC and the two lines of GRK2-overexpressing mice and GRK2 protein levels were assessed by protein immunoblot analysis. B, GRK2 kinase activity was assessed by phosphorylation of rhodopsin using total cellular protein isolated from NLC and GRK2-25 aorta smooth muscle cells. − lane, buffer only with no cellular protein; + lane, sample with pure GRK2 added. C, functional expression of the transgene was assessed by determining adenylyl cyclase activity in response to increasing doses of isoproterenol in aorta VSM cells isolated from NLC (○, n = 4) and SM22α-GRK2-25 (●, n = 7) mice. Conversion of [3H]adenine to [3H]cAMP was determined. The entire isoproterenol dose-response curve was statistically different in transgenic cells compared with NLC cells. Data are shown as mean ± S.E.M. ∗, P < 0.05, two-way ANOVA. D, ERK1/2 and JNK1/3 activity in response to graded βAR stimulation in NLC and GRK2-25 aorta smooth muscle. Autoradiogram of an experiment in which [γ-32P]ATP-mediated phosphorylation of MBP because of ERK1/2 activity or GST-c-jun because of JNK1/3 activity was assessed in 1 mg of cytosolic fraction isolated from cultured NLC and GRK2-25 aorta cells after stimulation for 5 min with increasing doses of isoproterenol (ISO). E, histogram of averaged data for ERK1/2 activity in NLC (n = 3–4) and GRK2-25 cells (n = 3–4) in response to stimulation with 10−7 to 10−5M isoproterenol (ISO) for 5 min using phospho-specific antibodies. Data indicate that there is a functional overexpression of GRK2 in vascular smooth muscle cells isolated from aorta of transgenic mice because the adenylyl cyclase and MAPK activation is attenuated in SM22α-GRK2 cells compared with NLC cells. Data shown as mean ± S.E.M. ∗, P < 0.05 two-way ANOVA.
To determine whether enhanced expression and activity of GRK2 seen in cultured VSM cells from SM22α-GRK2 mice could exert an effect on signaling, we examined intracellular cAMP accumulation in cultured aortic VSM cells in response to the βAR agonist isoproterenol. Intracellular cAMP accumulation in cells from SM22α-GRK2-25 mice showed significant desensitization as indicated by the minimal increase in cAMP generation in response to isoproterenol compared with cAMP generation induced by isoproterenol in VSM cells cultured from NLC mice (Fig. 2C). Similar results were observed in cells from SM22α-GRK2-10 mice (data not shown). Thus, it seems that increased GRK2 overexpression in the VSM of SM22α-GRK2 mice leads to enhanced desensitization and uncoupling of βARs demonstrating the potential for exerting an in vivo physiological effect on βAR as well as other GPCR mediated signaling. Importantly, there were no compensatory changes in the overall adenylate cyclase pathway because the response of NLC, SM22α-GRK2-10, and SM22α-GRK2-25 cells to 100 μM forskolin was similar (NLC, 58 ± 7% conversion of adenine to cAMP, n = 7; BK10, 44 ± 1%, n = 3; BK25, 47 ± 5%, n = 7).
We also investigated MAPK activity after βAR stimulation. Two MAPKs were studied, ERK1/2 (or p42/p44 MAPKs) and JNK. Fig. 2D is a representative autoradiograph of the ability of immunoprecipitated ERK1/2 or JNK1/3 to phosphorylate their in vitro substrates [myelin basic protein (MBP) or GST-c-jun, respectively] after the addition of different doses of isoproterenol to aortic VSM cells from NLC and SM22α-GRK2-25 mice. As shown, there is a dose-dependent increase in the phosphorylation of MBP in NLC cells, whereas phosphorylation is significantly attenuated in aorta VSM cells from GRK2 transgenic mice. This was also the case for the activity of JNK1/3 and its ability to phosphorylate GST-c-jun (Fig. 2D). We also performed studies in which we immunoblotted for the active phosphorylated form of ERK1/2 and normalized this data to total ERK levels after stimulation with 10−7 to 10−5M isoproterenol. There was a robust increase in induction of phosphorylation status of ERK1/2 in NLC VSM cells, whereas this activation was virtually abolished in SM22α-GRK2-25 VSM cells (Fig.2E). Thus, like βAR-mediated adenylate cyclase activation, MAPK activation is significantly attenuated in VSM cells isolated from GRK2 transgenic mice demonstrating that GRK2 overexpression does have significant effects on GPCR signaling.
Pharmacology.
To analyze the effect of VSM GRK2 overexpression independent of autonomic influences on the peripheral vasculature, responses of isolated thoracic aorta ring segments to the GPCR agonists isoproterenol (βAR) and phenylephrine (α1AR) were examined. Endothelial cell presence was first verified by relaxation responses to 10−5M acetylcholine (data not shown). Interestingly, phenylephrine-mediated constriction of aorta rings from both the NLC and SM22α-GRK2-25 mice were similar (Fig. 3A), indicating that GRK2 overexpression does not seem to alter α1AR-mediated signaling and in vivo function. Accordingly, pretension for the βAR studies was established using 3 × 10−7M phenylephrine, which corresponded to 60% maximum phenylephrine stimulation in both NLC and transgenic mice. VSM GRK2 overexpression was sufficient to significantly attenuate βAR-mediated vasodilation in the presence of endothelial cells (Fig. 3B). Release of nitric oxide from endothelial cells in response to isoproterenol stimulation can contribute to the βAR-mediated vasodilation; therefore, aorta rings were also preincubated in 100 μM l-NAME, a nitric-oxide synthase inhibitor, for 20 min. After inhibition of nitric-oxide synthase and relative endothelial cell influence, GRK2-overexpressing mice still exhibited an attenuated response to isoproterenol compared with NLC (Fig. 3C). Importantly, results were also similar if the endothelial cells were mechanically scraped using a thin wire as opposed to l-NAME pretreatment (data not shown). Our results indicate that approximately 40% of the NLC βAR-mediated vasodilation is caused by βAR on endothelial cells; this response is preserved in GRK2-overexpressing mice (Fig. 3B and C). Interestingly, the smooth muscle-mediated component to the vasodilation in GRK2 transgenic rings is almost completely abolished at the lower concentrations of isoproterenol and is only apparent at the higher doses (Fig. 3C). EC50 values for the relaxation of the rings to isoproterenol further illustrates that endothelial cell response to isoproterenol was masking the inhibitory effects of GRK2 overexpression on vascular βARs (Table1). EC50 values for isoproterenol were similar in transgenic and nontransgenic mice in the presence of endothelial cells but significantly greater in GRK2-overexpressing aorta when the endothelial cell influence was abolished (Table 1).

Response of thoracic aorta rings to phenylephrine (PE) and isoproterenol (ISO). Segments of thoracic aorta (2.5 mm) were isolated from NLC (○, n = 3) and SM22-GRK2-25 (●,n = 4) and hung on isolated ring baths. A, the vasoconstrictor phenylephrine increased tension in aorta rings in a similar dose-dependent manner for both NLC (○, n = 3) and SM22α-GRK2 mice (●, n = 4). B, cumulative dose response to isoproterenol was determined in the presence of endothelial cells. ∗, P < 0.05 versus NLC, two-way ANOVA. C, response of aorta rings to isoproterenol after pretreatment withl-NAME to prevent nitric oxide release from endothelial cells. Response of rings to isoproterenol after mechanical scraping of endothelial cells using a fine wire was also done and the results were similar to l-NAME pretreatment (data not shown). ∗,P < 0.05 versus NLC, two-way ANOVA.
Effect of endothelial cell (EC) presence or absence on response of isolated aortic ring segments to βAR agonist isoproterenol (ISO)
Hypertension in Mice with VSM GRK2 Overexpression.
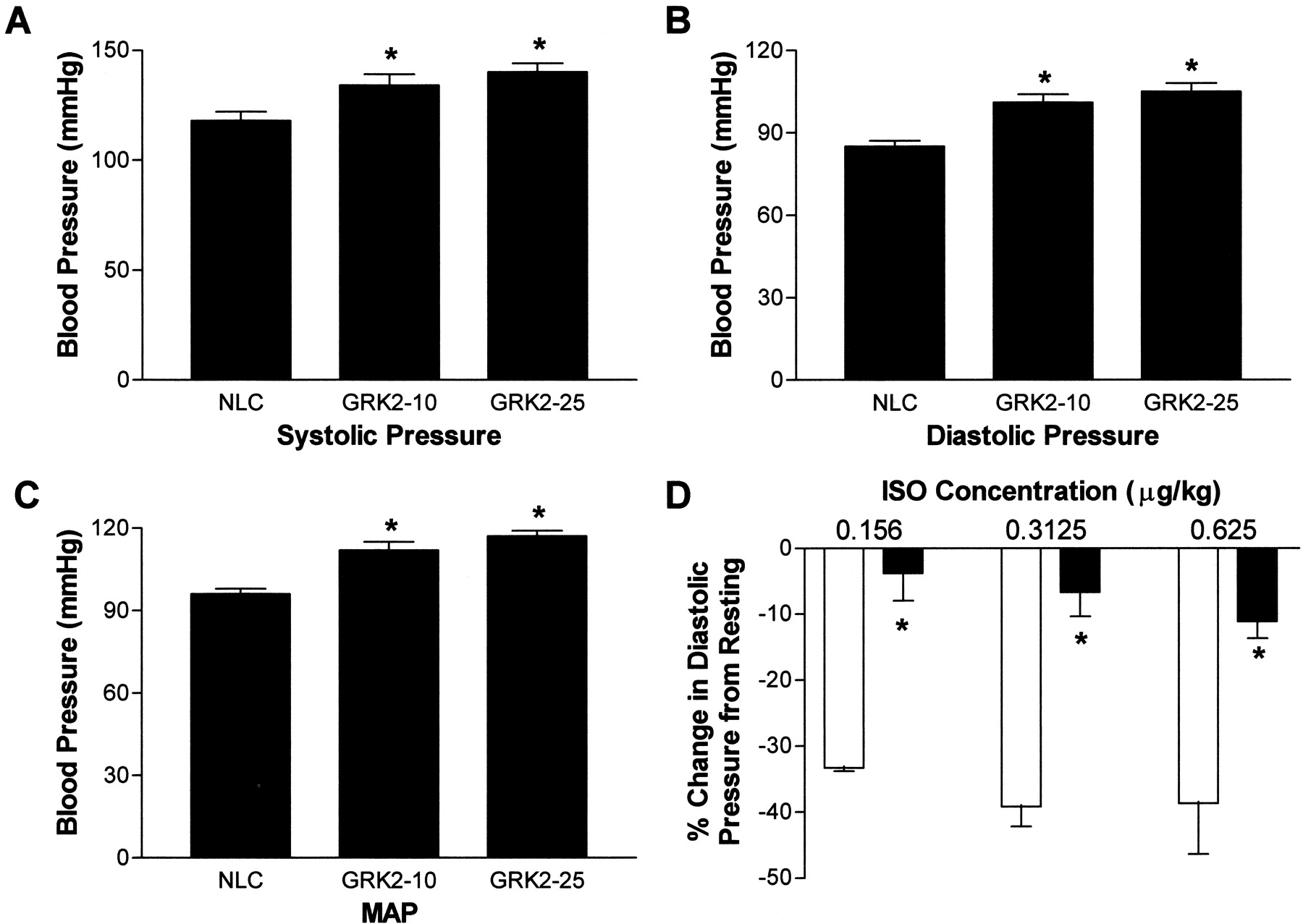
To investigate the effect of GRK2 on in vivo vascular function, we examined the blood pressure of conscious mice using an indwelling fluid-filled carotid artery catheter. Conscious systolic arterial pressure was increased in SM22α-GRK2-10 and GRK2-25 mice compared with NLCs [NLC, 118 ± 4 mm Hg (n = 9) versus SM22α-GRK2-10, 134 ± 5 mm Hg (n = 7) and GRK2-25, 140 ± 4 mm Hg (n = 5), p< 0.05] (Fig. 4A) as was diastolic pressure (NLC, 85 ± 2 mm Hg (n = 9); SM22α-GRK2-10, 101 ± 3 mm Hg (n = 7); GRK2-25, 105 ± 3 mm Hg (n = 5), p < 0.05] (Fig. 4B). MAP was also significantly increased by ∼20% in both lines of vascular GRK2-overexpressing mice compared with NLC animals (Fig. 4C). Heart rate was unchanged between the different groups (data not shown).
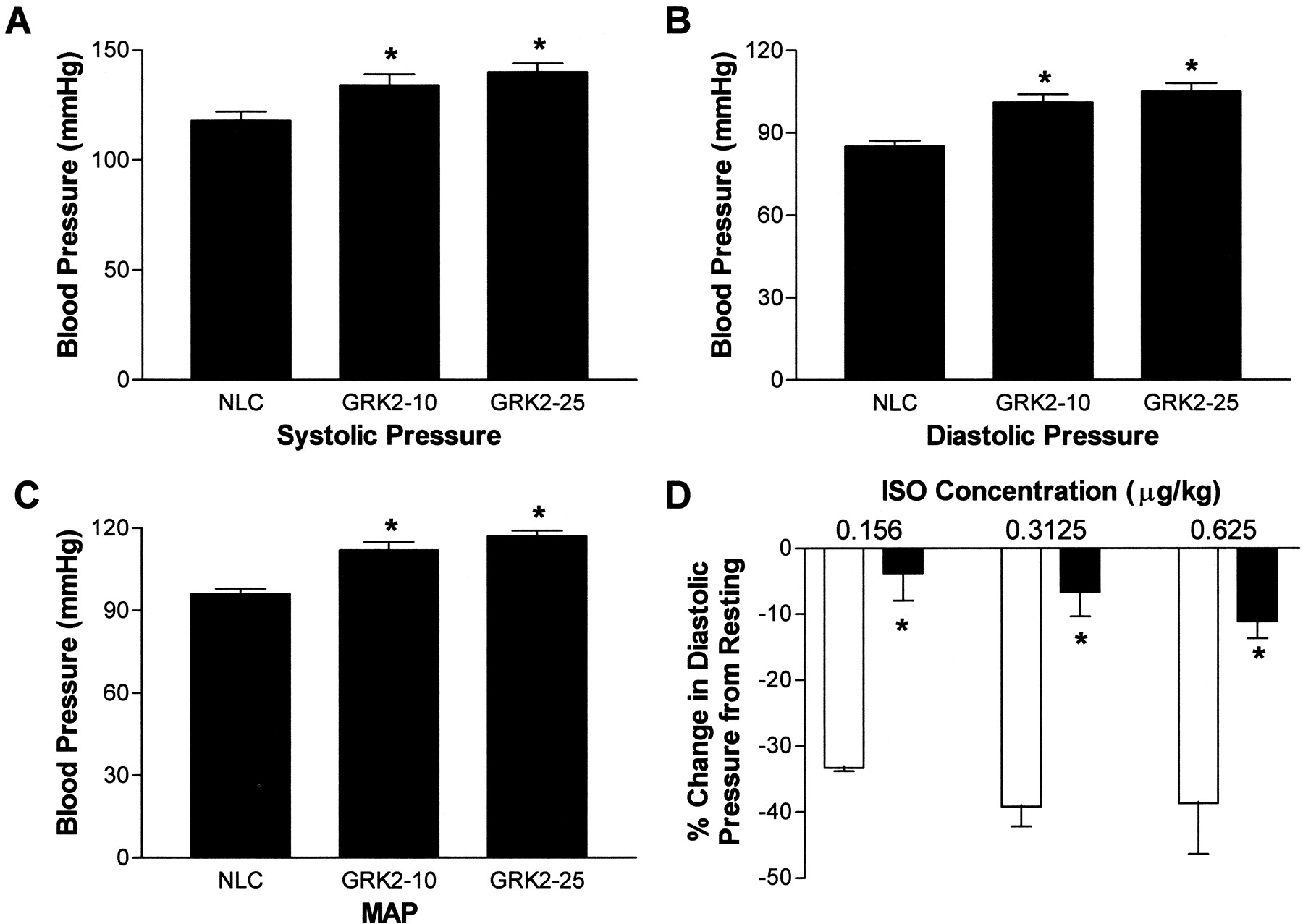
Conscious blood pressure in NLC and SM22α-GRK2 mice. A fluid-filled indwelling catheter was inserted into the left common carotid artery to determine blood pressure on conscious unrestrained mice 24 h after catheter insertion. Systolic (A) and diastolic (B) pressures were measured and MAP (C) was calculated from for NLC (n = 9), SM22α-GRK2-10 (n = 7), and SM22α-GRK2-25 mice (n = 5). ∗, P < 0.05 versus NLC, unpaired t test. D, at doses of isoproterenol (ISO) low enough not to effect heart rate, there was a significant attenuation in the increase in diastolic pressure compared with resting diastolic pressure in SM22α-GRK2 mice (▪; n = 3–5) versus NLC mice (■; n = 3–5). There was no difference in resting heart rate between NLC mice (192 ± 28 bpm) and SM22α-GRK2 mice (239 ± 17 bpm). ∗, P< 0.05 for transgene effect in two-way ANOVA comparison.
To understand the mechanisms involved in the GRK2-induced hypertension in these transgenic mice, we measured in vivo blood pressure responses to isoproterenol in anesthetized mice. As in the data obtained in conscious animals (Fig. 4A-C), resting MAP was significantly increased in anesthetized SM22α-GRK2-10 (89.3 ± 7.4 mm Hg,n = 9) compared with NLC mice (60.6 ± 3.2 mm Hg,n = 10) (p < 0.05, unpairedt test). First, we found a significant attenuation in the βAR-mediated decrease in MAP elicited by isoproterenol with the overexpression of GRK2 in VSM (data not shown). This depression was most significant at the lowest doses of isoproterenol (i.e., at 0.156 μg/kg isoproterenol: −30 ± 2.8 mm Hg decrease from resting MAP, n = 3, versus SM22α-GRK2, 4.7 ± 3.2 mm Hg,n = 3). More importantly, diastolic pressure, which is an essential component of peripheral vascular resistance, was also significantly attenuated in SM22α-GRK2-10 and GRK2-25 mice compared with the responses in diastolic pressure in NLC mice (Fig. 4D). Thus, in SM22α-GRK2 mice, there is a significant impairment in βAR-mediated diastolic blood pressure that correlates with the attenuated vasorelaxation described above in Fig. 3, resulting in impaired MAP responses to catecholamines with the overall result hypertension.
We also examined the in vivo response of SM22α-GRK2 transgenic mice to angiotensin II stimulation. Unlike phenylephrine-stimulated α1ARs, angiotensin II receptors are sensitive to GRK2 phosphorylation in vivo in the heart (Rockman et al., 1996;Eckhart et al., 2000; Koch et al., 2000). Therefore, we were interested in determining the effect of vascular GRK2 overexpression on the signaling through angiotensin II receptors. Similar to the anesthetized in vivo response to isoproterenol, angiotensin II-mediated MAP increase was partially attenuated in SM22α-GRK2 transgenic mice (Fig.5). There was a significant dose-dependent increase in MAP in both NLC and SM22α-GRK2 mice and the overall response was significantly different between the two types of mice (two-way ANOVA). However, in contrast to the complete attenuation of the βAR-mediated signal, the angiotensin signal was not completely abrogated. Interestingly, only at the highest dose of angiotensin II (1 mg/kg body weight) was there a significant difference in the MAP increase (NLC, 77.0 ± 7.9%, n = 11, versus SM22α-GRK2, 52.0 ± 6.8% over resting MAP,n = 9, P = 0.0312, two-tailed unpairedt test).

MAP response to angiotensin II. Increasing doses of angiotensin II (AngII) were administered in milligrams per kilogram of body weight to anesthetized NLC (○, n = 11) and SM22α-GRK2 mice (●, n = 9) mice via an indwelling jugular catheter and mean arterial blood pressure (MAP) was measured using a fluid-filled indwelling catheter within the carotid artery. ∗, P < 0.05 for both transgene and dose effect in two-way ANOVA comparison versus NLC mice.
As further evidence that SM22α-GRK2 transgenic mice have a phenotype of hypertension, we found aortic vascular wall thickness to be significantly increased by ∼30% in aortas isolated from perfusion-fixed GRK2-overexpressing mice (Fig.6). Importantly, this vascular hypertrophy is apparent only in the VSM layer and there do not seem to be any differences in collagen and elastin deposition between the NLC and SM22α-GRK2 lines (Fig. 6). Furthermore, we found that SM22α-GRK2 transgenic mice had significant myocardial hypertrophy probably as a result of the increased MAP as found by an increase in heart-to-body weight ratios (Fig. 7).
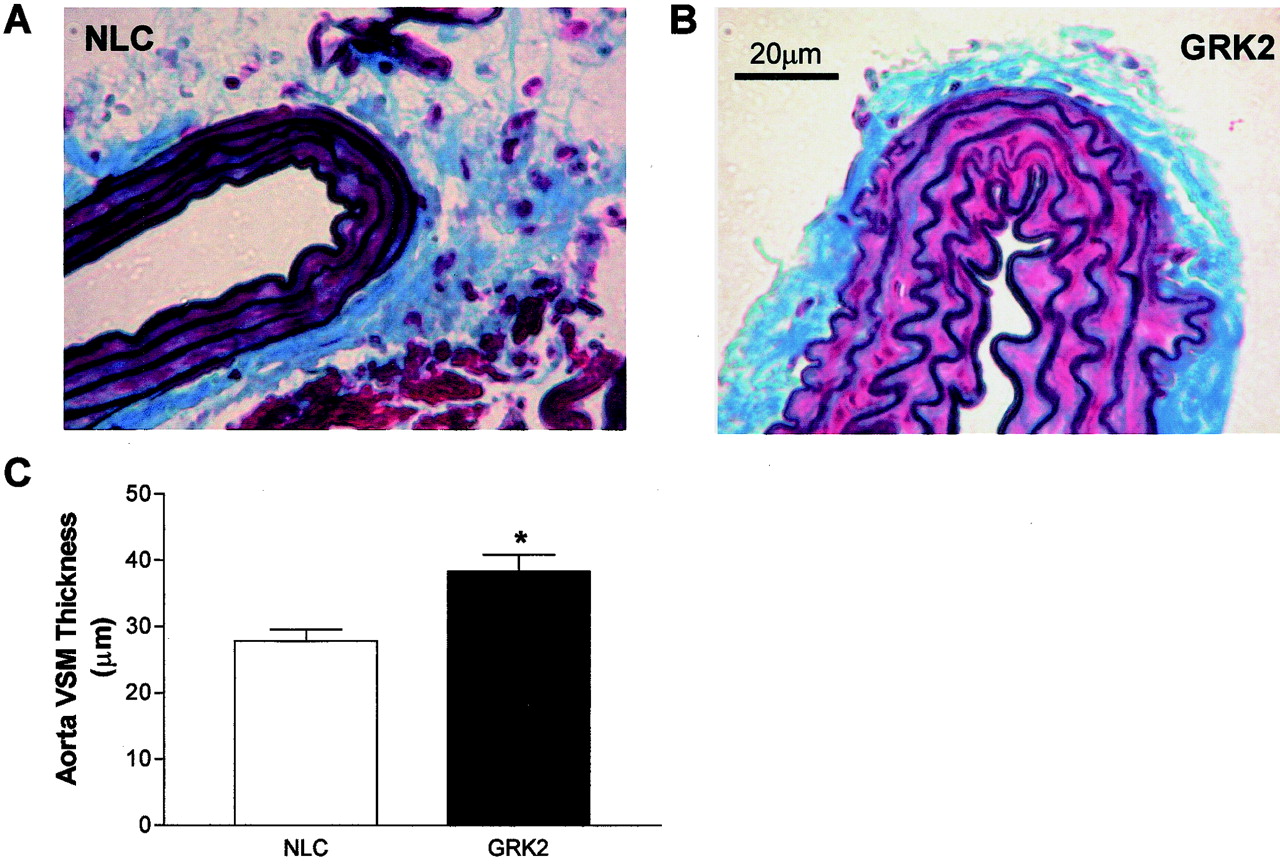
Aorta wall thickness in GRK2-overexpressing mice. Mice were perfusion fixed at 100 mm Hg with 10% neutral-buffered formalin. Thoracic aorta were then isolated from NLC (A) and SM22α-GRK2-10 mice (B), fixed, embedded in paraffin, and sliced. The slides were then stained using a Verhoeff Van Gieson/Masson's protocol. Scale bar, 20 μm; the same scale was used for each aorta. C, histogram of the aorta wall thickness for SM22α-GRK2 mice (▪,n = 6) compared with NLC mice (■, n =7). ∗, P < 0.05 versus NLC, unpairedt test.

.: Myocardial hypertrophy in GRK2-overexpressing mice. Heart weight-to-body weight ratio (milligrams per gram) in NLC (n = 7) and both lines of GRK2-overexpressing mice combined (n = 10). ∗, P < 0.05 versus NLC, unpaired t test.
Discussion
In this study, we have developed a transgenic mouse model with VSM-targeted overexpression of GRK2 to examine the influence of GRK2 activity on vascular βAR signaling. Importantly, these mice have attenuated βAR-mediated signaling in VSM cells and in vivo vasodilation. Moreover, we found that vascular GRK2 overexpression, even at modest levels, was sufficient to cause a significant elevation of resting blood pressure that was accompanied by vascular thickening and cardiac hypertrophy. Thus, these mice demonstrate that GRK2 activity in VSM is critically involved in the regulation of vascular βAR signaling and, consequently, blood pressure. Although other vascular GPCR signaling may be altered in these mice (see below for further discussion), impaired βAR signaling in VSM seems to be responsible for the hypertensive phenotype.
In the past, the β2AR has usually been designated as the βAR subtype mediating vasodilation; however, more recent studies using βAR knockout mice suggest that all three subtypes (β1-, β2-, and β3ARs) mediate vascular relaxation (Chruscinski et al., 1999; Rohrer et al., 1999). Although the β3AR is resistant to GRK-mediated phosphorylation, GRK2 is capable of desensitizing both β1- and β2ARs (Lefkowitz 1993; Koch et al., 2000). Interestingly, previous descriptive studies have uncovered abnormalities in vascular βAR signaling in hypertensive states (Feldman, 1990), including enhanced GRK2 activity (Gros et al., 1997; 2000). These findings may be of clinical significance because our mice indicate that vascular βAR signaling can have a critical influence on chronic resting blood pressure. Moreover, our relatively modest overexpression of GRK2 is in line with what has been described in human hypertensive patients (55% increase) (Gros et al., 1999). As further evidence of clinical significance, recent findings have discovered that polymorphisms present in the human β1- and β2AR genes seem to be genetic markers determining vascular reactivity and susceptibility to hypertension (Bray et al., 2000; Cockcroft et al., 2000; Hoit et al., 2000;Bengtsson et al., 2001). Thus, changes in βAR signaling, via either genetic variation that uncouples βARs from their signaling machinery (Bray et al., 2000; Cockcroft et al., 2000; Hoit et al., 2000) or increases in VSM GRK2 activity that enhance βAR desensitization (as in the SM22α-GRK2 mice), can lead to a hypertensive state.
The mechanism leading to the up-regulation of VSM GRK2 expression in hypertensive humans and animal models is not clear and certainly is a target for future studies. Nevertheless, our current data indicate that vascular GRK2 activity may be a key molecule in the pathogenesis of hypertension. Enhanced GRK2 activity, at least in the transgenic mice studied herein, primarily manifests itself as abnormal βAR-mediated vasorelaxation. Moreover, this attenuated vasodilation results in an increase in resting and βAR-stimulated diastolic pressure. Because diastolic pressure provides a rough estimate of systemic vascular resistance, this probably accounts for the increase in resting blood pressure of these mice. Further studies are warranted to verify this and to also examine any influences of other GPCR signaling pathways in addition to βARs that may be altered by increased vascular GRK2 activity.
Interestingly, transgene expression directed by the SM22α promoter was observed not only in arterial smooth muscle cells but also in cells isolated from vena cava, indicating that this promoter is more generalized in its vascular nature than previously thought. Importantly, it was found to be specific for directing transgene expression in the smooth muscle of blood vessels, which has been verified by others (Solway et al., 1995; Moessler et al., 1996; Kim et al., 1997; Imai et al., 2001; Ju et al., 2001). This is a finding that we plan to exploit in future studies; we can examine GPCR signaling differences in arterial versus venous VSM that have increased GRK2 expression, which could have important implications in specific vascular disorders.
Initial studies examining the properties of SM22α expression described the presence of the endogenous protein in smooth muscle that was in the “contractile” state, whereas expression was limited in the “synthetic” phenotype (Shanahan et al., 1994). Thus, limited expression of the GRK2 transgene may have been predicted in our VSM cell lines derived from the transgenic mice, because cells in culture are more similar to the “synthetic” phenotype. However, more recently, cell lines have been derived from atherosclerotic plaques and have been shown to express SM22α (Bonin et al., 1999). Moreover, at least using various portions of the SM22α promoter, different groups have been successful in detecting robust expression in cultured VSM cells (Ju et al., 2001; Solway et al., 1995; Strobeck et al., 2001). Consistent with these findings, we were able to detect VSM overexpression of GRK2 in vitro in VSM cells (aorta and vena cava), which was even greater that the overexpression observed in vivo (3-fold versus <2-fold). The reason for the discrepancy in overexpression with our in vivo and in vitro results is unknown; however, studies suggest that serum response factor is critically important in directing SM22α expression in VSM cells in culture (Kim et al., 1997; Chang et al., 2001; Strobeck et al., 2001). Thus, expression of the transgene may be more robust in vitro because of culture conditions and a high level of fetal bovine serum, directly in contact with the cells. Alternatively, the lower levels of GRK2 overexpression in vivo may be caused by the technical difficulties in acquiring adequate vascular tissue and the cell heterogeneity of tissue extracts.
A somewhat surprising yet interesting finding of this study was that the vascular walls (e.g., aorta) of the SM22α-GRK2 mice were hypertrophied. Although it would seem that this vascular thickening is the result of increased MAP, the possibility exists that enhanced vascular GRK2 activity may alter VSM growth. Interestingly, studies have suggested that GRK2 overexpression has the potential to enhance GPCR-stimulated MAPK activity by increasing β-arrestin–mediated mitogenic signaling (Luttrell et al., 1999). However, we found exactly the opposite, in that ERK1/2 and JNK1/3 activation is attenuated in GRK2-overexpressing VSM in response to βAR stimulation, indicating that increased desensitization decreases MAPK signaling in VSM and does not enhance it. Although other GPCRs (or GRK2 itself; see below) may be involved in the vascular hypertrophy, it seems that the vessel thickening is a consequence of chronic elevated blood pressure.
Not only is GRK2 capable of exerting its effects through βARs, but it can also affect signaling, via phosphorylation, of several other GPCRs in vivo including the vasoconstricting angiotensin II receptors (Rockman et al., 1996; Eckhart et al., 2000). We found that functional (i.e., MAP increases) angiotensin II signaling is partially attenuated in SM22α-GRK2 mice. However, because SM22α-GRK2 transgenic mice have the phenotype of high blood pressure, perhaps angiotensin II receptors do not contribute significantly to the establishment of resting tone. Interestingly, because in vivo angiotensin II signaling in the heart is completely uncoupled in cardiac-specific GRK2-overexpressing transgenic mice (Rockman et al., 1996; Eckhart et al., 2000), perhaps the major receptor subtype populations mediating the angiotensin II responses are disparate between the heart and vasculature. There are two major subtypes of angiotensin II receptors, AT1 (Gq/11 and Gi coupled) and AT2 (Gi coupled). In humans, there is a single AT1 receptor, whereas rodents express two AT1 receptor isoforms, AT1Aand AT1B (Brede and Hein, 2001). Studies from knockout mice suggest that the AT1A receptor is important for both maintenance of resting blood pressure and pressor responses to angiotensin II, although both receptors are expressed in vascular smooth muscle (Brede and Hein, 2001). In contrast, studies from knockout mice confirm presence of the AT1Bin vascular smooth muscle but suggest that this receptor plays a more prominent role in the heart (Brede and Hein, 2001). Furthermore, although AT2 receptor knockout mice were normotensive or slightly hypertensive at rest, they showed exaggerated pressor responses to angiotensin II, suggesting that the AT2 receptors oppose the actions of AT1 receptors on blood pressure (Brede and Hein, 2001). With the presence of these multiple angiotensin II receptor subtypes playing various roles within the vascular system, it is possible that they are not all equally susceptible to GRK2-mediated desensitization, therefore explaining the resting hypertension and attenuation, but not abrogation, of the angiotensin II response in these SM22α-GRK2 mice. This hypothesis remains to be determined and warrants further exploration.
It is important to note that additional GPCRs and/or effector systems downstream from GPCRs could also be involved in the phenotype because of enhanced VSM GRK2 activity. Accordingly, a recent study demonstrated that the loss (through gene knockout) of the β1 subunit of a calcium-dependent potassium channel led to increased blood pressure (Brenner et al., 2000). These types of channels are critical to normal vasoregulation and can be regulated by intracellular second messengers such as cAMP (Brenner et al., 2000). Thus, decreasing second messengers via enhanced desensitization of vascular βARs (or other cAMP-linked GPCRs) would effectively diminish the activity of this channel and potentially increase vascular tone. This is an interesting potential mechanism that will be explored in future studies.
The SM22α-GRK2 mice also represent an important model in which in vivo GRK selectivity can be investigated; in vitro experiments have been generally unsuccessful in uncovering GPCR selectivity among the different GRK family members. We have shown previously that in contrast to in vitro results, mice with cardiac-specific overexpression of GRK2 had maintained α1AR responsiveness (Eckhart et al., 2000). In the present study, in vivo and in vitro vascular responses to the α1AR agonist phenylephrine were not uncoupled, corroborating our previous cardiac results. Thus, although GRK2 is capable of phosphorylating and desensitizing α1ARs in vitro, GRK2, at least at the levels expressed in our cardiac and vascular transgenic mice, is not capable of performing this function in the cardiovascular system in vivo.
We have described a powerful transgenic mouse model of hypertension caused by vascular-targeted overexpression of GRK2. Interestingly, these mice verge on what would be predicted from selectively ablating βAR genes in the vasculature, although this needs to be verified using conditional knockout techniques, such as the one that has been described using the smooth muscle-specific myosin heavy chain promoter directing Cre-recombinase expression for use in a Cre-loxstrategy (Regan et al., 2000). Additionally, we can also use this strategy to ablate GRK2 expression specifically in the vasculature to further delineate its specific role in hypertension. Nevertheless, our data demonstrate that GRK2 activity and βAR regulation in the vasculature in vivo is critically involved in blood pressure control. Moreover, the enhanced GRK2 expression and activity seen in human hypertension seems to be clinically relevant suggesting that GRK2 inhibition may be a novel therapeutic strategy for hypertension.
Acknowledgments
We thank Rachel McAdam and Emily Greene for maintenance and screening of the mouse colony and Dr. Robert J. Lefkowitz for helpful discussions. We also thank Cheryl Bock and the Duke Comprehensive Cancer Center Transgenic Facility for creation of founder mice.
Footnotes
- Received August 7, 2001.
- Accepted January 16, 2001.
-
↵1 Current address: Fujisawa Pharmaceutical Co., Osaka, Japan.
-
This work was supported in part by National Institutes of Health grants HL61690, HL65360, and HL59533 (to W.J.K.).
Abbreviations
- GPCR
- G protein-coupled receptor
- AR
- adrenergic receptor
- MAPK
- mitogen-activated protein kinase
- VSM
- vascular smooth muscle
- GRK
- G protein-coupled receptor kinase
- ARK
- adrenergic receptor kinase
- PCR
- polymerase chain reaction
- SV40
- simian virus 40
- MAP
- mean arterial pressure
- RT
- reverse transcription
- PAGE
- polyacrylamide gel electrophoresis
- ERK
- extracellular signal-regulated kinase
- JNK
- c-Jun NH2-terminal kinase
- l-NAME
- Nω-nitro-l-arginine methyl ester
- MAP
- mean arterial pressure
- NLC
- nonlittermate control
- MBP
- myelin basic protein
- GST
- glutathione S-transferase
- ANOVA
- analysis of variance
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}