


Abstract
Homologous desensitization of the μ opioid receptor (μOR) can be resolved into distinct processes that include the uncoupling of the μOR from its G-protein effectors and internalization of cell surface receptors. Using electrophysiological recordings of μOR activation of G-protein-coupled K+ channels (Kir3) in Xenopus laevis oocytes and AtT20 cells, confocal microscopy of receptor localization, and radioligand binding of cell surface receptors, we resolved these desensitization mechanisms to determine the domain of μOR important for receptor uncoupling. Activation of μOR by saturating concentrations of [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin (DAMGO), methadone, or fentanyl, but not morphine, produced robust internalization of a green fluorescent protein-tagged μOR. A subsaturating concentration of DAMGO (100 nM) did not cause receptor internalization but markedly reduced the subsequent responsiveness of Kir3 by uncoupling μOR. μOR desensitization in AtT20 cells was confirmed to be homologous, because desensitization by 100 nM DAMGO was blocked by dominant-negative forms of either G protein-coupled receptor kinase (GRK) or arrestin, and pretreatment with DAMGO did not affect the Kir3 response to somatostatin receptor activation. Alanine substitution of a single threonine in the second cytoplasmic loop of the μOR (Threonine 180) blocked agonist-dependent receptor uncoupling without affecting receptor internalization. These results suggest that GRK-dependent phosphorylation of μOR required threonine 180 for uncoupling but that a different GRK and arrestin-dependent mechanism controlled μOR internalization in AtT20 cells.
Although the development of tolerance limits the clinical utility of opioid analgesic drugs, the underlying mechanisms responsible for tolerance are incompletely understood (Kieffer and Evans, 2002). Adaptive reductions in the responsiveness to opioids that produce tolerance have been attributed to homologous desensitization mechanisms (changes in signal transduction efficiency at the level of the receptor) and heterologous desensitization mechanisms (compensatory changes in effectors and neuronal circuits mediating the response to opioids). Examples of heterologous mechanisms include changes in adenylyl cyclase expression levels and cellular forms of learning and memory mediated by N-methyl-d-aspartate receptor-dependent synaptic plasticity (Trujillo, 2000; Williams et al., 2001). Homologous receptor desensitization mechanisms include processes initiated by receptor phosphorylation and β-arrestin binding (Law et al., 2000). Homologous receptor desensitization mechanisms can result from either uncoupling of receptor from G-protein effectors after β-arrestin binding or from a loss of cell surface receptors after receptor internalization (Ferguson, 2001). Binding of β-arrestin to a G-protein-coupled receptor sterically blocks G protein activation (uncoupling), thereby reducing the efficiency of signaling without loss of cell surface receptor. In addition, β-arrestin also serves as an adaptor to promote receptor internalization (Miller and Lefkowitz, 2001).
Homologous receptor desensitization mechanisms are also affected by opioid agonist efficacy (the efficiency with which the agonist-receptor complex activates the G protein). If the agonist receptor complex that most efficiently activates G protein is also most efficiently regulated by G-protein receptor kinase (GRK) and β-arrestin, one would predict that the efficiency of G protein activation would correlate with the ability to produce desensitization. For example, opioid agonists that more robustly activate μOR (have higher efficacies) would be predicted to more efficiently activate GRK2 and GRK3, enzymes activated by free Gβγ (DebBurman et al., 1996; Kovoor et al., 1998). However, the relationship between agonist efficacy and desensitization mechanisms is not simple, and some opiates, including methadone and morphine, have been reported to disproportionately activate desensitization mechanisms compared with their efficacy in activating μOR (Keith et al., 1996; Blake et al., 1997; Yu et al., 1997; Whistler and von Zastrow, 1998; Whistler et al., 1999). Furthermore, clear differences in the extent of tolerance development to opioids of different efficacies have been demonstrated in vivo and are inversely correlated to agonist efficacy (Stevens and Yaksh, 1989; Duttaroy and Yoburn, 1995). These findings have led investigators to propose a hypothesis in which tolerance and addiction depend on whether or not an opioid agonist produces receptor internalization (Whistler et al., 1999). For example, agonists, including morphine, that do not efficiently drive μOR internalization were predicted to have a longer duration of action, require greater compensation in neuronal circuits, and thus produce greater tolerance and addiction. In contrast, the reduction of morphine tolerance and the increase in morphine sensitivity in the β-arrestin2 knockout mice suggests a more direct and classic role of GRK and β-arrestin in influencing the development of opioid tolerance (Bohn et al., 2000, 2002). In the latter model, opioid tolerance depends on the efficiency of inducing homologous desensitization. Comparisons between in vivo and in vitro models of opiate tolerance, however, are further confounded by a large receptor reserve in vivo (spare receptors) (Chavkin and Goldstein, 1981, 1984), in which only a low fractional receptor occupancy is required to mediate the opioid analgesic response (Perry et al., 1982). In contrast, studies using in vitro expression systems usually compare saturating concentrations of agonists (Blake et al., 1997; Whistler et al., 1999). In addition, previous studies have rarely distinguished desensitization that results from either receptor uncoupling or receptor internalization. Thus, at the cellular level, determination of the extent of agonist dependent regulation, including receptor uncoupling and internalization of the μOR produced by equieffective, subsaturating concentrations of μOR agonists, may provide important information more consistent with the use of opioids in vivo.
These competing views of the role of receptor regulation by GRK and arrestin are not easily resolved without a better understanding of the underlying mechanisms of receptor uncoupling and internalization and the effect of agonist efficacy on each process. Several different residues in the μOR have been implicated as targets of GRK phosphorylation and have been shown to be important in receptor uncoupling or receptor internalization (see Chavkin et al., 2002). In mammalian expression systems, the μOR is rapidly internalized, and the removal of potential phosphorylation sites in the carboxyl terminal tail blocks μOR internalization (Deng et al., 2000; Wang, 2000; El Kouhen et al., 2001). In contrast, we previously demonstrated using the Xenopus laevis oocyte expression system that GRK3- and β-arrestin2–dependent uncoupling of μOR responses was strictly dependent on agonist efficacy (Kovoor et al., 1998) and was blocked by an alanine substitution of a single threonine in the second cytoplasmic loop, threonine 180, but was unaffected by carboxy terminal truncation (Celver et al., 2001). In the present study, we examined the role of threonine 180 in receptor uncoupling and internalization in two mammalian expression systems and compared the ability of agonists with different efficacies to produce receptor uncoupling and internalization.
Materials and Methods
Chemicals. DAMGO was obtained from Peninsula Laboratories (San Carlos, CA). [3H]CTAP was obtained from the National Institute on Drug Abuse. All other chemicals were from Sigma Chemical (St Louis, MO).
Complementary DNA Clones and cRNA Synthesis. The cDNA clones used and cRNA synthesis methods have been described previously (Kovoor et al., 1997). The rat μOR clone (GenBank accession no. L13069) was obtained from Dr. Lei Yu (University of Cincinnati College of Medicine, Cincinnati, OH). cDNA for the Kir3.1 (GIRK1) channel (GenBank accession no. U01071) was obtained from Dr. Henry Lester (Caltech, Pasadena, CA). The Kir3.4 (GIRK4) clone (GenBank accession no. X83584) was provided by Dr. John Adelman (Vollum Institute, Portland, OR). Each cDNA first amplified by the use of Taq DNA Polymerase (Promega, Madison, WI) in a standard polymerase chain reaction using oligonucleotides designed to add a T7 promoter region and a 45-base poly(A)+ tail. mMESSAGE MACHINE kits (Ambion, Austin, TX) were used to generate capped cRNA from the cDNA templates, which contained T7 or SP6 promoters to direct synthesis of sense transcripts. pHc-Red1 N1 was purchased from BD Biosciences Clontech (Palo Alto, CA). GRK2 K220R was obtained from Dr. Lefkowitz (Duke University, Durham, NC) and arr3 319–418 was obtained from Dr. Benovic (Thomas Jefferson University, Philadelphia, PA).
Generation of μOR-GFP Constructs. Restriction sites were added to the rat μOR wild type and μOR-T180A cDNAs through polymerase chain reaction (Celver et al., 2001). A 5′ oligonucleotide (gcagtatggtacctcatggacagcagcacc) introduced a KpnI site and a 3′oligonucleotide (gcagtgaattcgggcaatggagc) eliminated the μOR stop codon and also introduced an EcoRI site. The resulting polymerase chain reaction products were subcloned into the KpnI and EcoRI sites of the pcDNA3-GFP37 vector generated from pcDNA3 (Invitrogen) by Dr. Kenneth Mackie (University of Washington, Seattle, WA). This construct fused GFP-37 to the carboxyl-terminal tail of the μOR, including a 5-residue glycine linker.
Oocyte Culture and Injection. Stage IV oocytes were prepared as described previously (Kovoor et al., 1997). cRNA was injected (50 nl/oocyte) using a Drummond automatic microinjector, and then oocytes were incubated at 18°C for 3 to 7 days in normal oocyte saline buffer (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 5 mM HEPES, pH 7.5) supplemented with 2.5 mM sodium pyruvate and 50 μg/ml gentamycin.
Oocyte Electrophysiology. Oocytes were clamped at -80 mV with two electrodes filled with 3 M KCl having resistances of 0.5 to 1.5 MΩ using a Genclamp 500 amplifier and pCLAMP 6 software (Axon Instruments, Foster City, CA). Data were digitally recorded (Digidata 1200 from Axon Instruments and an IBM-compatible PC) and filtered. Membrane current traces also were recorded using a chart recorder. To facilitate the inward potassium current flow through the Kir3 channels, normal oocyte saline buffer was modified to increase KCl concentration to 16 mM. All drug responses were evaluated in these high K+-containing solutions. The concentration of NaCl was correspondingly decreased to maintain iso-osmolarity. Opioid activation of Kir3 conductance was measured as described previously (Kovoor et al., 1997).
Cell Culture and Microscopy. Human embryonic kidney (HEK) 293 cells were grown in Dulbecco's modified Eagle's medium/Ham's F12 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen). AtT20 cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal horse serum (Invitrogen). μOR GFP constructs were transiently transfected using Superfect (QIAGEN, Valencia, CA) with single colonies chosen and propagated in the selection media (G418; 800 μg/ml for HEK293 cells and 1000 μg/ml for AtT20 cells) to select for stable μOR-GFP cell lines. For microscopy, cells were grown on coverslips. Cells were treated with agonists as specified and then fixed in 4% formaldehyde in PBS. Images were acquired using a confocal microscope (MRC600; Bio-Rad, Hercules, CA) with COMOS 6.01 software. For Fig. 3, cells stably expressing μOR GFP were grown on coverslips and transiently transfected with either pHc Red1-N1 and pCDNA3.1, pHc Red1-N1 and arr3 318–418, or pHc Red1-N1 and GRK2 K220R at 2.5 μg of total DNA/well.

Effect of dominant-negative GRK2 and arrestin 3 on μOR GFP internalization. A, confocal images of AtT20 cells stably expressing μOR GFP transiently transfected with Hc Red and either pCDNA3, GRK2 K220R, or arr3 D319–418 as indicated, were treated for 1 h with DAMGO (2 μM). Confocal images represent GFP fluorescence, Hc Red fluorescence, or a merger of both channels as indicated. In the GFP fluorescent images, white arrows indicate significant intracellular fluorescence (internalized μOR), and red arrows indicate Hc Red-positive cells (transfected with plasmids as indicated). Transient transfection of GRK K220R or arr3 D318–418 but not pCDNA significantly reduced the internalization of μOR GFP as seen by the lack of intracellular GFP signal Hc Red-positive cells in images taken from cell transiently transfected with either the dominant-negative GRK2 or arr3. B, quantification of receptor internalization produced by agonist treatment was determined by measuring the average intracellular fluorescence intensity from confocal images. Each bar represents the average intracellular fluorescence intensity from 10 to 20 cells treated for 1 h with agonist DAMGO. Closed bars indicate the average intracellular GFP fluorescence intensity from Hc Red-positive cells in images taken from cells cotransfected the with DNA constructs indicated, open bars indicate the average intracellular fluorescence intensity from untransfected cells from the same images. Bars indicate mean ± S.E.; *, indicate p < 0.05 compared with untreated cells.
Whole-Cell [3H]CTAP Binding. AtT20 or HEK293 cells were treated with agonists at times specified under Results then washed three times with PBS. Cells (1 × 106) were harvested for radioligand binding. Cells were incubated in 50 mM Tris buffer with 10 nM [3H]CTAP for 30 min at 4°C. Cells were filtered through GF/C glass fiber filters (Whatman, Maidstone, UK) and washed five times with ice-cold buffer. Specific binding was determined by the difference in amount of binding in the presence or absence of 1 μM naloxone, and the average nonspecific binding, which accounted for less than 10% of total binding, was subtracted from each determination. Specific radioligand binding to intact, untreated cells was defined as 100% by dividing each specific binding determination by the average specific binding in untreated cells, including the specific binding determined in untreated cells.
Whole-Cell AtT20 Electrophysiology. Kir3 currents were recorded from AtT20 cells stably expressing wild-type μOR-GFP or μOR T180A GFP. In the whole-cell configuration of the patch clamp, cells were held at -45 mV and hyperpolarized to -100 mV for 50 ms every 5 s. The average current during the hyperpolarization was determined and plotted versus time. The pipette solution contained 130 mM KCl, 20 mM HEPES, 10 mM EGTA, 5 mM MgCl2, 3 mM Na2ATP, and 0.6 mM GTP, pH adjusted to 7.25 with KOH, whereas the external solution contained 40 mM KCl, 130 mM NaCl, 1 mM CaCl2, 25 mM HEPES, and 10 mM glucose, pH adjusted to 7.35 with NaOH. In receptor uncoupling experiments, the average Kir activation produced by 1 μM DAMGO in separate cell populations was compared before and after agonist treatments of 1 h. Cells were washed for 10 min after agonist pretreatment before the 1 μM DAMGO challenge. Similarly, possible heterologous desensitization of endogenous somatostatin receptors was determined by comparing the responses to 100 nM somatostatin with or without a 1-h pretreatment with 100 nM DAMGO.
Flow Cytometric Analysis. GFP fluorescence intensity in AtT20 cells stably expressing μOR GFP or μOR T180A GFP were analyzed using a FACScan Flow Cytometer (BD Immunocytometry Systems, San Jose, CA) running CellQuest software against untransfected controls.
Data Analysis. NIH Image version 1.62 software (National Institutes of Health, Bethesda, MD) was used to quantify the degree of receptor internalization in confocal images of cells expressing μOR GFP by measuring the average intensity of pixels within the cytoplasm of cells with or without agonist treatment. The average background pixel intensity from each image was subtracted from the average intracellular fluorescence intensity for each cell. In some experiments, intracellular fluorescence intensity was compared from cells that were transfected (Hc Red positive) and untransfected cells from the same slides. The Student's t test (with two-tailed p values) was used for comparison of the independent mean values. Dose-response curves were fitted to a simple Emax model using the nonlinear regression analysis PCNONLIN v4.2 (SCI Software Consultants, Lexington, KY) to determine the EC50 values and 95% confidence intervals.
Results
Effect of GFP Tag on μOR Function. To visualize cellular trafficking of the μOR in a mammalian cell expression system, we constructed a rat μOR with a carboxyl-terminal GFP tag. To determine whether the addition of the GFP tag disrupted the μOR functioning, we compared the dose response curves of wild-type μOR and μOR-GFP with DAMGO, methadone, and morphine in the activation of the G protein-gated inwardly rectifying potassium channel, Kir3, expressed in X. laevis oocytes. In oocytes injected with cRNA for either wild-type μOR or μOR GFP and Kir 3.1 with Kir 3.4, cumulative increases in agonist concentration of DAMGO, methadone, or morphine produced cumulative increases in Kir activation. Dose response curves for each of the agonists tested were not significantly different for wild-type μOR and μOR GFP, suggesting that addition of the carboxyl-terminal GFP tag did not significantly alter the function of μOR. The EC50 values (± S.E) at the wild-type μOR for DAMGO, methadone, and morphine were 105 ± 12.4, 288 ± 40.8, and 128 ± 20 nM, respectively; and 146 ± 18.6, 322 ± 23.6, and 135 ± 30.2 nM at μOR GFP. These data suggest that the addition of GFP to μOR did not significantly alter receptor functioning.
Furthermore, we confirmed that internalization of μOR-GFP stably expressed in HEK293 cells was agonist-specific as described previously for the wild-type μOR or epitope-tagged μOR (Keith et al., 1996; Blake et al., 1997; Whistler et al., 1999; Alvarez et al., 2002). Treatment for 30 min with saturating concentrations (5 μM) of DAMGO, fentanyl, methadone, but not morphine, induced robust μOR-GFP internalization as visualized by confocal microscopy (Fig. 1A). Receptor internalization was also quantified by cell-surface binding of [3H]CTAP to μOR. [3H]CTAP is a peptide antagonist that does not produce receptor internalization or cross the plasma membrane (Kramer et al., 1989); thus, it can be used to measure changes in cell surface μOR expression in live cells. A significant decrease in whole-cell, specific [3H]CTAP binding was produced by a 30-min treatment with 5 μM DAMGO, fentanyl, methadone, and morphine (Fig. 1B). However, the decrease in specific [3H]CTAP binding in whole cells produced by morphine pretreatment was significantly less than that produced by the other agonists. Internalization of μOR in HEK293 cells was previously shown to be blocked by dominant-negative GRK2 (Zhang et al., 1998; Wang, 2000). Thus, the agonist-specific internalization of μOR GFP in HEK293 cells suggests that GRK and β-arrestin regulation of μOR GFP, at least qualitatively, was not disrupted by the addition of a carboxy terminal GFP tag. The lack of an effect of a carboxy terminal GFP tag on the function of the human μOR has been reported previously (Sarramegna et al., 2002).
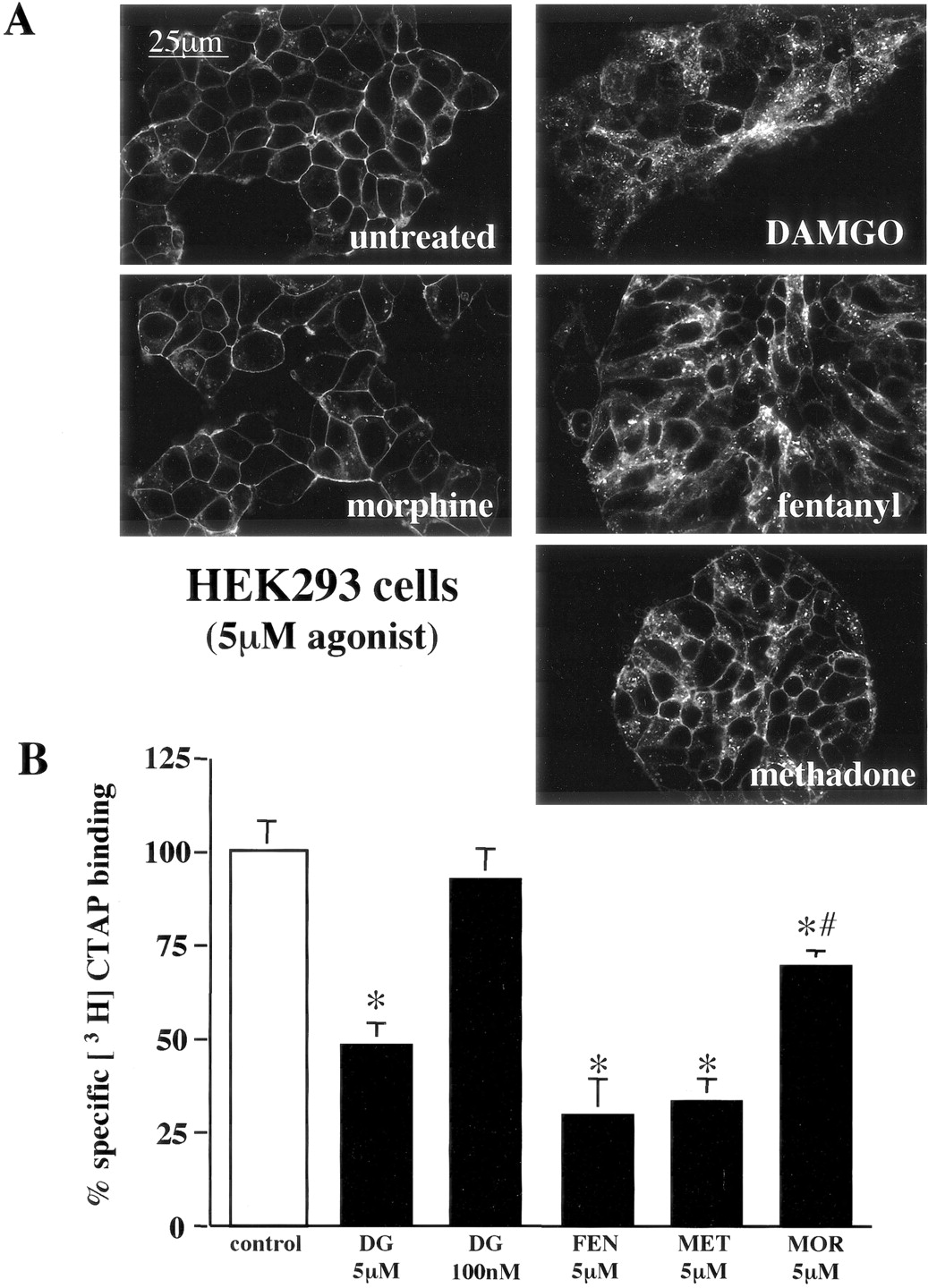
Agonist-specific internalization of the μOR in HEK293 cells. A, confocal images of HEK293 cells stably expressing μOR GFP treated for 30 min with the agonist indicated. 5 μM DAMGO, fentanyl, and methadone, but not morphine, caused an increase in intracellular GFP fluorescence in cells stably expressing μOR-GFP. B, receptor internalization measured by 10 nM [3H]CTAP binding to whole cells. HEK293 cells stably expressing μOR GFP were treated for 30 min with the agonist and concentration indicated: DAMGO (DG), fentanyl (FEN), methadone (MET), morphine (MOR), or untreated (control). After treatment, the cell monolayers were washed with PBS, and harvested for [3H]CTAP binding. Specific binding was defined by the difference in [3H]CTAP binding in presence and absence of 1 μM naloxone. The percentage reduction in specific radioligand produced by pretreatment with the opioid agonist indicated was calculated by comparing the specific [3H]CTAP binding in agonist treated and untreated control monolayers. Each bar represents the average specific [3H]CTAP binding in two separate experiments performed in triplicate. Bars indicate mean ± S.E.; *, p < 0.05 compared with untreated monolayers; #, p < 0.05 compared with cell monolayers pretreated with 5 μM DAMGO.
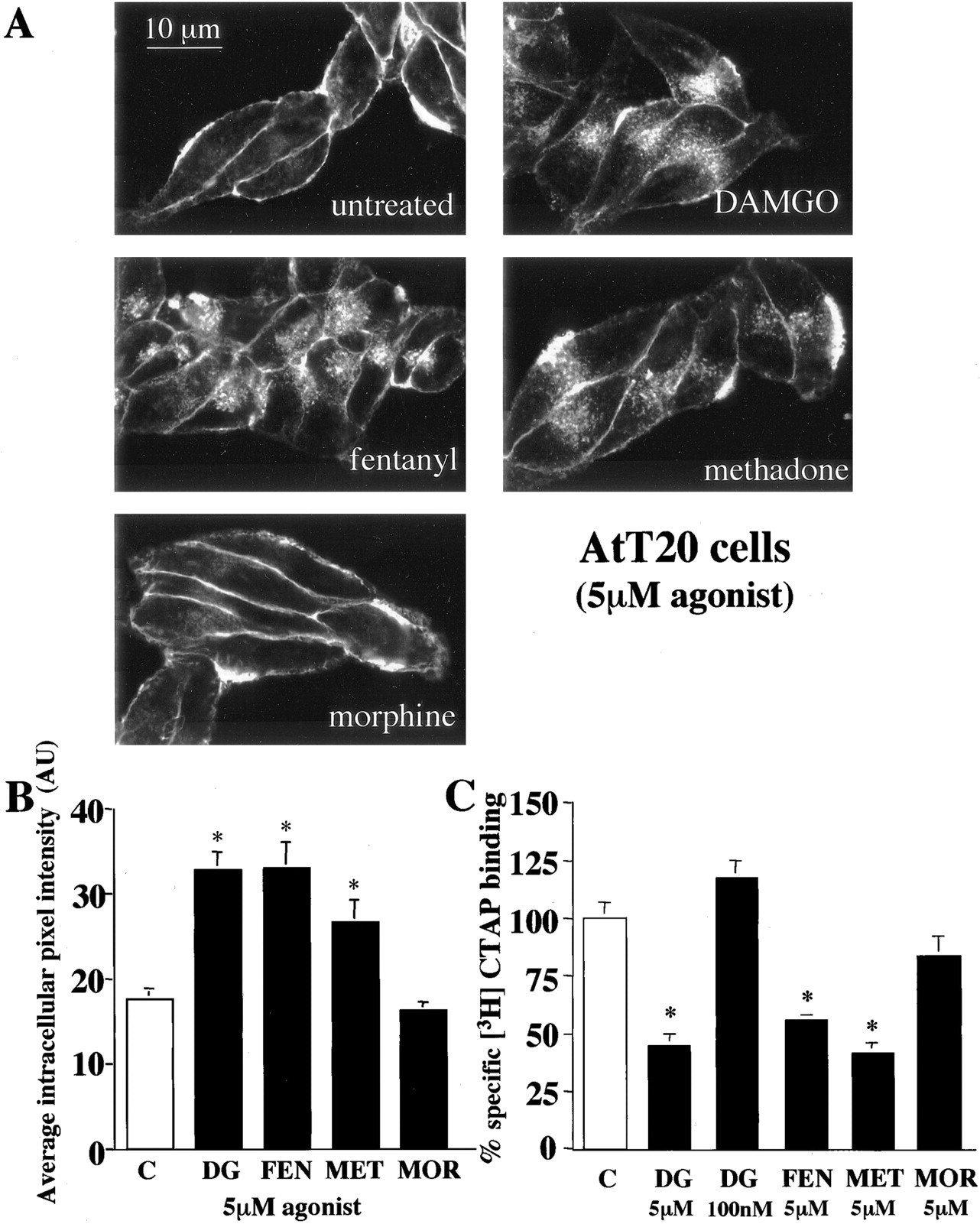
Agonist-Specific Internalization of μOR in AtT20 Cells. Endogenous expression of Kir3 channels in AtT20 cells, a neuronal cell line derived from mouse pituitary, provides a common signaling pathway for the μOR to monitor receptor activity (Mackie et al., 1995; Tallent et al., 1998). To determine whether we could also monitor receptor internalization in these cells, we stably expressed μOR-GFP in AtT20 cells and used confocal microscopy and radioligand binding to evaluate agonist induced receptor trafficking. Treatment for 1 h with saturating concentrations of DAMGO, fentanyl, methadone, but not morphine, induced μOR-GFP internalization as visualized by confocal microscopy (Fig. 2A). The amount of receptor internalization was quantified by analysis of intracellular fluorescence intensity from confocal images and a significant increase in the average intracellular fluorescence intensity was produced by DAMGO, fentanyl, and methadone, but not morphine, compared with untreated cells (Fig. 2B). Receptor internalization was also quantified by [3H]CTAP binding after treatment of AtT20 cells for 1 h with 5 μM DAMGO, fentanyl, methadone, or morphine. As evident in HEK293 cells, a significant decrease in whole-cell [3H]CTAP binding was produced by 1-h pretreatment of AtT20 cells with 5 μM DAMGO, fentanyl, or methadone, but not morphine (Fig. 2C). The lack of internalization evident after morphine pretreatment confirms prior work, but the short duration of exposure used does not distinguish whether morphine is unable to induce internalization or is simply slower.

Agonist-specific internalization of μOR GFP in AtT20 cells. A, confocal images of AtT20 cells stably expressing μOR GFP treated for 1 h with the agonist indicated (5 μM). DAMGO, fentanyl, and methadone, but not morphine, caused an increase in intracellular fluorescence in cells stably expressing μOR-GFP B, quantification of receptor internalization produced by agonist treatment indicated was determined by measuring intracellular fluorescence intensity from confocal images. Each bar represents the average intracellular fluorescence intensity from 10 to 12 cells treated for 1 h with agonist indicated. Bars indicate mean ± S.E.; *, p < 0.05 compared with untreated cells. C, receptor internalization measured by 10 nM [3H] CTAP binding to whole cells. Whole cell binding is the same as described in Fig. 1, except AtT20 cells stably expressing μOR GFP were used and agonist treatment was 1 h with the agonist and concentration indicated, DAMGO (DG), fentanyl (FEN), methadone (MET), or morphine (MOR). Specific [3H]CTAP binding in untreated cells (C) was defined as 100%. Each bar represents the average percentage reduction in specific [3H]CTAP binding in two separate experiments performed in triplicate. Bars indicate mean ± S.E.; *, p < 0.05 compared with untreated monolayers.
Internalization of μOR GFP in AtT20 Cells Is GRK- and Arrestin-Dependent. To determine the underlying mechanism responsible for μOR internalization in AtT20 cells, we transiently transfected AtT20 cells stably expressing μOR GFP with either the dominant-negative GRK2 lacking kinase activity, GRK2 K220R, or the dominant-negative form of arrestin, arr3 318–418, which is the sequence corresponding to the clathrin-binding domain of arrestin. Both GRK2 K220R and arr3 318–418 have been shown previously to disrupt the action of their endogenous wild-type counterparts in a variety of expression systems (Krupnik et al., 1997). To identify transfected cells, we cotransfected an expression plasmid for Hc Red along with an empty pCDNA vector or with constructs designed to drive expression of either GRK2 K220R or arr3 318–418 such that the total amount of DNA each slide received during the transfection was the same for each group. In the absence of agonist treatment, localization of μOR GFP was unaffected by expression of Hc Red, GRK2 K220R, or arr3 319–418 as visualized by confocal microscopy (data not shown). Confocal images in Fig. 3A demonstrate that Hc Red-positive cells, when cotransfected with pCDNA, readily internalized μOR GFP upon agonist stimulation. In contrast, Hc Red-positive cells in images from slides cotransfected with either GRK2 K220R or arr3 319–418 showed significantly reduced μOR GFP internalization compared with untransfected cells on the same slide. The average intensity of intracellular fluorescence was also analyzed from multiple images comparing untransfected cells and Hc Red-positive cells from the same images. Figure 3B shows that Hc Red and pCDNA transfection did not significantly alter the average intracellular fluorescence intensity of cells treated for 1 h with 2 μM DAMGO. In contrast, transient transfection of either GRK2 K220R or arr3 318–418 significantly reduced the average intracellular fluorescence intensity of cells expressing μOR GFP after agonist treatment. These data support the conclusion that the underlying mechanism responsible for μOR internalization in AtT20 cells was GRK- and arrestin-dependent. We demonstrated previously that μOR uncoupling in the absence of internalization was GRK and arrestin-dependent (Kovoor et al., 1997).
Effect of Extended Morphine Treatment on μOR Internalization in AtT20 Cells. To further characterize the agonist-dependent internalization of μOR GFP in AtT20 cells, we measured the amount of receptor internalization of μOR GFP after longer durations of DAMGO or morphine exposure. Importantly, extending the duration of morphine treatment increased the amount of μOR internalization. In AtT20 cells stably expressing μOR GFP, 2- and 4-h application of 5 μM morphine (a saturating concentration) produced a significant reduction in whole-cell [3H]CTAP binding compared with untreated cells. However, morphine was significantly less effective at producing receptor internalization than DAMGO at each time point (Fig. 4). These data suggest that morphine-activated receptors probably do not elude regulation necessary for μOR internalization; rather, they require longer agonist treatment for the regulation to occur. Similar findings were observed by Bushell et al. (2002) in single hippocampal neurons. Here, arrestin overexpression was required for agonist-dependent internalization to be produced by even high-efficacy agonists, and longer treatments were required to produce internalization with the low-efficacy agonist morphine. Our result is also consistent with the finding that increasing GRK or arrestin expression in HEK293 cells or primary hippocampal neurons also increases the internalization produced by morphine (Whistler et al., 1998; Zhang et al., 1998), because both treatments would increase the likelihood of interaction between the agonist activated receptor and these key cellular components that can mediate receptor internalization.

Effect of extended morphine treatment on μOR GFP internalization. Time course of morphine and DAMGO induced loss of cell surface μOR-GFP in AtT20 cells. Specific [3H]CTAP binding was determined as described in Fig. 1 in cell monolayers treated for 1, 2, or 4 h with 5 μM morphine or 5 μM DAMGO. Error bars indicate S.E. from two identical experiments performed in triplicate. *, p < 0.05 compared with cell monolayers treated for the same amount of time with DAMGO; #, p < 0.05 compared with specific [3H]CTAP binding in untreated cells.
Substitution of Threonine 180 to Alanine of μOR Does Not Effect Receptor Internalization. Previously, we demonstrated that GRK3- and arrestin-dependent desensitization of μOR expressed in X. laevis oocytes was dependent on threonine 180, a potential phosphorylation site in the second cytoplasmic loop of μOR (Celver et al., 2001). In this expression system, however, the desensitization was because of receptor uncoupling rather than receptor internalization (Celver et al., 2001). To determine whether substitution of threonine 180 of μOR to alanine affected μOR internalization, we stably expressed μOR T180A GFP in AtT20 cells. Comparison of the GFP fluorescence intensities between the stable cells lines used in this study expressing wild-type μOR-GFP and μOR T180A GFP suggested that the receptor expression levels were similar in the two lines. The geometric means for GFP fluorescence intensity in the μOR wt GFP cell line was 130, with a coefficient of variance of 45.5, and for the μOR T180A GFP cell line was 150, with a coefficient of variance of 46.7.
Treatment of AtT20 cells stably expressing μOR T180A GFP for 1 h with 5 μM DAMGO produced robust internalization of μOR T180A GFP as visualized by confocal microscopy (Fig. 5A) or measured by the reduction in whole-cell specific [3H]CTAP binding (Fig. 5B). In contrast, treatment with 100 nM DAMGO induce neither internalization nor reduction in specific cell surface binding of [3H]CTAP. Furthermore, the percentage reduction in specific [3H]CTAP binding after treatment of μOR T180A GFP expressing AtT20 cells for 1 h with 5 μM DAMGO was not significantly different from that produced by the same treatment of AtT20 cells expressing the μOR GFP (compare Figs. 2C and 5B). These data indicate that mutation of threonine 180 to alanine was not important for agonist-dependent internalization of μOR in AtT20 cells. Similarly, internalization of μOR in HEK293 cells was also not affected by mutation of threonine 180 to alanine (data not shown). Thus, if T180A mutation blocked μOR phosphorylation by GRK in these cells, phosphorylation of this site was not required for internalization.

Internalization of μOR T180A GFP in AtT20 cells. A, confocal images of AtT20 cells stably expressing μOR T180A GFP untreated or treated for 1 h with 5 μM DAMGO. B, receptor internalization measured by 10 nM [3H]CTAP binding to whole cells. The percentage reduction of whole cell [3H]CTAP binding in AtT20 cells stably expressing μOR T180A-GFP were determined as described in Fig. 1. One-hour treatment of 5 μM but not 100 nM DAMGO significantly decreased specific whole-cell [3H]CTAP binding. Each bar represents the average specific [3H]CTAP binding in two separate experiments performed in triplicate. Bars indicate mean ± S.E.; *, p < 0.05 compared with untreated monolayers.
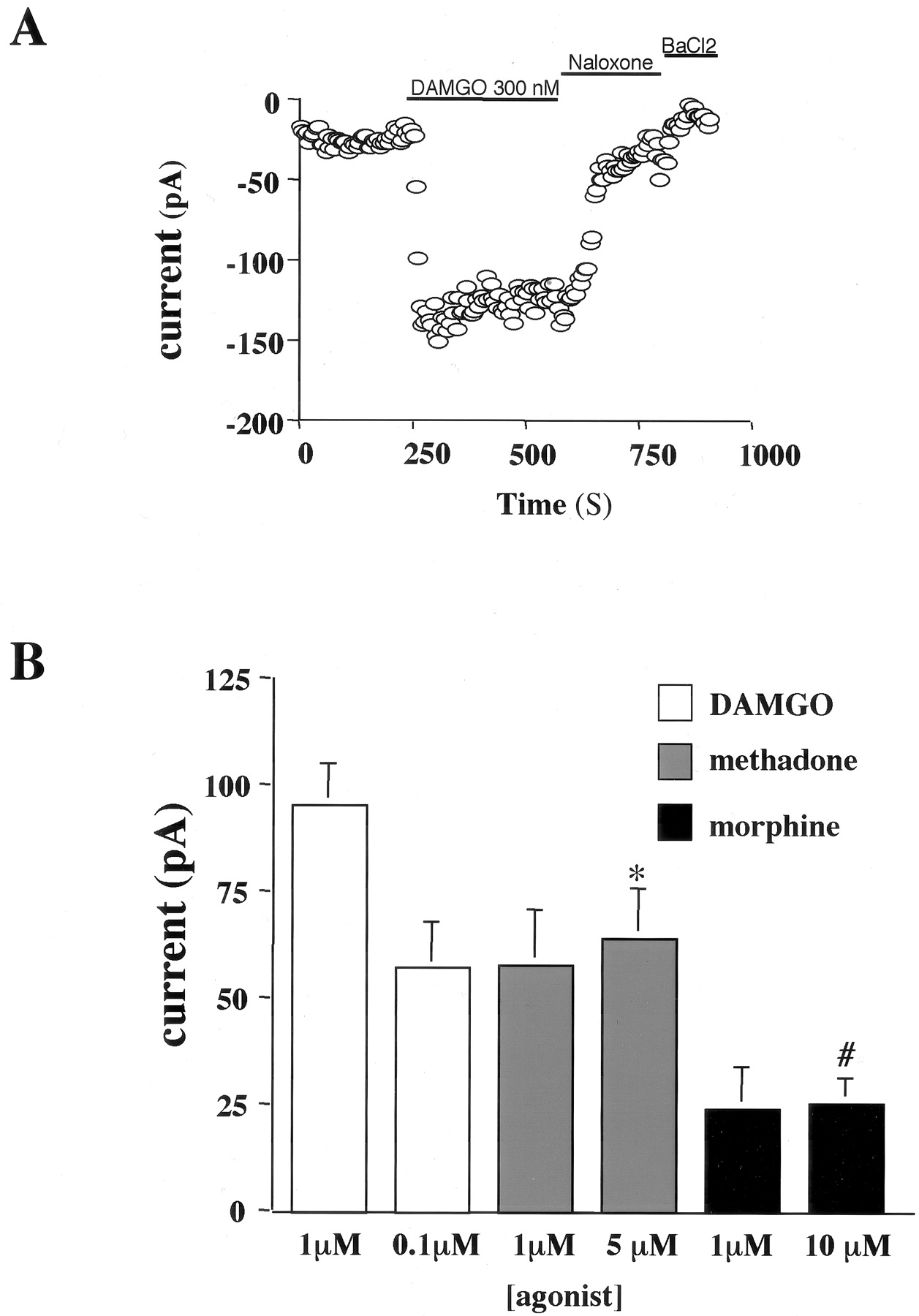
μOR GFP Mediated Kir3 Activation in AtT20 Cells. In AtT20 cells, Gi/o coupled receptors activate endogenously expressed Kir3 (Mackie et al., 1995; Tallent et al., 1998). As expected, application of DAMGO increased Kir3 currents recorded in AtT20 cells stably expressing μOR-GFP but not in untransfected cells. The DAMGO-induced current was reversed by an opioid antagonist, naloxone (1 μM) as well as by 1 mM BaCl2, a Kir3 channel blocker (Fig. 6A). To rank the relative activity of DAMGO, methadone, and morphine in this expression system, we separately measured the current response produced by saturating concentrations of each agonist. The average current response produced by DAMGO was greater than methadone, which was greater than morphine (Fig. 6B) [note that the responses shown represent the average agonist activated current in picoamperes rather than a normalized response]. The response produced by 1 μM morphine (24.0 ± 9.9 pA) was not significantly different from the response produced by 10 μM morphine (25.3 ± 5.9 pA). Similarly, the response produced by DAMGO at 300 nM (94 ± 19.9 pA) was not significantly different from the response to 1 μM (94.8 ± 10.4 pA), which indicates that 1 μM DAMGO was a saturating concentration. In addition, the response to 5 μM methadone (63.7 ± 12.2 pA) was not significantly different from that to 1 μM (57.5 + 13.1 pA). The dose response relationships presented show that AtT20 cells did not express spare receptors for methadone or morphine and that methadone was more efficacious than morphine and less than DAMGO. This efficacy ranking was consistent with the ranking of these agonists at the μOR expressed in Xenopus oocytes in the absence of a receptor reserve. In oocytes, the average maximal current response in the activation of the coexpressed Kir was 1519 ± 36, 865 ± 44, and 531 ± 53 nA for DAMGO, methadone, and morphine, respectively.

Relative activity of DAMGO, methadone, and morphine in AtT20 cells. A, representative trace from an AtT20 whole-cell recording. Activation of the stably expressed μOR-GFP with 300 nM DAMGO significantly increased the inward potassium conductance of the endogenously expressed Kir3, and the response was reversed by the opioid antagonist naloxone (1 μM). The Kir3 channel blocker BaCl2 blocked the basal potassium conductance. B, summarized average Kir3 current responses in AtT20 cells elicited by 1 or 0.1 μM DAMGO, 5 μM methadone, and 1 or 10 μM morphine. Agonist responses were defined as the current response above baseline in the presence of the high potassium-containing recording buffer. Bars indicate mean ± S.E. of four to six independent recordings. Statistical analysis showed that the response to methadone was significantly less than to DAMGO (*, p < 0.05) and that the response to morphine was significantly less than to methadone (#, p < 0.05).
Interestingly, concentrations of DAMGO and methadone that were equieffective at Kir3 activation in AtT20 cells produced different degrees of internalization (Fig. 2). The average μOR response elicited by 100 nM DAMGO was not significantly different from the average response elicited by 5 μM methadone (Fig. 6B), indicating that these concentrations of DAMGO and methadone were equieffective in the activation of Kir3. However, DAMGO (100 nM) did not induce significant μOR internalization in 60 min, whereas 5 μM methadone did.
μOR Uncoupling in AtT20 Cells. To determine whether the subsaturating concentration of DAMGO (100 nM) produced receptor uncoupling of the μOR in AtT20 cells, we compared the average Kir3 activation produced by 1 μM DAMGO in untreated cells and cells pretreated for 1 h with 100 nM DAMGO. DAMGO (100 nM) pretreatment significantly reduced the average peak response elicited by a subsequent 1 μM DAMGO challenge (Fig. 6B). Because 100 nM DAMGO treatment for 1 h did not produce receptor internalization, these data suggest that the desensitization was caused not by receptor internalization but instead by receptor uncoupling. Furthermore, Kir3 activation by 100 nM somatostatin via the endogenously expressed somatostatin receptor in AtT20 cells (Surprenant et al., 1992) produced an average response of 46.5 ± 2.4 pA. Pretreatment of cells expressing μOR GFP with 100 nM DAMGO for 1 h did not significantly reduce the somatostatin-mediated activation of Kir3 response (p < 0.005). These data suggest that the desensitization of the μOR-mediated Kir3 response by DAMGO pretreatment was specific, thus homologous. This finding differs from that of a previous report in AtT20 cells, where a more prolonged treatment with a much higher dose produced heterologous desensitization of both μOR and somatostatin receptors (Tallent et al., 1998).
Morphine (5 μM) did not produce μOR internalization within 60 min; thus, to determine whether morphine uncoupled μOR in the absence of internalization, we compared Kir3 currents produced by a 1 μM challenge of DAMGO in μOR-GFP expressing AtT20 cells that were pretreated for 1 h with 5 μM morphine. The average response to a 1 μM challenge of DAMGO was not significantly different after a pretreatment with 5 μM morphine for 1 h (Fig. 7). These results indicate that morphine did not produce a significant amount of μOR desensitization. This saturating concentration of morphine produced considerably less Kir activation than 100 nM DAMGO (Fig. 7). However, 1-h pretreatment of an equieffective concentration of DAMGO (40 nM) as a saturating concentration of morphine in Kir activation also failed to produce significant μOR uncoupling (data not shown). Thus, μOR expressed in AtT20 cells undergoes agonist specific internalization comparable with that produced in the more often used HEK293 cell expression system. Furthermore, by measuring μOR-mediated activation of the endogenously expressed Kir3 in AtT20 cells, opioid agonists of different efficacies can be distinguished, as well as their ability to produce receptor uncoupling and internalization measured in the same expression system.

μOR uncoupling in AtT20 cells. A, confocal images of AtT20 cells stably expressing μOR-GFP untreated or treated for 1 h with 100 nM DAMGO. 100 nM DAMGO treatment of μOR-GFP expressing cells failed to promote μOR internalization visualized by confocal microscopy (A) or radiolabeled [3H]CTAP binding (Fig. 2C). B, summarized desensitization of μOR activation of Kir3 current responses in AtT20 cells. Average responses elicited by 1 μM DAMGO (black bars) are summarized in AtT20 cells stably expressing μOR GFP or μOR T180A GFP as indicated. Pretreatment of AtT20 cells stably expressing μOR-GFP for 1 h with 100 nM DAMGO (□) but not (5 μM) morphine ( ) caused a significant decrease in μOR responsiveness. In AtT20 cells stably expressing μOR T180A GFP, the average response to a 1 μM challenge in cells was not significantly different after a 1 h, pretreatment of DAMGO (100 nM). Each bar represents four to six independent determinations and indicates mean ± S.E. *, p < 0.05 compared with morphine-treated cells.
) caused a significant decrease in μOR responsiveness. In AtT20 cells stably expressing μOR T180A GFP, the average response to a 1 μM challenge in cells was not significantly different after a 1 h, pretreatment of DAMGO (100 nM). Each bar represents four to six independent determinations and indicates mean ± S.E. *, p < 0.05 compared with morphine-treated cells.
Effect of Alanine Substitution of Threonine 180 on Agonist-Dependent μOR Uncoupling. To determine whether substitution of threonine 180 to alanine of the μOR affected receptor uncoupling, we compared the effect of 1-h 100 nM DAMGO pretreatment of AtT20 cells stably expressing μOR GFP or μOR T180A GFP. Before desensitization, the average responses of AtT20 cells stably expressing μOR T180A GFP was not significantly different from those elicited in cells expressing the wild-type μOR GFP (Fig. 7B). This is consistent with the equivalent fluorescent intensity measured in the two cell lines and suggests that the T180A substitution did not affect receptor efficacy. Pretreatment of μOR GFP expressing cells with 100 nM DAMGO for 1 h significantly decreased the average current response to a subsequent 1 μM DAMGO challenge (Fig. 7B). In contrast, equivalent pretreatment of μOR T180A GFP-expressing cells with DAMGO did not significantly decrease the average current response to a subsequent 1 μM DAMGO challenge (Fig. 7B). The block of homologous desensitization by T180 mutation of μOR was consistent with the effects previously reported in X. laevis oocytes (Celver et al., 2001). These data support the hypothesis that phosphorylation of threonine 180 was critical for agonist-dependent desensitization of μOR in AtT20 cells. Furthermore, these data suggest that the desensitization of μOR in these cells produced by the 100 nM DAMGO pretreatment was the result of receptor uncoupling rather than internalization.
Discussion
The principal finding of this study was that distinct domains of μOR mediate receptor uncoupling and internalization. Substitution of a single threonine in the second cytoplasmic loop of μOR blocked agonist-dependent uncoupling without affecting μOR internalization. This finding was aided by resolving homologous receptor desensitization into two processes: receptor uncoupling and receptor internalization. Furthermore, equieffective concentrations of DAMGO and methadone in μOR-mediated Kir activation did not produce the same degree of receptor internalization. These data offer further support to the conclusion that the efficiency of μOR activation of G proteins is not necessarily directly correlated to the efficiency of which a receptor is internalized (Whistler et al., 1999). In contrast, G protein activation more closely correlated with the degree of receptor uncoupling produced by equiactive concentrations of high- and low-efficacy agonists.
Importantly, the relative efficacy of the μOR agonist methadone and morphine in the activation of Kir3 in X. laevis oocytes and AtT20 cells reported in this study differ from previous reports (Yu et al., 1997; Whistler et al., 1999), but are consistent with a recent report also using transfected AtT20 cells (Borgland et al., 2003). In AtT20 cells, morphine, and methadone were clearly partial agonists producing approximately 25% and 75% of the maximum Kir activation as DAMGO, respectively. In several opioid responses measured in other systems, morphine has been shown to be equally efficacious as DAMGO (Keith et al., 1996; Blake et al., 1997; Whistler et al., 1999; Bushell et al., 2002). This difference is probably resolved by considering the presence or absence of a receptor reserve (Kovoor et al., 1998; Borgland et al., 2003). In the X. laevis oocyte system receptor and effector expression can be tightly controlled, and the activity of an agonist can be tested under both conditions (Kovoor et al., 1998). In this system, increasing receptor expression compared with the effector expression, Kir3, leads to increases in the amplitude of μOR-activated K+ conductance. This increase in current amplitude can be seen as the cRNA injection for μOR is increased as much as 10-fold, until further increases in cRNA injection no longer increase the peak Kir3 activation by even low efficacy agonists indicating a large receptor reserve (Kovoor et al., 1998; Celver et al., 2001). In the absence of a receptor reserve in the X. laevis oocyte expression system, the relative efficacy of DAMGO, methadone, and morphine were similar to that seen in the activation of Kir in AtT20 cells expressing μOR. Although the difference in the efficacies reported for morphine in different systems is most probably explained by the presence or absence of a receptor reserve, methadone has been reported to have an even lower relative efficacy than morphine (Yu et al., 1997; Whistler et al., 1999) in contrast to our findings. A higher efficacy of methadone compared with morphine, however, is consistent with the relative ability of these agonists to promote receptor phosphorylation (Yu et al., 1997; Whistler et al., 1999). In addition, a greater analgesic efficacy of methadone than morphine in guinea pig and mouse pain assays has also been reported previously (Ivarsson and Neil, 1989; Adams et al., 1990).
Understanding the role of agonist efficacy on opioid tolerance in vivo is complicated because at analgesic doses, agonists of different efficacies would have different fractional μOR occupancies. Data from the present study suggest that the degree of G protein activation, measured by Kir3 activation correlates better with the degree of receptor uncoupling than with the extent of receptor internalization. In the present study, we extended the analysis by also showing that with equally effective concentrations of DAMGO and methadone, methadone more readily induced receptor internalization. Furthermore, we found that a concentration of DAMGO that had equivalent activity to the maximal effect of morphine also did not produce either uncoupling or internalization in 1 h. Thus, we suggest that the ineffectiveness of morphine results from its lower efficacy.
Our findings in present study demonstrate that agonist dependent receptor uncoupling and internalization of μOR can be dissociated. This finding is consistent with previous reports showing that distinct sites mediate receptor internalization and uncoupling in the CB1 cannabinoid receptor (Jin et al., 1999) and the D1 dopamine receptor (Lamey et al., 2002). The μOR is different in that sites mediating receptor uncoupling and internalization are present in different cytoplasmic domains, namely the second cytoplasmic loop and the carboxy terminal tail. Importantly, although the substitution of threonine 180 to alanine blocks GRK3- and arrestindependent uncoupling of MOR in X. laevis oocytes (Celver et al., 2001) and the uncoupling of MOR in AtT20 cells, these findings do not constitute direct evidence that this residue is phosphorylated. In fact, the substitution of threonine 180 to alanine does not affect GRK- and arrestin-mediated internalization in AtT20 cells. Similarly, the carboxyl-terminal tail may have no effect on receptor desensitization in the absence of receptor internalization (Celver et al., 2001). Thus, receptor uncoupling and internalization can be dissociated not only pharmacologically but also biochemically, and the distinct domains responsible for each may serve as useful markers for the determination of role of uncoupling and internalization in vivo. With respect to threonine 180 serving as a potential phosphorylation site, recent reports argue that agonist-dependent phosphorylation of μOR is restricted to the carboxyl-terminal tail (Qiu et al., 2003). However, the time course and agonist treatment required for phosphorylation of threonine 180 may be different from that which leads to phosphorylation of sites in the carboxy terminal tail. Thus, further investigation is needed to determine whether substitution of threonine 180 to alanine disrupts receptor uncoupling by disrupting an important phosphorylation site or by an alternative mechanism.
The ultimate goal is to understand the processes resulting in analgesic tolerance to opioids that most investigators acknowledge is a complex and multifaceted process. The contribution of cellular events including receptor uncoupling and internalization to the total process of behavioral tolerance is still unresolved. The contributions of other components to the tolerance process including NMDA-dependent learning mechanisms (Siegel 1976), compensatory changes in other endogenous peptide systems (Vanderah et al., 2000; Kim and Siegel, 2001) and other adaptive processes cannot be ignored. Because multiple adaptive processes underlying tolerance are likely to be differentially activated, the adaptive mechanisms produced by one opioid may be different from another that has a different efficacy. The results from this study show that even at the cellular level, tolerance is complex and may be uniquely determined by multiple processes.
Acknowledgments
We thank Dr. Terri L. Gilbert for helpful discussion and FACS expertise. We thank Ms. Abigail Parsley for technical assistance.
Footnotes
-
This study was supported by National Institute on Drug Abuse grants DA11672 and training grant T32-DA07278.
-
ABBREVIATIONS: GRK, G protein-coupled receptor kinase; μOR, μ-opioid receptor; DAMGO, [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin; CTAP, d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2; HEK, human embryonic kidney; GFP, green fluorescent protein; PBS, phosphate-buffered saline.
- Received July 22, 2003.
- Accepted November 21, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}