


Abstract
G-protein-coupled receptors (GPCRs) serve as catalytic activators of heterotrimeric G-proteins (Gαβγ) by exchanging GTP for the bound GDP on the Gα subunit. This guanine nucleotide exchange factor activity of GPCRs is the initial step in the G-protein cycle and determines the onset of various intracellular signaling pathways that govern critical physiological responses to extracellular cues. Although the structural basis for many steps in the G-protein nucleotide cycle have been made clear over the past decade, the precise mechanism for receptor-mediated G-protein activation remains incompletely defined. Given that these receptors have historically represented a set of rich drug targets, a more complete understanding of their mechanism of action should provide further avenues for drug discovery. Several models have been proposed to explain the communication between activated GPCRs and Gαβγ leading to the structural changes required for guanine nucleotide exchange. This review is focused on the structural biology of G-protein signal transduction with an emphasis on the current hypotheses regarding Gαβγ activation. We highlight several recent results shedding new light on the structural changes in Gα that may underlie GDP release.
Many key extracellular signals, including hormones, neuro-transmitters, growth factors, and sensory stimuli, relay information intracellularly by activation of plasma membrane-bound receptors. The largest class of such receptors is the superfamily of heptahelical G-protein-coupled receptors (GPCRs). In many genomes, GPCRs are encoded by the largest gene family; in humans, >1% of the genome is dedicated to producing hundreds of these critical signal detectors (Takeda et al., 2002; Fredriksson et al., 2003). Genetic studies have highlighted the physiological importance of GPCRs, with knockout models revealing pathological phenotypes involving the cardiovascular, nervous, endocrine, and sensory systems (Rohrer and Kobilka, 1998; Yang et al., 2002; Karasinska et al., 2003). Several hereditary diseases have also been linked to mutations within the genes encoding specific GPCRs (Spiegel and Weinstein, 2004). Indeed, GPCRs represent a major therapeutic target, giving rise to the largest single fraction of the prescription drug market, with annual sales of several billion dollars (Overington et al., 2006). Therefore, a complete mechanistic understanding of how GPCRs communicate extracellular signals into the cell would be extremely valuable for the continued development of novel therapeutics that target this family of receptors and the signaling cascades they modulate.
G-Protein Signaling and the Guanine Nucleotide Cycle
GPCRs transduce signals by activating heterotrimeric G-proteins that normally exist in an inactive state of Gα-GDP bound to Gβγ subunits (Fig. 1). Agonist activation of GPCRs induces a conformational change within the receptor, which subsequently catalyzes the exchange of GDP for GTP on the Gα subunit (Gilman, 1987). In this way, GPCRs serve as guanine nucleotide exchange factors (GEFs) for Gα-GDP/Gβγ complexes (Fig. 1). Although the exact mechanism by which GPCRs exert their GEF activity remains to be fully elucidated, this action is critical to the commencement of G-protein signaling, because GDP release is the rate-limiting step of the Gα guanine nucleotide cycle (Ferguson et al., 1986). The subsequent binding of GTP induces a conformational change in three flexible “switch regions” of the Gα subunit, leading to dissociation of the Gβγ dimer and adoption of the conformation capable of interacting with effectors (Hamm, 1998). Activated Gα-GTP and liberated Gβγ each relay signals to several downstream effectors, including ion channels, adenylyl cyclases, phosphodiesterases, and phospholipases, giving rise to respective second messenger molecules intimately involved in regulating physiological processes (Offermanns, 2003; McCudden et al., 2005) (Fig. 1). Based on their sequence homology and differential regulation of effectors, G-proteins are grouped in four classes: Gαs, Gαi/o, Gαq, and Gα12/13 (Offermanns, 2003). GPCRs have the ability to couple selectively to members of one or more of these G-protein subfamilies, thus allowing selective modulation of signaling cascades by particular GPCR ligands. Deactivation of G-protein signaling occurs by the intrinsic hydrolysis of GTP to GDP by the Gα subunit, which occurs at a rate that varies among the G-protein subfamilies. Hydrolysis rates can be dramatically enhanced by members of a superfamily of “regulators of G-protein signaling” (RGS) proteins (Siderovski et al., 1996; Ross and Wilkie, 2000; Siderovski and Willard, 2005) that serve as GTPase-accelerating proteins (GAPs) (Fig. 1). The inactivated, GDP-bound Gα subsequently reassociates with Gβγ to complete the cycle. Given that this represents a true cycle of activation (by nucleotide exchange and subunit dissociation) and deactivation (by GTP hydrolysis and subunit reassociation), heterotrimeric G-proteins serve as molecular switches and are critical to defining the spatial and temporal aspects of cellular responses to external stimuli.

The guanine nucleotide cycle of heterotrimeric G-proteins. Seven TM GPCRs bind, via their intracellular loops, to the heterotrimeric G-protein consisting of Gα (with bound GDP) associated with the Gβγ dimer. The isoprenylated Gβγ dimer aids in association of the heterotrimer with the plasma membrane, participates in receptor coupling, and serves as a guanine nucleotide dissociation inhibitor (GDI) preventing spontaneous activation of the Gα subunit. Agonist-bound receptors act as GEFs by provoking conformational changes in Gαβγ resulting in the release of GDP and binding of GTP by Gα. Binding of GTP induces changes in three conformationally flexible switch regions within Gα, leading to the dissociation of Gβγ. Both Gα-GTP and freed Gβγ can subsequently regulate downstream effector molecules alone or in a coordinated fashion. The system returns to the inactive state by intrinsic GTP hydrolysis activity of the Gα subunit, cleaving the terminal γ-phosphate from GTP [note the loss of inorganic phosphate (Pi)] and rendering Gα again bound to GDP and reassociated with Gβγ, thus mutually terminating the signaling capacity of both subunits. The GTP hydrolysis reaction is greatly enhanced by the RGS family of proteins, which serve as GAPs.
Biochemical and structural analyses over the past 2 decades have advanced our understanding of the mechanics underlying G-protein regulation and the guanine nucleotide cycle (Sprang, 1997). Despite these extensive and formidable efforts, the precise molecular details of how GPCRs activate Gα subunits remain elusive. The remainder of this review therefore focuses on G-protein structure and current perspectives regarding receptor-mediated activation of heterotrimeric G-proteins to highlight recent findings that are helping to shape a contemporary structural view of this process.
G-Protein and Receptor Structure
Structures of GDP-bound G-proteins (both as isolated Gα and Gαβγ), as well as guanosine 5′-O-(3-thio)triphosphate-bound and transition-state GDP-AlF-4-bound G-proteins (Table 1), have detailed the secondary (Fig. 2) and tertiary structures of both Gα and Gβγ, how the heterotrimeric complex is formed, the conformational changes induced by GTP binding, and the mechanism of intrinsic GTP hydrolysis (Noel et al., 1993; Coleman et al., 1994; Lambright et al., 1994; Sondek et al., 1994; Mixon et al., 1995; Wall et al., 1995; Lambright et al., 1996; Sunahara et al., 1997). Subsequent analyses have defined the structural basis for engagement of several Gα subunits with their specific downstream effectors (Tesmer et al., 1997b; Slep et al., 2001; Chen et al., 2005; Tesmer et al., 2005). In addition, the interaction of Gα with RGS proteins and the mechanism of GAP activity have been extensively characterized structurally (Tesmer et al., 1997a; Slep et al., 2001; Chen et al., 2005). Finally, recent efforts have resulted in structures of dark-adapted (inactive) and light-activated rhodopsin, the archetypical GPCR of visual phototransduction most amenable to structural interrogation (Palczewski et al., 2000; Salom et al., 2006). Outlined below are aspects of this impressive collection of work especially germane to our understanding of Gα activation and deactivation.
Milestone GPCR/heterotrimer structures obtained by crystallography
The Gα Subunit. The nucleotide-binding pocket of the Gα subunit resides between two distinct domains: a Ras-like domain (named given its structural resemblance to the Ras superfamily of monomeric GTPases) and an additional, all α-helical domain composed of a structurally distinct six-helix bundle (Figs. 2A and 3A). Binding of GTP causes a structural rearrangement within three segments of Gα, called “switch” regions (I-III), resulting from favorable interactions with the γ-phosphate of the newly bound GTP (Lambright et al., 1994; Wall et al., 1998). Switch I serves as one of two connections between the Ras-like and α-helical domains. Switch II assumes a partially helical conformation in the active state and governs many of the interactions of Gα with Gβγ, effectors, RGS proteins, GoLoco motifs, and other nucleotide-state-selective binding partners (e.g., Kimple et al., 2002; Johnston et al., 2005; Johnston et al., 2006). Switch III assumes a loop structure found ordered only in the active conformation of Gα.
The structural conformations adopted by switches I-III upon GTP binding allows the Gα subunit to specifically recognize downstream effectors. Structures of Gα/effector complexes [Gαs/adenylyl cyclase, Gαt/PDEγ, Gα13/p115-RhoGEF, and Gαq/GRK2 (Tesmer et al., 1997b, 2005; Slep et al., 2001; Chen et al., 2005)] have revealed that a highly conserved hydrophobic cleft within GTP-bound Gα, formed by the α2 and α3 helices (Fig. 3A), serves as a universal site for effector engagement (for review, see Johnston et al., 2006). Additional effector-binding regions are formed by the α2/β4 and α3/β5 loops of Gα (Itoh and Gilman, 1991; Tesmer et al., 1997b; Slep et al., 2001). However, the precise nature of effector specificity remains unclear (Tesmer et al., 2005; Johnston et al., 2006).
The mechanism of intrinsic GTP hydrolysis and RGS protein-mediated acceleration of this activity have been delineated from structures of the GTPase transition state (GαGDP-AlF-4) in isolation (Fig. 3A) and bound to RGS proteins (Coleman et al., 1994; Sondek et al., 1994; Tesmer et al., 1997a; Slep et al., 2001), as well as reaction intermediates such as Gα bound to 5′-guanylylimidodiphosphate or GDP plus inorganic phosphate (Raw et al., 1997; Coleman and Sprang, 1999). Intrinsic GTP hydrolysis is mediated by a triad of conserved Gα residues (numbered as in Gαi1; Fig. 2A): Thr181 in switch I coordinates a Mg2+ ion that helps stabilize the γ-phosphate ion; Arg178 in switch I also aids in the stabilization of the leaving γ-phosphate ion; and Glu204 in switch II coordinates the critical nucleophilic water molecule responsible for hydrolysis of the γ-phosphate (Coleman et al., 1994; Sondek et al., 1994) (Fig. 3A). Because these residues are both necessary and sufficient for GTP hydrolysis, RGS protein binding does not introduce additional catalytic residues; rather, it stabilizes the transition state conformation, thus lowering the activational free energy required for the hydrolysis reaction (Berman et al., 1996; Tesmer et al., 1997a; Srinivasa et al., 1998).
The Gβγ Subunit. Dimerization between the Gβ and Gγ subunits of the G-protein heterotrimer is obligate in nature; Gβ requires Gγ to fold properly (Higgins and Casey, 1994). The Gβγ dimer only dissociates under denaturing conditions (Schmidt et al., 1992). The Gβ subunit begins with an extended N-terminal α-helix and mainly comprises a β-propeller fold (Fig. 3B), a structural motif found in many other proteins unrelated to the Gβ family (Neer et al., 1994; Li and Roberts, 2001). The β-propeller domain of Gβ is formed by seven individual segments of a ∼43-amino acid sequence known as a WD repeat motif (Fig. 2B). An arrangement of seven four-stranded antiparallel β sheets forms the β-propeller; however, a given WD repeat motif does not correspond exactly to any one blade. The β-propeller is completed by the connection of strands from the first and seventh WD repeats (Fig. 3B), with hydrophobic packing between blades contributing to the overall architecture. The Gγ subunit is an extended stretch of two α-helices joined by an intervening loop (Fig. 2C). Assuming no significant tertiary structure on its own, the N terminus of Gγ forms a coiled-coil interaction with the N-terminal α-helix of Gβ (Fig. 3, B and C); much of the remainder of Gγ binds along the outer edge of the Gβ toroid (Wall et al., 1995; Sondek et al., 1996). More recently, structures of Gβγ bound to phosducin (a regulatory protein), GRK2 (an effector), and SIRK (a non-natural peptide capable of disrupting effector activation) have defined the mode of Gβγ/effector interactions (Gaudet et al., 1996; Lodowski et al., 2003; Davis et al., 2005). It is noteworthy that the effector-binding site on Gβγ overlaps significantly with the region responsible for binding switch II of Gα near the central pore of the Gβ toroid (Fig. 3B). Additional interaction sites exist for each specific complex (Gaudet et al., 1996; Lodowski et al., 2003; Davis et al., 2005).
The Gαβγ Heterotrimer. The G-protein heterotrimer is formed by two principal sites of interaction between Gα and Gβγ (Fig. 3C). First, extensive burial of the β3/α2 loop and α2 helix (switch II) of Gα within six of the seven WD repeats (specifically the DA and BC loops) of Gβ, with a majority of interactions governed by a hydrophobic core centered around Trp211 of Gα (positioned deep within the α2/α3 cleft) and Trp99 of Gβ(numbered as in Gβ1) (Fig. 3B). This interaction buries approximately 1800 Å2 of solvent-accessible surface and forms the basis for Gβγ-mediated guanine nucleotide dissociation inhibitor activity (Higashijima et al., 1987) and competition for Gβγ binding between Gα-GDP and Gβγ-effectors. The structures of Gβγ bound to the effector-competing, non-natural peptide SIRK and a bona fide effector, GRK2, have now firmly established this region of Gβ as critical to effector recognition (Lodowski et al., 2003; Davis et al., 2005). Second, a Gα/Gβ interaction surface occurs between the side of the first β-propeller blade of Gβ (WD1 and -2; specifically the D strands and CD loops) and the extended N-terminal helix of Gα (Fig. 3, B and C), the latter being normally disordered in structures of isolated Gα subunits (Wall et al., 1995; Lambright et al., 1996; Wall et al., 1998). This interaction buries an additional ∼900 Å2 of solvent-accessible surface. Figure 3C illustrates the structure of Gαi1β1γ2 in its predicted membrane orientation with the Gα N terminus and Gγ C terminus, both sites of critical lipid modifications (Wedegaertner et al., 1995; Manahan et al., 2000), juxtaposed to the plasma membrane.

Heterotrimeric G-protein subunit secondary structure. Amino acid sequence alignments and secondary structure features from high-resolution structures of Gα (A), Gβ (B), and Gγ (C) subunits. Secondary structure assignments (α-helices, β-strands) and ruler numbering are derived from Gαi1 (PDB code 1GFI) (A), Gβ1 (PDB code 1TBG) (B), and Gγ1 (PDB code 1TBG) (C) and color-coded to match tertiary and quaternary structural representations of Fig. 3. A, the three conformationally flexible switch regions of Gα (including the entire α2 helix within switch II) are indicated by green dots within the ruler line; residues in Gα that contact Gβ are marked with red dots above the ruler line. Conserved Gα guanine base and phosphate contact positions are highlighted in purple (GAGE and DVGGQ motifs) and red (TCAT motif); the conserved arginine, threonine, and glutamate residues involved in GTP hydrolysis are highlighted in orange. B, the four β-strands that comprise each of the seven WD repeat segments within Gβ subunits are color-coded to match the tertiary structure of Gβ1 as represented in Fig. 3B. Residues in Gβ that contact Gγ or the switch regions of Gα are marked with gray or green dots, respectively; additional Gα contacts are marked with blue dots. C, residues in Gγ that contact Gβ are marked with red dots.
Rhodopsin. A crystal structure of a prototypical GPCR, bovine rhodopsin, was first determined in its inactive, dark-adapted state (Palczewski et al., 2000). This structure provided the first glimpse into the arrangement of the seven transmembrane (TM) architecture of GPCRs and has provided an excellent tool for homology model-based studies involving other GPCRs (e.g., Mehler et al., 2006; Zhang et al., 2006). The observed orientation of the TM helices within rhodopsin positions specific residues, previously identified as important for ligand binding in other GPCRs (e.g., the β2-adrenergic receptor), toward the central core of the seven transmembrane-domain topology. The highly conserved E/DRY motif, involved in the activation mechanism of many rhodopsin-like (class A) GPCRs (for review, see Flanagan, 2005; Rovati et al., 2007), was found within the rhodopsin structure to be engaged in an ion pair interaction between Glu134 and Arg135 residues, supporting the hypothesis that disruption of this bond and movement of TM6 is involved in receptor activation (Palczewski et al., 2000; Ballesteros et al., 2001). The remainder of the intramolecular interactions within the seven TM core of rhodopsin are dominated by hydrophobic interactions. Biophysical studies have suggested that this network of interactions must rearrange (probably involving movements of TM3, TM5, and TM6) during activation to allow G-protein coupling (Swaminath et al., 2005). However, the structure of light-activated rhodopsin has now been solved and, contrary to previous thought (Altenbach et al., 2001; Ghanouni et al., 2001), demonstrates only minor conformational changes within the TM helices (Salom et al., 2006). In contrast, alterations in the cytoplasmic loops that contact the rhodopsin-specific G-protein (transducin; Gαtβ1γ1) were noticed, most notably with the paths of ic2 and ic3, which become largely disordered upon activation. These results suggest that receptor activation leads to a relaxation within the intracellular loops allowing for an induced fit with the G-protein heterotrimer. It must be stressed, however, that the low resolution in these structures prevents a precise definition of the molecular determinants for G-protein coupling and activation (Salom et al., 2006); furthermore, neither the dark-adapted nor light-activated rhodopsin structures (Palczewski et al., 2000; Salom et al., 2006) were obtained in the presence of G-protein heterotrimer. Thus, hypothetical models based on these and other experimental results have been put forth to describe the molecular mechanism of receptor-mediated G-protein activation. Below, we detail two of these prevailing hypotheses and recent evidence in their favor.

Heterotrimeric G-protein tertiary and quaternary structure. A, the tertiary structure of Gαi1 (PDB code 1GFI), composed of a Ras-like domain (blue) and an all α-helical domain (yellow), is shown in a transition-state mimetic form bound to a molecule of GDP (magenta), magnesium ion (red), and tetrafluoroaluminate (AlF-4) ion (gray/blue sticks). The three critical switch regions (numbered SI to SIII) are colored green. Three essential catalytic residues that participate in the water-mediated GTP hydrolysis reaction are shown in orange: Arg178 and Thr181 in switch I as well as Gln204 in switch II. B, the structure of Gβ1γ2 (PDB code 1GP2) reveals the prototypical β-propeller fold of the Gβ1 subunit and the partially α-helical nature of the Gγ2 peptide (black); note the coiled-coil interaction between the N termini of these subunits. The Gβ1 subunit is colored according to the seven WD repeat segments (N-terminal helix, red; WD1, brown; WD2, yellow; WD3, magenta; WD4, teal; WD5, orange; WD6, gray; WD7, wheat). Under this color scheme, notice that each four-bladed propeller segment is composed of three blades from one WD repeat and one blade from the preceding WD repeat. The tryptophan (Trp99) in WD2 critical for interaction with Gαi1 is shown in sticks. Also shown are the two significant contact regions from Gαi1: the Gαi1 N-terminal α-helix (“N-end Gα”; blue) interacts along the outer edge of the Gβ1 toroid, whereas the switch II helix (green; Trp211 shown in sticks) interacts in the center and makes critical contacts with six of the seven WD repeats. C, the structure of the Gαi1·GDP/Gβ1γ2 (PDB code 1GP2) illustrates the molecular basis for the formation of a heterotrimeric G-protein. The heterotrimer is depicted in its proposed membrane-bound orientation with the Gβ1γ2 heterodimer. Gαi1 is colored as in A, with bound GDP in magenta and several key switch II residues that interact with Gβ1 represented as sticks (Lys209, Trp211, Ile212, and Phe215; green). The Gβ1 subunit is shown in red and the Gγ2 in gray. Key Gβ1 residues interacting with the Gαi1 switch II helix are represented in sticks (Trp99, Asp228, and Asp246; red). The C terminus of Gγ2 terminates with a CAAX motif that is isoprenylated (depicted as saw-tooth line) to increase association with plasma membrane.
Mechanism of Receptor-Mediated G-Protein Activation
Despite the immense efforts and resounding successes described above in discerning the structural aspects of G-protein signal transduction, the structural basis for heterotrimeric G-protein activation by GPCRs remains largely unknown. This deficit has arisen mostly from the inherent obstacles to purification and crystallization of receptors and receptor/Gαβγ complexes (for review, see Sarramegn et al., 2006). In lieu of such structural insights, biochemical approaches, such as site-directed mutagenesis and the use of synthetic peptides and protein chimera, have been used to identify regions within both receptors and Gαβγ heterotrimers critical to the activation process. Although these studies have mapped the receptor contact interface to the N terminus, C terminus, and the α4/β6 loop of Gα (Hamm et al., 1988; Onrust et al., 1997; Grishina and Berlot, 2000) along with the C termini of both Gβ and Gγ (Hou et al., 2000, 2001) (Fig. 4), they have provided little direct evidence for the actual mechanism of receptor-mediated activation of Gαβγ. Thus, these studies have led to hypothetical, and somewhat conflicting, models for receptor-mediated G-protein activation (Rondard et al., 2001; Cherfils and Chabre, 2003; Johnston et al., 2005; Van Eps et al., 2006). It is clear from the structures of dark-adapted (inactive) rhodopsin and Gαβγ, and their predicted orientations at the plasma membrane, that the receptor must act “at a distance” to invoke GDP release by communicating structural changes through the G-protein toward its GDP-binding pocket that resides ∼30 Å from the intracellular surface of the receptor (Bourne, 1997). To date, three distinct models have been proposed to describe how this process of long-range structural changes might occur. Below, we discuss two of these models: the “C-terminal latch” and “Gβγ lever” models (Rondard et al., 2001; Nanoff et al., 2006). We detail the foundations of each model and how each proposes distinct regions of the G-protein to be critical to activation. Finally, with recent results from our laboratory, we remark on the potential that these two models are indeed complementary with one another, rather than competing, in assembling the overall mechanism of receptor action. It is noteworthy that the third proposed model, termed the “gear-shift” model (Cherfils and Chabre, 2003), will not be discussed in detail here, because significant experimental results in its favor are currently lacking.

Regions within the G-protein heterotrimer critical to the receptor-mediated activation process. Several regions within both Gα and Gβγ have been implicated in receptor coupling and the activation process. The Gαi1·GDP/Gβ1γ2 structure (PDB code 1GP2) is depicted in the proposed membrane-bound, receptor-associated conformation from two different perspectives. A, lateral view of the heterotrimer highlights critical Gαi1 regions discussed and referenced in detail in the text. The coloring scheme is as follows: Gα N terminus (teal), β1-β3 strands including the β2/β3 loop (orange), α3/β5 loop (cyan), α4/β6 loop (yellow), β6 strand (blue), β6/α5 loop (green), and α5 helix (red). A molecule of GDP is depicted as sticks (magenta). B, rotation 90° about the y-axis reveals additional regions, including potential receptor contacts within Gβ1γ2. The Gαi1 switch II and β3/α2 and α2/β4 loops are colored brown. Within Gβ1γ2, proposed receptor contact sites of Gβ1 are colored salmon (including residues 31-39 in the N terminus as well as residues 280 to 340 of WD6 and WD7). The C terminus of Gγ2 (residues 60-71) is colored pink.
C Terminus “Latch” Hypothesis. One of the first regions within Gα identified as being critical to receptor-promoted activation was the extreme C terminus. Hamm et al. (1988) first demonstrated that synthetic peptides corresponding to the C terminus of Gαt could block rhodopsin-promoted activation, suggesting that the C terminus of Gα is a critical receptor-binding site. Additional peptides corresponding to the α4/β6 loop region of Gαt resulted in a similar attenuation, suggesting multiple sites of Gα/receptor contact. Alanine-scanning experiments confirmed these two regions (i.e., C terminus/α5 helix and α4/β6 loop) were essential for rhodopsin-promoted activation of Gαt (Onrust et al., 1997) (Fig. 4). Several subsequent studies have suggested that the extreme C terminus communicates through the extended α5 helix of Gα to invoke the structural changes necessary for GDP release. Marin et al. (2001) have shown that mutations to several residues in an inward-facing, buried surface of the α5 helix cause a dramatic increase in the basal and receptor-promoted nucleotide exchange rates of Gα. Moreover, disruption of the α5 helix (via insertional and deletional mutagenesis) results in a loss of rhodopsin-mediated activation of Gαt (Marin et al., 2002). For example, insertion of a five-glycine flexible repeat sequence in Gα between the α5 helix and extreme C terminus dramatically reduces receptor-promoted activation with little effect on receptor coupling (Natochin et al., 2001). Electron paramagnetic resonance (EPR) studies have suggested that the Gα C terminus moves into a more hydrophobic environment after AlF-4-mediated activation, perhaps resulting in an interaction with the α2/β4 loop (Yang et al., 1999). Together, these results suggest that activated receptor uses critical contacts with the C-terminal tail of Gα to elicit conformational changes in the α5 helix during nucleotide exchange (Fig. 5).
The α5 helix extends to the nucleotide binding pocket and connects with the β6 strand through the β6/α5 loop—a loop that makes several contacts to the guanine ring of the bound GDP molecule (Bohm et al., 1997; Sprang, 1997) (Fig. 4A). Within the β6/α5 loop resides a conserved threonine-cysteinealanine-threonine (TCAT) motif that mediates key contacts with GDP that are believed to stabilize the binding of GDP within Gα. Indeed, mutations within this region (Gαs-A366S, Gαi-A326S, Gαo-C325S) result in dramatically enhanced spontaneous nucleotide exchange rates (Thomas et al., 1993; Iiri et al., 1994; Posner et al., 1998) and are clinically manifested in pseudohypoparathyroidism and gonadotropin-independent precocious puberty in the case of Gαs-A366S (Iiri et al., 1994, 1998). The effect of the alanine-to-serine mutation on nucleotide exchange is thought to result from introduction of steric clash between the extended side chain of serine and the guanine ring of GDP. Thus, the TCAT motif within the β6/α5 loop may serve as a conserved regulator of nucleotide exchange. Overall, this prevailing model suggests that the receptor contacts the Gα C terminus and communicates structural changes through the α5 helix to modulate the conformation of the β6/α5 loop and its TCAT motif, ultimately resulting in the release of GDP via an exit route thought to be away from the “Gβγ face” of Gα (Kisselev et al., 1998; Oldham et al., 2006) (Figs. 5 and 6).
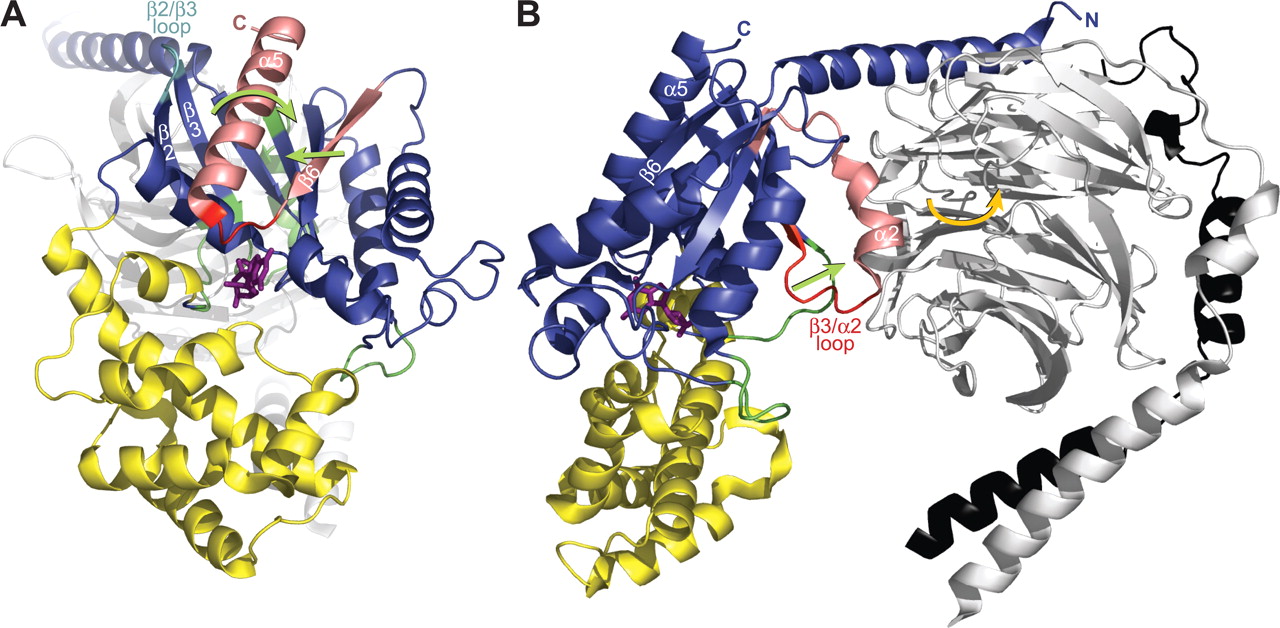
Proposed receptor-induced conformational changes in the G-protein heterotrimer leading to GDP release. Although the precise mechanism of receptor-catalyzed activation of heterotrimeric G-proteins remains incompletely resolved, several conformational changes within the G-protein have been proposed to occur as the result of agonist activation of the receptor. As detailed in the text, these conformational changes are thought to result in the release of GDP, the rate-limiting step in the activation process. A and B each represent a component movement, with the critical regions of Gα engaged in each movement highlighted in salmon and red. A, lateral view of the Gαi1·GDP/Gβ1γ2 heterotrimer (PDB code 1GP2) highlighting the Gαi1 subunit, which is colored similarly to Fig. 3. The critical β6 strand and α5 helix are colored salmon, and the β6/α5 loop connecting them is colored red. The bound GDP (sticks) is colored magenta. Green arrows indicate the proposed receptor-induced conformational changes in these regions of Gα. Work from our laboratory suggests a displacement in the β6 strand upon binding of a receptor-derived peptide with GEF activity (Johnston and Siderovski, 2007). Work from several laboratories suggests a receptor-induced rotation in the α5 helix (Kisselev et al., 1998; Marin et al., 2002; Oldham et al., 2006). Both of these conformational changes are thought to propagate into a movement of the β6/α5 loop, thus weakening the stabilizing bonds it makes with GDP and resulting in a decreased affinity of the heterotrimer for nucleotide. Through direct contacts with the α5 helix, the β2/β3 loop (cyan) may serve as a critical regulator of the basal state of the α5 helix (Marin et al., 2001). B, rotation of the heterotrimer 90° about the y-axis reveals another proposed mechanism of receptor-mediated activation. In this view, the switch II (α2) helix is colored salmon and the β3/α2 loop red. The Gβ1 subunit is colored gray and the Gγ2 black. As the receptor is thought to make contacts with both Gα and Gβγ (see Fig. 4), conformational changes in the receptor are proposed to translate into a tilting (orange arrow) of the Gβγ subunit relative to Gα (Iiri et al., 1994; Rondard et al., 2001). This tilting (or translation) of G-protein subunits relative to one another may displace switch II and result in the β3/α2 loop being pulled away (green arrow) from the GDP-binding pocket (Johnston et al., 2005). Because the β3/α2 loop is thought to serve as an occlusive barrier or “lip” to the release of GDP, this conformational change would create a more feasible exit route for the nucleotide in a direction that is toward the Gβγ subunit (Rondard et al., 2001; Johnston et al., 2005). The proposed mechanisms depicted in these two panels may work together (and perhaps synergistically) to regulate nucleotide exchange by Gα: the mechanism shown in A would weaken the affinity for bound GDP, and that described in B would create a pathway for GDP release (Onrust et al., 1997; Van Eps et al., 2006; Johnston and Siderovski, 2007).
A recent study by Hamm and colleagues (Oldham et al., 2006) has added further support for the “latch” hypothesis and the involvement of the α5 helix in transmitting structural changes to the GDP binding pocket. By examining the dynamics of an EPR probe systematically attached to several individual Gα residues, the authors demonstrated that labeled residues within the α5 helix undergo specific receptor-mediated changes in EPR spectra, indicative of a perturbation in the conformation of this helix. The authors suggest that the receptor induces a rigid-body movement, specifically a rotation-translation function, in the α5 helix that preserves its overall helical structure (Oldham et al., 2006) (Fig. 5A). Further experiments suggested that this effect constitutes a ∼5Å change in the distance distribution in the α5 helix. Moreover, insertion of a flexible glycine linker between the α5 helix and C terminus reduced the receptor-mediated changes in EPR spectra of specific α5 helix residues (Oldham et al., 2006). These results strongly suggest the receptor uses contacts with the extreme C terminus to communicate structural changes through the α5 helix presumably to the β6/α5 loop to induce the release of GDP. However, these studies also revealed changes in the EPR spectra of labeled residues in the β2/β3 loop and the β6 strand, suggesting that these regions are mobile during receptor activation and thus may also play a crucial role in receptor-mediated activation of Gα (Fig. 5A).
Beyond the α5 helix, several studies have implicated other regions of the Gα subunit in transmitting the necessary conformational changes to the β6/α5 loop for GDP release. The α3 helix, which connects the α3/β5 loop to switch III, was found by Berlot and colleagues (Marsh et al., 1998; Grishina and Berlot, 2000) to be important for receptor activation of Gαs. A network of β-strands within Gα (β1, β2, β3; Fig. 4A), which connect the N terminus, P-loop, and switch I-II regions, has been shown to regulate the intrinsically slow rate of spontaneous nucleotide exchange in Gαt (Thomas et al., 2001). Finally, the β6 strand, through results of mutational studies (Onrust et al., 1997), is also considered an essential component of rhodopsin-mediated activation of Gαt (Figs. 4, 5 and 6).
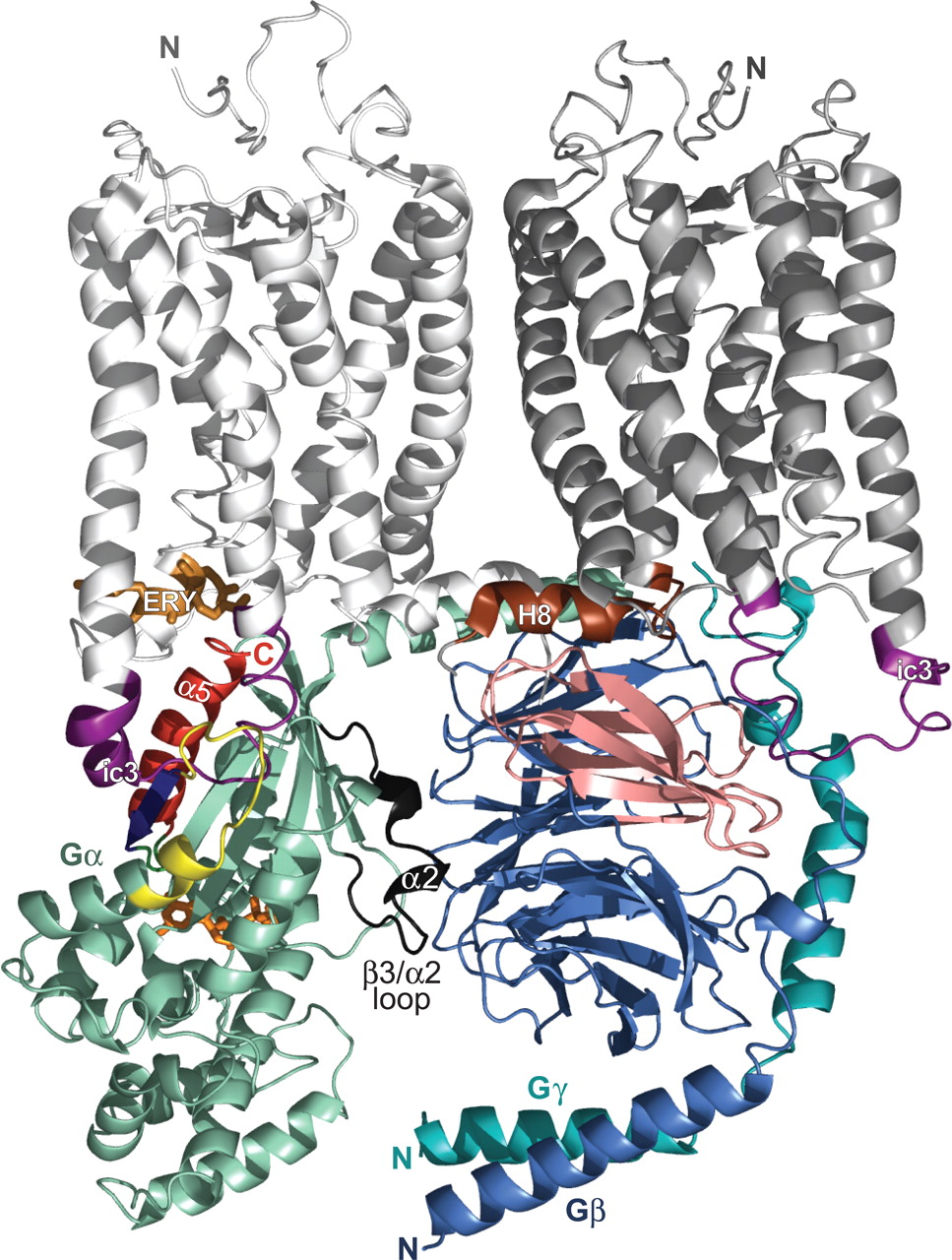
Relative orientation and interactions of the transducin G-protein heterotrimer with a rhodopsin dimer. A recently determined structure of dark-adapted rhodopsin in a trigonal crystal form (PDB code 2I36) revealed a dimeric interface proposed to be a physiologically functional entity (Salom et al., 2006). This structure is depicted with protomer 1 colored white and protomer 2 colored gray. The ic3, universally critical to receptor activity (Bourne, 1997), is colored purple in both protomers. Peptides isolated from the ic3 loop of several receptors have been found to directly interact with the Gα subunit and elicit modest activation (Nanoff et al., 2006; Johnston and Siderovski, 2007). The ic3 loop has also been demonstrated to interact with the Gβγ dimer (Taylor et al., 1996; Wu et al., 2000). The C-terminal tail [including “helix 8” (H8) of rhodopsin (brown)] has recently been implicated in direct interaction with the Gβ subunit (Mahon et al., 2006). The Gαtβ1γ1 heterotrimer (PDB code 1GOT) has been positioned in its presumed orientation at the receptor/membrane interface. The color scheme for the transducin heterotrimer follows that of Fig. 4 with slight modifications: the Gα subunit is colored greencyan (with its bound GDP as orange), the Gβ is marine-blue (with its proposed receptor-interaction site colored salmon), and the Gγ is teal. In addition, switch II and the β3/α2 loop are colored black and the α4/β6 loop is colored yellow. Although this “docking” is mostly artistic in nature, its position is loosely constrained by our recent identification of molecular determinants within the N terminus of the dopamine D2-receptor ic3 loop binding to the α4/β6 region of Gαi1 (Johnston and Siderovski, 2007). When constrained by this particular interaction site, our rendition of the GPCR/G-protein interaction positions the heterotrimer in a way that satisfies many of the previous results discussed in the text. It is noteworthy that the α5 helix and C terminus of Gα (red) are positioned within reasonable distance to the ERY motif (copper sticks) of the rhodopsin protomer 1 that is essential for receptor activity (Palczewski et al., 2000; Rovati et al., 2006). The proposed Gβ contact site (salmon) (Taylor et al., 1996; Hou et al., 2001) is positioned within bonding distance to the two receptor regions previously proposed to directly interact with Gβγ: the ic3 loop and the C-terminal region (Wade et al., 1996; Wu et al., 1998; Mahon et al., 2006). In addition, in this orientation, the lipid modifications (not depicted) on the Gα N terminus (myristol and/or palmitoyl groups) and the Gγ C terminus (isoprenyl group) are positioned to be inserted within the plasma membrane.
Recent results from our laboratory have added more direct evidence for the involvement of the β6 strand in receptor-mediated Gα activation. We determined the structure of Gαi1 bound to a peptide (D2N) corresponding to the N-terminal portion of the third intracellular loop (ic3) of the dopamine D2-receptor (Johnston and Siderovski, 2007). D2N, in common with several other receptor loop peptides, exhibits modest GEF activity on Gα subunits in vitro with a selectivity profile analogous to the cognate full-length receptor (Nanoff et al., 2006). We found that D2N binds to the α4/β6 loop region of Gα, previously identified as a critical receptor contact site important for Gα-coupling selectivity (Hamm et al., 1988; Onrust et al., 1997; Slessareva et al., 2003; Oldham et al., 2006) (Figs. 4 and 6). Binding of D2N results in a displacement of the β6 strand compared with the native Gαi1 structure (Johnston and Siderovski, 2007). These results suggest that the receptor uses the β6 strand, perhaps in combination with the α5 helix, to communicate structural changes to the β6/α5 loop and thereby disrupt contacts to GDP, resulting in nucleotide release (Fig. 5A).
Gβγ “Lever” Hypothesis. Whereas the above model of receptor-catalyzed nucleotide exchange relies solely on receptor/Gα contacts, an alternative model has been proposed that evokes Gβγ as an active participant in the exchange reaction (Iiri et al., 1998; Rondard et al., 2001; Johnston et al., 2005; Johnston and Siderovski, 2007). In the previously considered “latch” model, Gβγ may serve merely to aid in heterotrimer association with the plasma membrane [via Gγ prenylation (Iñiguez-Lluhi et al., 1992; Muntz et al., 1992)] and/or direct interaction with the receptor (Kisselev et al., 1999), thus playing only a passive role in the actual activation event. However, several observations would suggest that Gβγ indeed has a more active role in the activation mechanism. The requirement of Gβγ for proper receptor coupling and Gα activation has long been established (Fung, 1983; Fung and Nash, 1983). Receptor contacts established with both Gα and Gβγ (Figs. 4 and 6) could be used to transmit conformational changes in both subunits relative to one another to establish a GDP exit route.
As detailed above, several regions of the Gα subunit have been proposed to directly contact receptor (Fig. 4A). Likewise, efforts have been made to determine direct interactions between receptor and the Gβγ subunit. Intracellular regions of GPCRs [i.e. the third intracellular loop (ic3) and C-terminal tail], have been implicated in direct interaction with Gβγ (Taylor et al., 1996; Wu et al., 1998; Mahon et al., 2006), and may engage the N terminus and sixth WD repeat segment of Gβ (Fig. 4B), both of which are located on the outer surface and contained within the inferred receptor contact face (Hou et al., 2001). In this way, the activated receptor would undergo a conformational change that, in turn, would use contacts with Gβγ as a “lever” to indirectly induce conformational changes in Gα (Fig. 5B). Mutational experiments using Gαt have demonstrated that alanine substitution at several Gα/Gβγ contact sites in the switch II/Gβ interface attenuate rhodopsin-promoted activation without affecting Gαβγ heterotrimer formation (Ford et al., 1998). In addition, disruption of a salt bridge mediated by Lys206 (Gα switch II) and Asp228 (Gβ) completely abolishes β-adrenergic-mediated activation of Gαs without disrupting heterotrimer formation (Rondard et al., 2001). Together, these results suggest that an activation model evoking Gβγ may indeed apply universally to all Gα families. Finally, the established mechanism of action of GEFs for monomeric GTPases involves direct reorientation of switch I and II to establish a feasible GDP exit route (Kawashima et al., 1996; Cherfils and Chardin, 1999; Rossman et al., 2005). Because the receptor cannot rationally be in direct contact with the Gα switch regions while Gα is ensconced within the heterotrimer (Fig. 6), Gβγ would have to serve as a surrogate contact site for receptor-mediated manipulation of these key regions within Gα. The model described herein has been referred to as the “Gβγ lever” hypothesis, in which the receptor actively uses Gβγ as an “adjuvant catalyst” in the nucleotide exchange reaction (Rondard et al., 2001). Gβ contacts with Gα switch II (Fig. 3B) are proposed to lever open switch II away from the GDP binding site after a receptor-mediated tilt of Gβγ away from Gα. Reorientation of switch II (α2 helix) in this way would, by necessity, also reorient the β3/α2 loop thought to serve as an “occlusive lip” normally preventing GDP release (Iiri et al., 1994). GDP would, in turn, be more efficiently released, probably with an ejection path toward the “Gβγ face” of Gα (Fig. 5B).
Previous work from our laboratory using a phage display-derived peptide (KB-752) with inherent GEF activity on isolated Gαi subunits provided direct biochemical and structural evidence for the “Gβγ lever” hypothesis. KB-752, originally identified as a GDP-selective Gα-binding peptide, binds within a hydrophobic cleft created by the α2 and α3 helices of Gα, the same site used for binding Gβγ (Johnston et al., 2005). Binding of KB-752 causes a dramatic displacement of switch II compared with its orientation with the heterotrimeric complex (Johnston et al., 2005). Furthermore, this movement in switch II results in a concurrent displacement of the “occlusive” β3/α2 loop away from the GDP binding pocket—a displacement that is stabilized by contacts between this loop and KB-752. Thus, the KB-752 GEF peptide seems to serve as a Gβγ “surrogate” in mimicking the proposed Gβγ-mediated displacement of the β3/α2 loop upon receptor activation (Johnston et al., 2005; Johnston and Siderovski, 2007).
Studies from Hamm and colleagues, again using EPR analysis, have recently added further support to structural changes within the Gα-Gβγ interface (i.e., β3/α2 loop, α2 helix, α2/β4 loop) underlying receptor-mediated activation (Van Eps et al., 2006). When complexed with activated rhodopsin, spin-labeled residues within Gα at the interface with Gβγ underwent dramatic rotational changes, suggesting that this region of Gα is conformationally altered during receptor-mediated activation. The most dramatic changes were seen in the α2/β4 loop, whereas more moderate changes occurred in the β3/α2 loop and switch II (α2) helix (Van Eps et al., 2006). Because these regions on Gα are not considered viable receptor contact sites per se, rhodopsin-induced changes here suggest an allosteric regulatory mechanism, possibly through the proposed Gβγ “levering.”
Model Convergence. The two models detailed above represent the prevailing thought regarding the mechanism of receptor-promoted activation of heterotrimeric G-proteins, although other hypotheses have also been presented in the literature (Remmers et al., 1999; Cherfils and Chabre, 2003). Although these models are often presented as starkly opposing mechanisms, they may not actually be mutually exclusive. Indeed, Bourne and colleagues (Onrust et al., 1997), in particular, while championing the idea of the “Gβγ lever” have suggested that multiple mechanisms may play complementary roles in the overall action of activated receptors.
Recent results from our laboratory now lend experimental evidence for such a case of model convergence. As previously mentioned, we determined the structure of Gαi1 bound to D2N, a receptor-derived peptide with demonstrated GEF activity (Johnston and Siderovski, 2007). This structure also included the KB-752 peptide described above. It is noteworthy that, whereas D2N and KB-752 each possess modest GEF activity alone, a combination of both peptides yields a synergistic GEF activity on Gαi1 (Johnston and Siderovski, 2007). The D2N peptide binds to and displaces the β6 strand, which connects to the α5 helix via the β6/α5 loop; simultaneously, KB-752 “pulls” the β3/α2 loop away from the GDP binding pocket. These findings suggest that activated receptors could use a similar multipronged approach to cause structural changes in several Gα regions acting together to invoke GDP release. In accordance with the overwhelming data supporting a role for the α5 helix in this process, we hypothesize that the receptor uses contacts with both the β6 strand and the α5 helix to ultimately disrupt the critical contacts between the β6/α5 loop and GDP. However, the synergistic GEF activity observed by concomitant application of D2N and KB-752 peptides onto Gα suggests that modulation of the β6/α5 loop alone is insufficient for maximal GEF activity; therefore, modulation of the β3/α2 loop serves as a second key determinant of GDP release. Thus, we suggest that the receptor uses direct contacts with the β6 strand and α5 helix to release guanine base contacts with the β6/α5 loop (Fig. 5A), coincident with Gβγ-mediated levering of the β3/α2 loop to remove the occlusive lip blocking GDP release (Fig. 5B), thereby causing maximally efficient nucleotide release.
Receptor Dimerization. The precise mechanism of receptor-catalyzed G-protein activation is likely to be quite complex. Another feature of this process that remains intensely debated is the existence of receptor dimerization (Prinster et al., 2005; Fotiadis et al., 2006). In the past, a single receptor was thought sufficient to activate a G-protein heterotrimer. However, determination of the rhodopsin structure, as a dimer, illustrated the G-protein-facing surface to be relatively narrow compared with the width of the Gαβγ heterotrimer (Palczewski et al., 2000; Salom et al., 2006) (Figs. 5 and 6), adding support to the argument that a receptor dimer is necessary for efficient G-protein activation (Angers et al., 2002). Dimerization can clearly be critical for certain aspects of receptor function such as membrane targeting or ligand recognition (e.g., White et al., 1998; Waldhoer et al., 2005); however, whether the receptor dimer is absolutely required for G-protein activation remains controversial (Chabre and le Maire, 2005). Although far from conclusive, our low-resolution model depicting key Gα and Gβγ interaction sites occurring within the rhodopsin dimer (Fig. 6) supports a role for dimerization in proper G-protein coupling and activation. We do not intend to draw conclusions regarding any requirement for both receptor protomers to bind agonist or the allosteric regulation (e.g., cooperativity) that may result (Schwartz and Holst, 2006); rather, we suggest that each receptor protomer underlies specific contacts with Gα and/or Gβγ that may not be achieved by a monomeric receptor and are therefore required for efficient activation.
Concluding Remarks and Future Directions
The importance to human physiology of GPCR signaling through heterotrimeric G-proteins cannot be overstated. These receptors and the pathways regulated by activated G-proteins are crucial to a wide variety of cellular responses, underlie the etiology of many serious pathophysiologies, and represent the molecular target for many pharmacotherapeutic agents. Although an amazing amount of work has led to our current understanding of the structural basis for much of the G-protein signaling cycle, the precise mechanism of receptor-mediated activation remains incompletely defined. Several models, described herein, have been proposed to depict this event.
Continued efforts should one day yield the “holy grail” of a high-resolution structure of a receptor/Gαβγ complex, ultimately revealing to atomic resolution the structural basis for this ubiquitous event of receptor GEF activity. Such a feat will undoubtedly greatly enhance continued drug discovery and design with GPCR targets, although prospects for achieving such a monumental accomplishment might be limited given the inherent conformational flexibility of these receptors and the dynamic nature of the G-protein activation process. Certain technical “tricks” could enhance efforts to trap the receptor/Gα-GDP/Gβγ complex into a state of non-productive or “stalled” activation that would be more stable and thus suitable for crystallization: application of stabilizing ligands such as inverse agonists (Kenakin, 2004), mutation to the receptor/Gα or receptor/Gβγ interface to increase the affinity of the overall complex, or mutation to Gα (e.g., Wall et al., 1998) or Gβγ (e.g., Rondard et al., 2001) to limit conformational changes in the heterotrimer but preserve receptor association. Such analyses would probably reveal the structural determinants for complex formation between receptor and heterotrimer, allowing one to manipulate one or more aspects of their interface for subsequent studies—perhaps even rational drug design.
However, to provide the full picture of the structural rearrangements induced by receptor that provoke GDP release, one would require the structural determinants of the transition-state reaction intermediate as represented by a receptor-bound heterotrimer depleted of nucleotide. Because the activation process is undoubtedly highly dynamic in nature, with the transition state probably a highly transient event, capturing such a conformation in a crystal structure may prove more difficult yet. Our work using both non-native and receptor-derived peptide GEFs (Johnston et al., 2005; Johnston and Siderovski, 2007) have been unsuccessful in promoting a crystallization-compatible, nucleotide-free state of the Gα subunit and thus have not yet revealed profound structural alterations within the nucleotide binding pocket per se. The crystal structure of a Gαi1 mutant (A326S) with a dramatically enhanced intrinsic nucleotide exchange rate revealed a state only partially occupied with GDP; however, no significant alterations in the overall nucleotide binding pocket were observed either (Posner et al., 1998). Although these crystallography results suggest that gross alterations in the nucleotide binding pocket may not be required for nucleotide release, recent NMR analysis of a receptor-bound, “nucleotide empty” Gα (under activation conditions) has revealed significant line broadenings in the obtained spectra (Abdulaev et al., 2006), suggesting that the nucleotide-free form represents a dynamic intermediate state. It is noteworthy that these structural changes were specific to conditions incorporating activated receptor, highlighting the critical role of receptor-induced conformational changes. Strategies designed to trap stable, receptor-bound, and nucleotide-free Gαβγ complexes suitable for crystal formation will thus be crucial to ultimately understand the precise conformational changes induced by activated receptor leading to nucleotide exchange.
Acknowledgments
We thank Dr. Francis Willard for critical appraisal of this review and the on-going support of the UNC Biomolecular X-ray Facility and UNC Structural Bioinformatics Core.
Footnotes
- Received January 20, 2007.
- Accepted April 10, 2007.
The writing of this review was made possible by generous funding support (F32-GM076944, R01-GM062338, and R01-GM074268) from the National Institute of General Medical Sciences.
ABBREVIATIONS: GPCR, G-protein-coupled receptor; GEF, guanine nucleotide exchange factor; GAP, GTPase-accelerating protein; RGS, regulator of G-protein signaling; TM, transmembrane domain; EPR, electron paramagnetic resonance; ic3, third intracellular loop; PDB, Protein Data Bank.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}