


Abstract
A fundamental question regarding receptor-G protein interaction is whether different agonists can lead a receptor to different intracellular signaling pathways. Our previous studies have demonstrated that although most β2-adrenoceptor agonists activate both Gs and Gi proteins, fenoterol, a full agonist of β2-adrenoceptor, selectively activates Gs protein. Fenoterol contains two chiral centers and may exist as four stereoisomers. We have synthesized a series of stereoisomers of fenoterol and its derivatives and characterized their receptor binding and pharmacological properties. We tested the hypothesis that the stereochemistry of an agonist determines selectivity of receptor coupling to different G protein(s). We found that the R,R isomers of fenoterol and methoxyfenoterol exhibited more potent effects to increase cardiomyocyte contraction than their S,R isomers. It is noteworthy that although (R,R)-fenoterol and (R,R)-methoxyfenoterol preferentially activate Gs signaling, their S,R isomers were able to activate both Gs and Gi proteins as evidenced by the robust pertussis toxin sensitivities of their effects on cardiomyocyte contraction and on phosphorylation of extracellular signal-regulated kinase 1/2. The differential G protein selectivities of the fenoterol stereoisomers were further confirmed by photoaffinity labeling studies on Gs,Gi2, and Gi3 proteins. The inefficient Gi signaling with the R,R isomers is not caused by the inability of the R,R isomers to trigger the protein kinase A (PKA)-mediated phosphorylation of the β2-adrenoceptor, because the R,R isomers also markedly increased phosphorylation of the receptor at serine 262 by PKA. We conclude that in addition to receptor subtype and phosphorylation status, the stereochemistry of a given agonist plays an important role in determining receptor-G protein selectivity and downstream signaling events.
Differential activation of receptors to specific signaling pathways has evolved to be a paradigm in pharmacological theory that can be translated into clinical relevance (Kenakin, 2004, 2007; Mailman, 2007; Urban et al., 2007; Violin and Lefkowitz, 2007). As an archetypical member of the G protein-coupled receptor (GPCR) superfamily, β2-adrenergic receptor (β2-AR) couples dually to Gs and Gi proteins, resulting in opposing effects on cardiac myocyte contractility and viability (Xiao et al., 1995, 1999; Zhu et al., 2001). In congestive heart failure, impaired β-AR response is often associated with increased Gi signaling (Feldman et al., 1988; Bohm et al., 1994) and selective down-regulation of β1-AR (higher β2/β1 ratio) (Bristow et al., 1986, 1993). Previous studies have demonstrated that disrupting Gi signaling with pertussis toxin (PTX) restores the markedly depressed β2-AR contractile response in two rat heart failure models, and that a full β2-AR agonist, fenoterol, which selectively activates β2-AR-coupled Gs signaling, reverses the diminished β2-AR inotropic effect in myocytes from failing spontaneously hypertensive rat hearts in the absence of PTX (Xiao et al., 2003). In vivo studies have further demonstrated that prolonged use of fenoterol not only improves cardiac function but also retards cardiac maladaptive remodeling, and that the overall beneficial effects of fenoterol with β1-AR blockade are greater than the salutary effects of β1-AR blockade alone in a rat chronic heart failure model-induced by myocardial infarction (Ahmet et al., 2004, 2005, 2008). These studies suggest that selective activation of the β2-AR-coupled Gs signaling may provide a useful therapeutic target for the treatment of congestive heart failure.
Although the pharmaceutical preparation of fenoterol is a racemic mixture of its R,R and S,S enantiomers (rac-fenoterol), our recent studies have shown that the R,R enantiomer is the only active isoform in receptor binding and cardiomyocyte contraction assays (Beigi et al., 2006; Jozwiak et al., 2007). A cohort of fenoterol derivatives including the R,R, R,S, S,R, and S,S isomers of fenoterol were synthesized (Beigi et al., 2006; Jozwiak et al., 2007). Using some of these compounds, we attempted to examine the hypothesis that the stereochemistry of an agonist determines functional selectivity of a given receptor coupling to different G protein(s) and resultant activation of subset(s) of downstream signaling pathways.
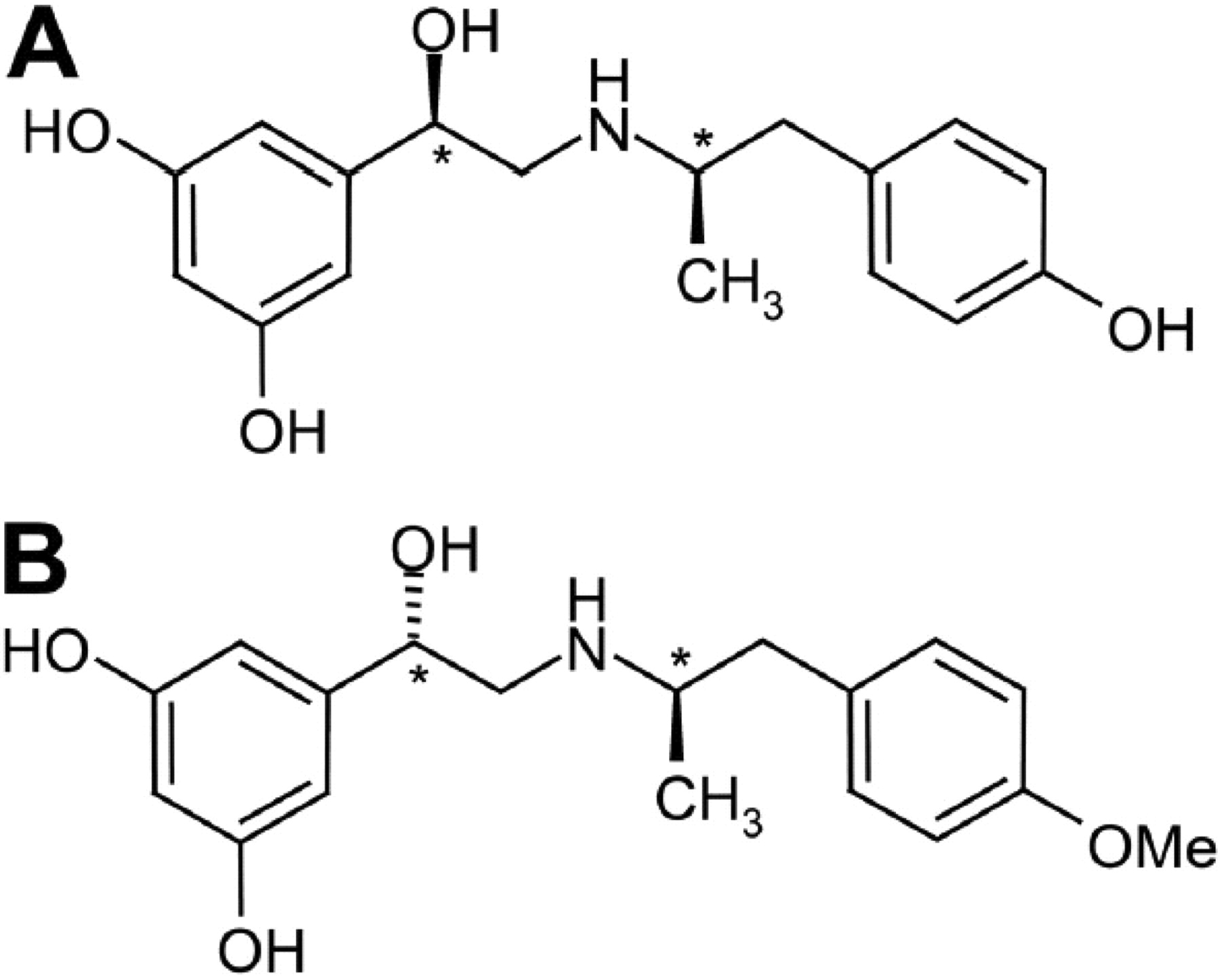
Chemical structures of fenoterol and methoxyfenoterol. A, (R,R)-fenoterol. B, (S,R)-methoxyfenoterol. Chiral centers are indicated with asterisks.
Materials and Methods
Compounds and Reagents. A series of stereoisomers and derivatives of fenoterol, including the R,R and S,R isomers of fenoterol and methoxyfenoterol (see Fig. 1 for structures), were synthesized into enantiomeric purity. The detailed procedures of the chemical synthesis and the receptor binding affinities of the compounds have been reported previously (Beigi et al., 2006; Jozwiak et al., 2007). Zinterol was kindly supplied by Bristol-Myers (Evansville, IN). ICI 118,551, (-)-isoproterenol (ISO), PTX, and other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cardiomyocyte Isolation, Cell Culture, and Adenoviral Infection. Cardiac myocytes were isolated from 2- to 4-month-old male Sprague-Dawley rats using a standard enzymatic technique and then were cultured in medium 199 containing 5 mM creatine, 2 mM l-carnitine, 5 mM taurine, 25 mM HEPES, 0.01% insulin-transferrin-selenium X, and 1% penicillin-streptomycin on dishes precoated with laminin. Human embryonic kidney (HEK) 293 cells purchased from the American Type Culture Collection (Manassas, VA) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Cells were incubated at 37°C under an atmosphere of 5% CO2. Adenovirus-mediated gene transfer was implemented by infecting the cells with 100 multiplicity of infection of adenovirus carrying the human β2-AR gene (Zhu et al., 2001).
Cardiomyocyte Contractility. Freshly isolated cardiac myocytes were perfused with a buffer containing 137 mM NaCl, 4.9 mM KCl, 1.2 mM MgCl2,1 mM NaH2PO4, 1 mM CaCl2, 20 mM glucose, and 20 mM HEPES, pH 7.4, and electrically stimulated at 0.5 Hz at 23°C. Cell length was monitored by an optical edge-tracking method (Beigi et al., 2006; Jozwiak et al., 2007). Measurements were made under steady-state conditions before and after exposure of the myocyte to a single concentration of agonist. The specificity of the agonist toward β2-AR was assessed by the ability of the response to be blocked by ICI 118,551 (10-7 M), a selective β2-antagonist. In a subset of experiments, aliquots of cells were incubated with PTX (0.75 μg/ml at 37°C for 3 h) to block Gi signaling as described previously (Xiao et al., 1995). The complete disruption of Gi function was confirmed by the insensitivity of the β1-adrenergic stimulatory response to carbachol.
Photoaffinity Labeling of G Proteins. Photoaffinity labeling of G proteins was performed as described previously (Xiao et al., 1999) with minor modifications. In brief, rat heart membranes were prepared by differential centrifugation and stored in a buffered sucrose solution at -80°C. The labeling reaction was allowed to proceed at 25°C for 7 to 20 min in a mixture containing 30 mM HEPES, pH 7.4, 30 mM NaCl, 0.1 mM EGTA, 1 mM benzamidine, 1 mM reduced glutathione, MgCl2 (5 mM for Gs and 1 mM for Gi), GDP (0.001 mM for Gs and 0.003 mM for Gi), unstimulated or agonist-prestimulated membranes (150-200 μg), and [γ-32P]GTP-azidoanilide (2-3 μCi; ALT Bioscience, Lexington, KY) and stopped by cooling on ice. After removing the unbounded labels, the membranes were irradiated with UV light (254 nm, 100 W, 6 cm, 1 min) and solubilized in 2% SDS. The clarified supernatants were incubated with rabbit antisera (4-10 μl) raised against Gαs (United States Biological, Swampscott, MA), Gαi2 (Abcam, Cambridge, MA), and Gαi3 (Santa Cruz Biotechnology, Santa Cruz, CA) in standard radioimmunoprecipitation assay buffer overnight at 4°C and subsequently with protein-A/G agarose (Calbiochem, La Jolla, CA). Beads were washed according to standard procedures and then boiled for 10 min in Laemmli sample buffer. The resultant supernatants were subjected to Urea/SDS-polyacrylamide gel electrophoresis. Radiolabeled proteins were visualized by autoradiography.
Western Blot Analysis. Solubilized proteins were analyzed in a denaturing polyacrylamide gel, electrotransferred to a polyvinylidene difluoride membrane, and immunoblotted with the appropriate primary antibody, followed by incubation with a peroxidase-conjugated secondary antibody. Films were exposed to enhanced chemiluminescence (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) reaction. The intensities of the gel bands were quantified using the NIH ImageJ software (http://rsbweb.nih.gov/ij/). Extracellular signal-regulated kinase (ERK) 1/2 activation was detected using the phospho-p44/42 mitogen-activated protein kinase (Thr202/Tyr204) antibody (1:1000; Cell Signaling Technology, Danvers, MA). Total ERK1/2 was detected by reprobing the membrane with the p44/42 mitogen-activated protein antibody (1:1000; Cell Signaling Technology). Phosphorylation of the PKA site serine 262 on β2-AR was detected using an anti-p-Ser262 monoclonal antibody at 1 μg/ml (generously provided by Dr. Richard B. Clark of The University of Texas, Houston, Medical School, Houston, TX; Tran et al., 2004). Total β2-AR was detected with the anti-C-tail antibody (1:500, Santa Cruz) recognizing the carboxyl terminus of β2-AR.
Statistical Analysis. Results are expressed as mean ± S.E. Student's t test was performed to compare the means between two groups and one-way analysis of variance for multiple group comparison followed by post hoc analysis with Dunnett's test. For each treatment group in the contractility study, the concentration-response curves were prepared using the Prism software (GraphPad Software Inc., San Diego, CA), followed by statistical analysis with curve-fitting using the three parameter logistic fixed-bottom model with constraints (bottom = 100; top < 360; Hill slope, <1.8) and calculation of the logEC50 ± S.E. and the P values. A value of P < 0.05 was considered statistically significant.
Results
In our previous studies, we characterized the binding affinities to β-ARs for the 26 stereospecific fenoterol derivatives (Jozwiak et al., 2007). The binding affinities reported previously to β1- and β2-AR for the R,R and S,R isomers of fenoterol and methoxyfenoterol are shown in Supplementary Table 1. Here, the specificity of these compounds was further assessed pharmacologically using cardiomyocyte contractility. Preliminary experiments have shown that near maximal contractile response could be achieved by the R,R isomers at 0.5 μM and by the S,R isomers at 10 μM. The selectivity of these compounds toward β2-AR was assessed by the inhibitory effect of ICI 118,551 (10-7 M). As shown in Fig. 2, all of these compounds could produce contractile responses that were blocked by ICI 118,551, suggesting that they are selective to β2-AR. These results are consistent with the binding affinity data.
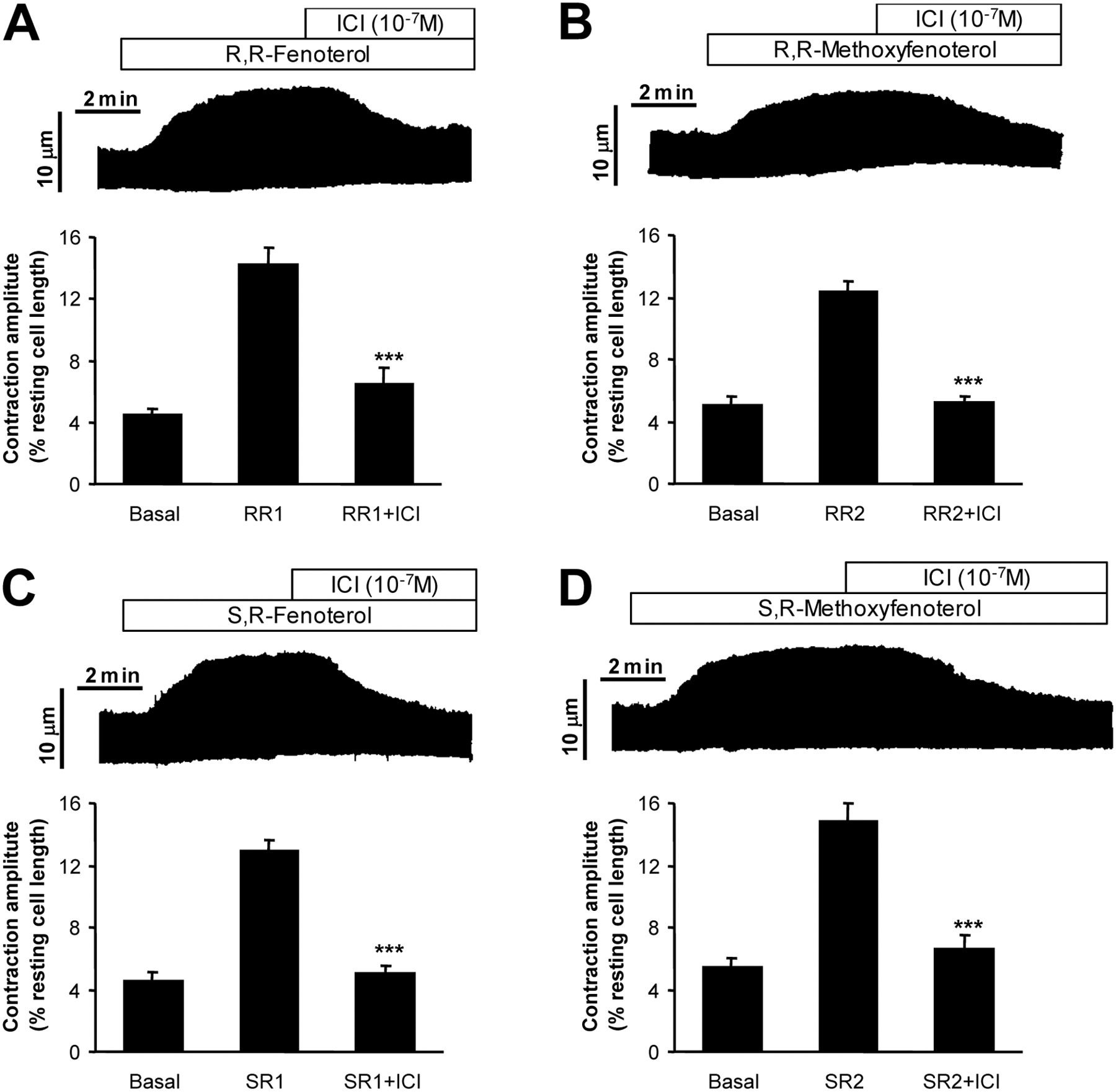
β2-AR selectivity of the fenoterol compounds in myocyte contraction. Single ventricular myocytes from rats were stimulated electrically. The contraction amplitude of the cell in response to the fenoterol compound: (R,R)-fenoterol (RR1, 5 × 10-7 M) (A), (S,R)-fenoterol (SR1, 10-5 M) (B), (R,R)-methoxyfenoterol (RR2, 5 × 10-7 M) (C), and (S,R)-methoxyfenoterol (SR2, 10-5 M) (D) followed by ICI 118,551 (ICI, 10-7 M) were recorded. The top part for each panel depicts a typical chart recording, and the averaged data (n = 6-9 independent observations on cells from 4-6 hearts for each data point) are shown at the bottom. ***, P < 0.001 compared with the corresponding agonist-stimulated contraction amplitude under the steady-state condition without ICI 118,551.
The full concentration-response profiles for the fenoterol compounds stimulated contraction with or without PTX treatment are shown in Fig. 3. All four stereoisomers of fenoterol and methoxyfenoterol caused a concentration-dependent increase in the contraction amplitude of adult rat cardiomyocytes with similar maximal responses (∼3-fold increase in cell contractility) (Fig. 3), whereas the S,S isomers had no detectable effect (data not shown), consistent with our previous report (Beigi et al., 2006). Disruption of Gi signaling by PTX had only minor effects on the contractility profiles of the R,R isomers (Fig. 3, A and B), as indicated by the insignificant changes in the EC50 values (in terms of log [M]: -7.11 ± 0.17 for the control versus -7.44 ± 0.07 for the PTX-treated group in (R,R)-fenoterol, P = 0.085; and -6.76 ± 0.15 versus -6.84 ± 0.10 in (R,R)-methoxyfenoterol, P = 0.579). On the contrary, PTX treatment caused a clear leftward shift of the concentration-response curves of the S,R isomers (Fig. 3, C and D) and significantly decreased the EC50 values (from -5.63 ± 0.21 to -6.08 ± 0.03 for (S,R)-fenoterol, P < 0.01; and from -5.50 ± 0.03 to -5.96 ± 0.09 for (S,R)-methoxyfenoterol, P < 0.05). These results suggest that the R,R isomers evoke Gs-selective β2-AR signaling, whereas the S,R isomers allow the receptor to activate both Gs and Gi pathways.
In HEK293 cells, treatment with agonists triggers the phosphorylation of ERK1/2, which peaks at 5 min (Daaka et al., 1997). Based on the concentration-response relationships obtained above for fenoterol and its derivatives to induce cardiomyocyte contraction, preliminary experiments were conducted to determine the concentrations of the compounds to be used for agonist-induced ERK1/2 activation in HEK293 cells. We found that treatment of cells with a nonselective β-AR agonist, ISO (10-6 M), resulted in a 5-fold increase in phosphorylation of ERK1/2 over the control and that treatment of cells with PTX reduced ERK1/2 activation to approximately 2-fold of the control level (Fig. 4, A and C), which is similar to the previous notion (Daaka et al., 1997). It is interesting that the S,R compounds at 10-6 M, a concentration with minimal effects on myocyte contraction (Fig. 3, C and D), were able to induce a full activation of ERK1/2, an effect comparable with that induced by ISO treatment (Fig. 4, C and D) and also completely inhibitable by ICI 118,551 (data not shown). The R,R isomers at this concentration (10-6 M) also increased ERK1/2 activation to a similar extent (Fig. 4, C and D). It is noteworthy that treatment of cells with PTX largely abrogated ERK1/2 activation induced by the S,R isomers but had minimal effects on ERK1/2 activation induced by the R,R isomers (Fig. 4).
The lack of PTX-sensitivity in the myocyte contractile responses and in ERK1/2 activation in cells stimulated with (R,R)-fenoterol and (R,R)-methoxyfenoterol suggests that the R,R isomers selectively stimulate β2-AR-coupled Gs signaling. To directly assay G protein activation, we measured photoaffinity labeling of the α subunits of G proteins with the photoreactive GTP analog [γ-32P]GTP-azidoanilide in response to the fenoterol derivatives. Subsequent immunoprecipitation with specific antisera was carried out to determine the amount of the activated G protein(s). The top of Fig. 5A shows that at the same concentration (10-6 M), fenoterol compounds and the nonspecific β-AR agonist ISO increased the incorporation of [γ-32P]GTP-azidoanilide in Gαs. Both the short and long isoforms of Gαs (which have approximate molecular masses of 45 and 47 kDa, respectively) are activated similarly in response to the stimuli. Moreover, the Gαs labeling induced by (R,R)-fenoterol is significantly greater than that induced by (S,R)-fenoterol (P < 0.05, Fig. 5B). As a positive control, zinterol (10-5 M), a selective β2-AR partial agonist, was able to activate both Gαi2 and Gαi3 (Fig. 5A, bottom), consistent with our previous notion (Xiao et al., 1999). It is interesting that the fenoterol compounds (10-6 M) exhibited diverse effects on Gi proteins (Fig. 5A, bottom). In particular, activation of Gα-i2, the predominant Gi protein in the heart, was observed only if β2-AR was stimulated with (S,R)-fenoterol, but not by (R,R)-fenoterol, (R,R)-methoxyfenoterol, or (S,R)-methoxyfenoterol (Fig. 5C). The activation effects of (S,R)-fenoterol and (R,R)-fenoterol on Gαi2 are also statistically different (P < 0.01; Fig. 5C). In addition, activation of Gαi3 in response to S,R isomers was significantly greater than that induced by R,R isomers of the fenoterol derivatives (Fig. 5D; P < 0.01 for fenoterol and P < 0.05 for methoxyfenoterol).

Effect of PTX treatment on the contractile responses to the fenoterol compounds. Concentration-response profiles of cardiomyocyte contractility subjected to (R,R)-fenoterol (A), (R,R)-methoxyfenoterol (B), (S,R)-fenoterol (C), and (S,R)-methoxyfenoterol (D) with and without PTX treatment. Contractile response to the agonist is expressed as a percentage of the basal contractility (n = 9-14 cells from 7-9 hearts for each data point). Basal contraction amplitude is 5.46 ± 0.14% (n = 138 cells). PTX did not alter basal contraction (5.33 ± 0.14%, n = 130 cells).

Chirality determines the PTX sensitivities of fenoterol derivatives in activating ERK1/2. HEK293 cells were grown to confluence in growth medium in six-well plates before deprivation of the serum for 7 h. Treatment with PTX (0.5 μg/ml) was implemented during serum-starvation. The cultured cells were then stimulated with agonists for 5 min followed by cell lysis. After adjusting the protein concentration of the resultant cell lysates, the extent of ERK1/2 phosphorylation was analyzed by Western blotting. A and B, representative Western blots of phospho-ERK1/2 and total ERK1/2 (as protein loading control) after stimulation with ISO (10-6 M) or the fenoterol compounds (10-6 M) with or without PTX as indicated. C and D, averaged data from three to four independent experiments. The data are presented as the -fold increase over the -PTX control. #, P < 0.01 compared with the -PTX control; *, P < 0.05; **, P < 0.01 compared with the -PTX group in the same agonist treatment group.

Differential activation of Gs and Gi proteins by fenoterol stereoisomers. Rat heart membranes were labeled with [γ-32P]GTP-azidoanilide in the presence of ISO (10-6 M), zinterol (10-5 M), or fenoterol compounds (10-6 M). The radiolabeled G protein α subunits Gαs,Gαi2, and Gαi3 were immunoprecipitated with the corresponding subunit-specific rabbit polyclonal antibodies and then subjected to electrophoresis. A, representative autoradiographs of the resolved G proteins. B to D, averaged data of the densitometric analysis of the labeled G protein bands from three experiments. Data are presented as percentages of the control. *, P < 0.05; **, P < 0.01 compared with the control; †, P < 0.05; ‡, P < 0.01 compared with the corresponding diastereomer.
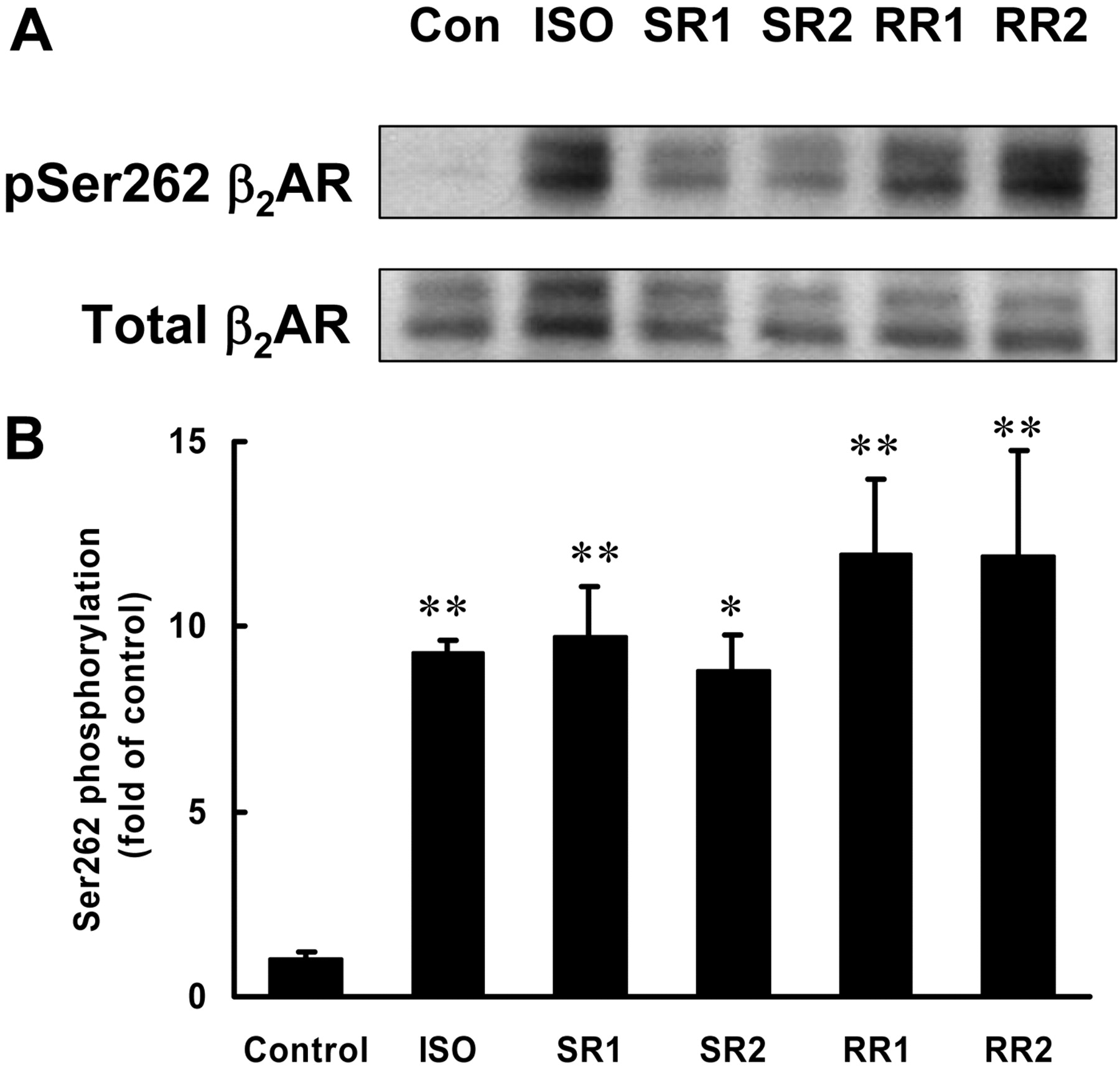
It has been proposed previously that PKA-mediated phosphorylation of β2-AR is necessary and sufficient for the switch of the receptor coupling from Gs to Gi (Daaka et al., 1997). To determine whether PKA-mediated phosphorylation of β2-AR is affected by the chirality of the fenoterol compounds, cultured rat cardiomyocytes overexpressing human β2-AR were stimulated with ISO (10-6 M) and the four fenoterol compounds (10-6 M), and the extent of PKA-mediated receptor phosphorylation was then detected by Western blotting using the anti-p-Ser262 antibody (Tran et al., 2004). The levels of the total β2-AR in the cell lysates were also determined in parallel using an anti-C-tail antibody. As expected, treatment with ISO increased the PKA-mediated phosphorylation of β2-AR by 9-fold after adjusting for the amount of total β2-AR (Fig. 6), a result confirming those obtained from another study (Iyer et al., 2006). The effects of the S,R isomers on receptor phosphorylation at Ser262 are comparable with that of ISO in magnitude. It is interesting that the R,R isomers also evoked an approximately 12-fold increase in phosphorylation of the receptor at Ser262. The differences in the potencies of these agonists are not significant. These results suggest that the degree of β2-AR-Gi coupling in response to the fenoterol isomers is not proportional to the phosphorylation status of β2-AR by PKA. Thus, PKA-mediated phosphorylation of β2-AR at Ser262 is insufficient for β2-AR-Gi coupling.

Fenoterol compounds trigger phosphorylation of β2-AR at the PKA site Ser262. Rat cardiac myocytes were cultured in 35-mm dishes and infected with adenovirus to overexpress human β2-AR. After 24 h, the myocytes were stimulated with ISO (10-6 M) and the fenoterol compounds (10-6 M) for 10 min, followed by cell lysis. The resultant cell lysates were subjected to Western blot analysis using the anti-p-Ser262 β2-AR antibody to detect for the phosphorylation of β2-AR at the PKA site and the anti-C-tail β2-AR antibody to detect for total β2-AR. A, representative Western blots of the phosphorylated Ser262 β2-AR and the total β2-AR. B, averaged data from three to four independent experiments. The data are presented as the -fold increase over basal phosphorylation of β2-AR after normalization with the amount of total β2-AR.
Discussion
As a prototypical Gs-coupled GPCR, β-AR stimulation activates the well established Gs-adenylyl cyclase-cAMP-PKA signaling cascade, which increases cardiac contractility via PKA-mediated phosphorylation of a panel of proteins involved in cardiac excitation-contraction coupling. Whereas β1-AR couples only to the Gs signaling pathway, β2-AR couples dually to Gs and Gi proteins (Xiao et al., 1995, 1999). As a result, stimulation of the β2-AR by most β2-AR agonists results in increased cardiomyocyte contractility that can be augmented by the inhibition of Gi signaling by PTX (Xiao et al., 1995, 2003). A major finding of the present study is that the positive inotropic effects and activation of ERK1/2 induced by (R,R)-fenoterol and (R,R)-methoxyfenoterol are insensitive to PTX treatment, suggesting that the R,R isomers selectively direct β2-AR to the Gs, bypassing the Gi coupling. It is interesting that alteration of their stereochemistry from the R,R to the S,R configuration is able to change this property. Apart from reducing the potencies, the contractile responses become sensitive to PTX treatment (Fig. 3, C and D).
β2-AR Activates ERK1/2 Signaling Via Gi-Dependent and Independent Mechanisms. The β2-AR-mediated activation of ERK1/2 in HEK293 cells expressing endogenous β2-AR has been shown previously to be Gi-dependent (Daaka et al., 1997), although alternative Gi-independent mechanisms (Schmitt and Stork, 2000; Friedman et al., 2002) have been proposed. Some G protein-independent mechanisms have been described previously (Shenoy et al., 2006; Sun et al., 2007). We have demonstrated here that activation of ERK1/2 by (R,R)-fenoterol and (R,R)-methoxyfenoterol is insensitive to PTX treatment (Fig. 4), suggesting the involvement of a Gi-independent mechanism. On the other hand, activation of ERK1/2 by (S,R)-fenoterol and (S,R)-methoxyfenoterol is PTX-sensitive (Fig. 4), suggesting the Gi-dependence of these effects. The lack of PTX-sensitivity in their effects on ERK1/2 activation further suggests that (R,R)-fenoterol and (R,R)-methoxyfenoterol selectively activate the β2-AR-coupled Gs pathway, whereas (S,R)-fenoterol and (S,R)-methoxyfenoterol can activate both the Gs and Gi proteins, as manifested by the robust PTX sensitivities in their responses.
Substitution and Chirality Confer the Fenoterol Compounds Different Effectiveness in Activating Gs and Gi Proteins. In the present study, we compared different fenoterol compounds on their effectiveness in activating Gs and Gi proteins by means of direct labeling of the G proteins on isolated heart membranes with a radioactive GTP analog (Fig. 5). We found that substituting the hydroxyl group with the methoxy group has a smaller impact on the G protein-selectivity of (R,R)-fenoterol than changing its configuration to the S,R form in terms of Gs and Gi2 stimulation (Fig. 5, B and C). This is consistent with the functional data on cardiomyocyte contraction (Fig. 3) and ERK1/2 activation (Fig. 4). On the other hand, substituting the hydroxyl group with the methoxy group increases selectivity of fenoterol to Gi3 while maintaining the differential coupling efficiency between the R,R and S,R isomers (Fig. 5D). Thus, the different effectiveness of (R,R)- and (S,R)-fenoterol in activating Gs and Gi proteins echoes the different PTX sensitivities of their functional effects. The distinction of the G protein activation effects and their correlation with functional data for the methoxyfenoterol isomers are less obvious compared with the fenoterol isomers (Fig. 5, B-D), suggesting that substitution can also affect the coupling and functional selectivity of the agonist.
PKA-Mediated Phosphorylation of β2-AR Is Insufficient to Cause Receptor Coupling to Gi Protein. Desensitization of β2-AR is triggered by the phosphorylation of the receptor by a combination of actions of PKA and G protein-coupled receptor kinases. In particular, phosphorylation of β2-AR at the PKA sites is suggested to switch the receptor coupling from Gs to Gi (Daaka et al., 1997). The subsequent binding of β-arrestin-2 to the phosphorylated receptor uncouples Gs from β2-AR and triggers receptor internalization. Here, we have demonstrated that (R,R)-fenoterol and (R,R)-methoxyfenoterol induce robust phosphorylation of β2-AR at its PKA site located at the third intracellular loop of the receptor, and this action is similar to that induced by their S,R isomers or the nonselective β-AR agonist ISO (Fig. 6). It is noteworthy that R,R isomer-induced β2-AR phosphorylation by PKA is not accompanied by activation of Gi2 proteins (Fig. 5C), the predominant Gi proteins in the heart. In addition, R,R isomer-induced β2-AR phosphorylation is associated with significantly less potent effects on Gi3 compared with those induced by S,R isomers (Fig. 5D). Taken together, our pharmacological and biochemical data suggest that the degree of PKA-mediated phosphorylation of β2AR is not proportional to their ability to induce Gi coupling. Thus, PKA-dependent phosphorylation of β2-AR is insufficient to trigger the receptor coupling to Gi proteins in the physiologically relevant setting, adult rat cardiac myocytes.
Molecular Nature of (R,R)-Fenoterol-Activated Gs Signaling. Ligand-induced different G protein coupling has been described in various GPCRs (Akam et al., 2001; Cordeaux et al., 2001, 2004; Gazi et al., 2003; Beyermann et al., 2007). The present study is the first to suggest the role of chirality in the functional selectivity of an agonist. Differential activation of receptors to specific signaling pathways might be explained by the fact that agonists can stabilize a receptor into multiple conformational states (Gether et al., 1995; Ghanouni et al., 2001; Swaminath et al., 2004; Granier et al., 2007), and each of which can trigger a distinct pluridimensional functional outcomes, including G protein coupling, receptor phosphorylation, receptor dimerization/oligomerization, receptor desensitization, receptor internalization, and/or β-arrestin-dependent ERK activation (Kenakin, 2007; Kobilka and Deupi, 2007; Urban et al., 2007). Further studies using these compounds to characterize the receptor conformation that confers selectivity to G protein coupling are highly warranted. The data thus obtained could be applied to the development of assay systems for future drug-screening.
Potential Clinical Implications of Gs-Signaling Selective β2AR Activation. Because enhanced Gi signaling is involved in the dysfunction of both β1-AR and β2-AR in the failing heart (Sato et al., 2004; Xiao and Balke, 2004; He et al., 2005; Zhu et al., 2005), selective activation of the β2-AR-Gs coupling may provide an effective means to improve contractile function of the failing heart without β1-AR detrimental effects (Zheng et al., 2005). We have reported previously the contractility stimulatory effects of a number of β2 agonists on rat cardiomyocytes (Xiao et al., 2003). Although most β2-agonists increased myocyte contraction in a PTX-sensitive manner, the contractility stimulatory effect of rac-fenoterol was insensitive to PTX (Xiao et al., 2003). We have concluded that rac-fenoterol selectively activates the β2-AR-Gs pathway and further postulated that this special property of rac-fenoterol might have potential therapeutic implications in heart failure. This idea is supported by recent in vivo studies in an ischemic rat heart failure model (Ahmet et al., 2004, 2005, 2008). Because (S,S)-fenoterol is inactive (Beigi et al., 2006), the pharmacological effects observed for rac-fenoterol are exerted by (R,R)-fenoterol. In the present study, we have provided multiple lines of evidence to demonstrate that (R,R)-fenoterol and its derivative (R,R)-methoxyfenoterol selectively activate the β2-AR-coupled Gs signaling and largely spare the β2-AR-coupled Gi pathway. It is noteworthy that the selectivity for Gs activation is determined by the chirality of the compounds because this selectivity is lost in the S,R isomers of these fenoterol compounds. This finding may have broad-reaching implications in GPCR biology and signaling pathway-targeted drug development.
We conclude that, in addition to receptor subtype and phosphorylation status, the stereochemistry of an agonist plays a role in the selectivity of the receptor-G protein interaction and downstream signaling. Moreover, different stereoisomers of an agonist can selectively activate different post-receptor events, presumably by stabilizing the receptor in multiple conformational states, which possess distinctly different G protein selectivity.
Acknowledgments
We thank Dr. Richard B. Clark of The University of Texas, Houston, Medical School (Houston, TX), for providing the anti-p-Ser262 monoclonal antibody for the present study. The skilful technical assistance of Dr. Olga Fedorova, Dr. Harold Spurgeon, and Bruce Ziman is gratefully acknowledged.
Footnotes
-
This work was supported by the intramural research program of the National Institutes of Health, National Institute on Aging (to A.Y.H.W., D.R.A., I.W.W., R.P.X., T.B.W., W.Z., X.Z.).
-
A.Y.H.W. and T.B.W. should both be regarded as first authors.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; β2-AR, β2-adrenoceptor; ERK, extracellular signal-regulated kinase; HEK, human embryonic kidney; ISO, isoproterenol; PKA, protein kinase A; PTX, pertussis toxin; ICI 118,551, (±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Received August 5, 2008.
- Accepted September 18, 2008.
- U.S. Government work not protected by U.S. copyright




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}