


Abstract
Agonist-stimulated desensitization of the β2-adrenergic receptor (β2AR) is caused by both a potent cAMP-dependent protein kinase (PKA)-mediated phosphorylation and a less potent, occupancy-dependent, G protein-coupled receptor kinase (GRK)-mediated phosphorylation that leads to β-arrestin binding and internalization. In this study the kinetics of phosphorylation of the third intracellular loop PKA site Ser262 and the putative C-tail GRK sites Ser355, Ser356 of the human β2AR overexpressed in human embryonic kidney (HEK) 293 cells were characterized using phosphoserine-specific antibodies. Specificity of the antibodies was shown by their lack of reactivity with mutant β2ARs lacking the respective sites. In addition, overexpression of GRK2 and GRK5 increased basal levels of phosphorylation of the GRK sites Ser355, Ser356 in both COS-7 and HEK 293 cells. Epinephrine, prostaglandin E1, and forskolin at maximum concentrations stimulated phosphorylation of the β2AR PKA site (Ser262) by 4-fold, whereas PMA stimulated it by 2-fold. Epinephrine stimulated PKA site phosphorylation with an EC50 of 20 to 40 pM. In contrast, epinephrine stimulated GRK site phosphorylation (Ser355,Ser356) with an EC50 of 200 nM (1-min treatments), which is more than 4000-fold higher relative to PKA site phosphorylation, consistent with an occupancy-driven process. After 10 to 30 min, the EC50 for epinephrine stimulation of GRK site phosphorylation was reduced to 10 to 20 nM but was still ≈200-fold greater than for the PKA site. The EC50 for internalization correlated with GRK site phosphorylation and showed a similar shift with time of epinephrine stimulation. The kinetics of epinephrine-stimulated GRK site phosphorylation were not altered in a mutant of the β2AR lacking the PKA consensus sites. The initial levels (2 min) of a range of agonist-stimulated GRK site phosphorylations were correlated with their efficacy for activation of adenylyl cyclase, namely epinephrine ≥ formoterol = fenoterol > terbutaline = zinterol = albuterol > salmeterol >> dobutamine ≥ ephedrine. However, after 20 to 30 min of treatment, agonists with intermediate strengths, such as albuterol and salmeterol, stimulate GRK site phosphorylations that are approximately equal to that produced by epinephrine, and the correlation breaks down. The GRK and PKA site antibodies were also effective in detecting phosphorylation of the endogenous β2AR expressed in A431 human epidermoid carcinoma cells. To summarize, our results show a remarkable amplification of PKA site phosphorylation relative to the putative GRK site phosphorylation, heterologous stimulation of the PKA site phosphorylation, no dependence of GRK site phosphorylation on PKA sites, and a reasonable correlation of initial levels of GRK site phosphorylation with the strength of a range of agonists.
The β2-adrenergic receptor (β2AR) plays significant roles in relaying signals from the autonomic sympathetic nervous system to the cardiovascular and pulmonary systems in particular. There are currently a number of clinically important drugs used in the treatment of asthma, chronic obstructive pulmonary disease, and hypertension that stimulate or inhibit β2ARs. The goal of our group has been to quantitatively define the molecular actions of these drugs in cultured human cells, and to correlate the measurements of agonist strength in activation of adenylyl cyclase with their downstream effects on desensitization, internalization, down-regulation, GRK and PKA site phosphorylation, and extracellular signal-regulated kinase activation (January et al., 1997, 1998; Clark et al., 1999; Seibold et al., 2000; Friedman et al., 2002). Defining these parameters will aid in the clinical application of these drugs in various disease states and provide insight into the molecular mechanisms involved.
Agonist-induced desensitization of the β2AR is caused by a combination of PKA phosphorylation of the third intracellular loop PKA consensus site and, at higher concentrations, GRK-mediated phosphorylation, β-arrestin binding, and internalization (Clark et al., 1999). In a recent study, we identified three sites that seem to be associated with GRK phosphorylation of the β2AR, serines 355, 356, and 364 (Seibold et al., 2000). These assignments were based on mutagenesis of various combinations of these sites in a receptor (PKA-) lacking the two PKA consensus sites (S261,262A and S345,346A) to eliminate the contribution of PKA to the desensitization. A substitution mutant with serines 355, 356, and 364 converted to alanine essentially eliminated receptor-level desensitization in the PKA- background and caused an 80 to 90% reduction in phosphorylation assessed by 32P-prelabeling. However, there are several problems associated with assignment of these GRK sites based on these standard approaches. First, background levels of phosphorylation in the basal state were a particular problem in comparisons of mutant β2ARs with WTβ2AR based on 32P-prelabeling of cells. Second, site-directed mutagenesis creates the possibility of de-localized changes in the receptor that may confound interpretation of results (Hausdorff et al., 1991).
To circumvent these problems our group initiated studies to quantitate phosphorylation of the putative GRK sites and the third intracellular loop PKA consensus sites of the β2AR by developing phospho-specific monoclonal antibodies and assessing commercially available antibodies to the putative phosphorylation sites involved in desensitization. The success of this approach for GPCRs was recently demonstrated in studies of the PKC and GRK site phosphorylation of the CCR5 receptor (Pollok-Kopp et al., 2003). Using site-specific anti-phosphoserine antibodies, it was shown that regulated on activation normal T cell expressed and secreted (RANTES) stimulation of PKC-mediated phosphorylation of serine 337 was far more potent (EC50 of 0.05 nM) than GRK phosphorylation of serine 349 (EC50 of 5 nM). Although the assignment of GRK to serine 349 was tentative and based on use of protein kinase inhibitors, the study illustrates the utility of this approach. With the exception of the identification of the light-stimulated rhodopsin kinase sites in rhodopsin in vivo by mass spectrometry (Ohguro et al., 1995), quantitation of either second messenger- or GRK-mediated phosphorylations in the absence of confounding phosphorylations has been a major obstacle.
In the present study, we used phosphospecific antibodies to GRK and PKA sites in the β2AR to answer a number of questions. First, we established the kinetics and concentration-dependence of epinephrine-induced phosphorylation of the wild-type (WT) β2AR and showed a remarkable amplification of PKA site phosphorylation relative to GRK site phosphorylation and internalization. Second, the possibility of hierarchical phosphorylation of the GRK site (Moffett et al., 1996) was probed in a mutant lacking the PKA consensus sites, and no dependence on PKA site phosphorylation was observed. Third, we demonstrated that the GRK and PKA site antibodies were sufficient to measure phosphorylation of the endogenous receptor in A431 cells. Fourth, we demonstrated heterologous phosphorylation of the PKA site after stimulation with PGE1, forskolin and PMA. Finally, we showed a strong correlation of the ability of a number of partial agonists to stimulate the initial levels of phosphorylation of the GRK sites with their efficacies for activation of adenylyl cyclase.
Materials and Methods
Materials. Enhanced chemiluminescence reagents and Hyperfilm were from Amersham Biosciences (Piscataway, NJ). BA85 nitrocellulose was from Schleicher & Schuell (Keene, NH). Cell culture reagents were purchased from Mediatech. β2AR agonists were purchased from Sigma (St. Louis, MO), except for RR formoterol, which was provided by Sepracor (Marlborough, MA), and salmeterol, a gift from GlaxoSmithKline (Uxbridge, Middlesex, UK). The GRK2 and GRK5 cDNAs cloned into pcDNA3 were kindly provided by Dr. Jeffrey Benovic (Thomas Jefferson University, Philadelphia, PA). Antibodies to GRK2, GRK5, the C-tail of the β2AR, and pSer(355,356) and the C-tail affinity resin were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Cell Culture. HEK 293 and COS-7 cells, purchased from the American Type Culture Collection (Manassas, VA), were grown in 5% CO2 at 37°C in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. A431 human epidermoid carcinoma cells were a kind gift from Dr. Craig Malbon (State University of New York at Stony Brook) and were grown as described above for HEK 293 cells. HEK 293 cells stably transfected with wild-type or mutant β2ARs (Seibold et al., 2000) were grown in the same medium as above with the addition of 600 μg/ml G418. WT and mutant β2ARs were tagged with a hemagglutinin antigen (HA) at the N terminus and 6 histidines at the C terminus as described previously (January et al., 1997; Seibold et al., 2000) and were designated Hβ2ARH for the WT β2AR and PKA- for the mutant β2AR lacking both PKA consensus sites (S261,262,345,346A). Mutants lacking either the third intracellular loop PKA consensus site [Hβ2ARH(S261,262A)] or the carboxyl-tail PKA consensus site [Hβ2ARH(S345,346A)] were used for screening the PKA site monoclonal antibodies. PKA- (S364A), PKA- (S355,356A), and PKA- (S355,356,364A) were used for screening the GRK site antibodies. β2AR levels in these stably transfected cell lines were from 1.5 to 3.0 pmol/mg of membrane protein.
Transient Coexpression of β2ARs and GRKs. COS-7 cells growing in six-well plates were transfected with 0.6 μg of a plasmid expressing Hβ2ARH and 2.4 μg of either empty vector, GRK2, or GRK5 using FuGene 6 transfection reagent (Roche Molecular Biochemicals, Basel, Switzerland) according to the manufacturer's instruction, with a DNA to FuGene 6 ratio of 3 μg/4.5 μl. The transfection mixture remained on cells for 48 h, at which time the cells were treated and harvested.
Cell Treatment and Preparation of Solubilized Extract. Cells were grown to confluence in growth medium in six-well plates coated with poly-l-lysine. Cells were treated with β2AR agonists dissolved in the carrier, 0.1 mM ascorbate/1 mM thiourea pH 7 (AT), or AT alone as indicated at 37°C. PGE1 and forskolin were dissolved in ethanol, and PMA in dimethyl sulfoxide. These compounds were diluted 1000-fold into the cell incubations, and control cells received the appropriate concentration of carrier. To terminate all treatments, the medium was removed, the cells were washed once rapidly with 2 ml of 20 mM HEPES, pH 8, and 1 mM EDTA, pH 7, at 0 to 4°C. Cells were solubilized by addition to each well of 1 ml of solubilization buffer at 0 to 4°C [20 mM HEPES, pH 7.4, 150 mM NaCl, 1% dodecyl-β-maltoside (DBM), with 20 mM tetrasodium pyrophosphate, 10 mM NaF, 0.1 μM okadaic acid (for inhibition of phosphatase activity) and 10 μg/ml benzamidine, 10 μg/ml trypsin inhibitor, and 10 μg/ml leupeptin (for inhibition of protease activity)]. The plates were placed on ice, the cells were scraped, and the solubilized cells were pipetted into 1.5-ml microcentrifuge tubes. The tubes were rocked at 4°C for 30 min and then centrifuged at 15,000 rpm for 15 min. The supernatants were removed and frozen at -80°.
Determination of β2AR Phosphorylation with Phosphoserine-Specific Antibodies. Commercially prepared antibodies to the C terminus of the β2AR and to the phosphorylated serines pSer(355,356) were from Santa Cruz Biotechnology. Antibody to the hemagglutinin antigen was purchased from Berkeley Antibody Co. (Berkeley, CA). Custom monoclonal antibodies were obtained from A and G Pharmaceutical, Inc. (Baltimore, MD). These antibodies were raised against the peptides C-DRTGHGLRRSpSKF (pSer262 PKA site), C-NGNTGEQpSGYH (pSer364 GRK site), and C-KAYGNGYSpSNGN (pSer356 GRK site). Monoclonal antibodies were used either as hybridoma supernatants without purification or after purification over protein A Sepharose, and results were identical with either preparation.
Monoclonal antibodies were initially screened against the peptides by enzyme-linked immunosorbent assay and subsequently against immunoblots of purified β2AR derived from control or epinephrine-stimulated HEK 293 cells expressing WT or mutant β2ARs. For β2AR purification, solubilized extracts were applied to Talon Co2+-carboxymethylaspartate-agarose columns (0.125-ml packed resin) that had been prewashed with Talon buffer (0.05% DBM, 20 mM HEPES, pH 7.4, and 150 mM NaCl). The columns were washed once with 2 ml of Talon buffer, followed by a 2-ml wash with 10 mM imidazole in Talon buffer. β2ARs were eluted with 0.4 ml of Talon buffer containing 100 mM imidazole. The eluates were digested with 250 units of N-glycosidase F (N Gly F; New England Biolabs, Beverly, MA) for 2 h at 37°C and heated at 60°C for 15 min in SDS-sample buffer. Aliquots (20 μl) of samples (≈20–40 fmol) were resolved on SDS-polyacrylamide 12% gels and transferred to nitrocellulose for immunoblotting. The membranes were probed with the polyclonal β2AR anti-phosphoserine-specific antibody pSer(355,356) at a dilution of 1:500. The PKA site anti-pSer262 (clone 2G3) monoclonal antibody hybridoma media was used at a 1:10 dilution for the majority of experiments, although similar results were obtained using protein A Sepharose-purified monoclonal antibody (mAb) 2G3 at 0.5 μg/ml. Blots were washed twice and incubated with the appropriate secondary antibodies [goat anti-rabbit IgG horseradish peroxidase (Bio-Rad, Hercules, CA) at a 1:5000 dilution for polyclonal antibodies and goat anti-mouse IgG horseradish peroxidase at a 1:3000 dilution for monoclonal antibodies] and detected by ECL Plus chemiluminescence reagents. Although this purification procedure was followed for all of the PKA site phosphorylation data shown, we have determined that it is not necessary for the GRK site phosphorylations, and identical results for the latter are found with or without purification. After quantitation of β2AR phosphorylation, the immunoblots were stripped and reprobed with a β2AR-specific rabbit anti-C-tail antibody at a dilution of 1:5000 to determine β2AR levels. Western blots were quantitated using Scion Image software (Scion Corporation, Frederick, MD). All results with the anti-phosphoserine-specific antibodies were first normalized to the corresponding levels of β2AR measured with the anti-C-tail antibody. To compare the data from multiple experiments, further normalization was performed as described in the legends to the various figures. For most experiments, the data were normalized to the maximal epinephrine stimulations in each experiment. Significance of the results (p-values) for the data shown in Figs. 3 and 7, A and B, were calculated with Prism software (ver. 4; Graph Pad Software, San Diego, Ca) using an unpaired t test.

Effect of time and concentration of epinephrine stimulation on phosphorylation of Ser262. Hβ2ARH cells in six-well dishes were stimulated for from 1 to 30 min with the various concentrations of epinephrine or carrier AT as indicated. Cells were solubilized, and the Hβ2ARH purified over Talon affinity columns, treated with N Gly F, and immunoblotted with anti-pSer262 mAb 2G3. The arbitrary units derived from the Western blots were first normalized to the corresponding measure of the receptor levels by the C-tail antibody. Results were further normalized in each experiment to the mean of the values at 10 and 20 min for the 0.3 and 3.0 nM epinephrine treatments. Data shown are the mean ± S.E.M. (n = 4–6). A, effect of time of treatment with epinephrine on Ser262 phosphorylation; B, the same data plotted against the epinephrine concentration. A representative immunoblot after the 2-, 5-, and 20-min treatments is shown.

Initial levels of GRK site phosphorylation in response to stimulation of Hβ2ARH cells with a range of β2AR agonists of varied efficacy. Hβ2ARH cells were stimulated for 2 min with a range of β2AR agonists at near-maximal concentrations as indicated that gave occupancies of the Hβ2ARH of 95 to 98%. Hβ2ARH cells were solubilized and the receptor was purified over Talon affinity columns, treated with N Gly F and immunoblotted with anti-pSer(355,356) antibody. Phosphorylation data were first normalized to receptor levels using the anti-C-tail antibody and expressed as a percentage of the maximal epinephrine stimulation in each experiment. A, data shown are the means ± S.E.M. (n = 7). B, the phosphorylation data from A were plotted as a function of the efficacy of the various agonists determined as discussed under Materials and Methods. Efficacy measurements shown are the means ± S.E.M. (n = 3–5). Best fit of the data were with a hyperbolic function (Prism 4). C, Hβ2ARH cells were stimulated for the indicated times with either carrier (AT), 10 μM albuterol, 100 μM ephedrine, 50 nM salmeterol, 15 μM dobutamine, or 10 μM epinephrine. The cells were solubilized, purified over Talon affinity columns, treated with N Gly F, and immunoblotted with anti-pSer(355,356) antibody and anti-C-tail antibody. Data are the means ± range (n = 2).
For the measurement of GRK site phosphorylation in A431 cells, solubilization and Western blots were performed as given above. However, for the measurement of PKA site phosphorylation, the solubilized samples were first subjected to purification on C-tail affinity resins as described previously (Seibold et al., 2000). Briefly, the solubilized samples were added to 100 μl of C-tail resin, recycled twice, and then washed with 1.5 ml of 10 mM phosphate buffer, pH 6.8, containing 0.05% DBM. β2AR was eluted with 500 μl of glycine buffer, pH 2.5, also in 0.05% DBM and neutralized with 150 μl of 1 M phosphate buffer, pH 8.0. Samples were concentrated by microcon filtration to 100 μl, and 20-μl aliquots were removed, treated with N-Gly F, combined with SDS sample buffer (2×), and then Western blots were performed and quantitated as described above.
Internalization. The procedure for measuring internalization of the β2AR has been described in detail previously (Seibold et al., 2000). Cells in 12-well plates were treated with epinephrine or AT for 5, 10 and 30 min. Cells were then washed 5 times with ice-cold serum-free DMEM, placed on ice then incubated with 10 nM [3H]CGP-12177 with or without 1 μM alprenolol in serum-free DMEM. Dishes were then incubated for 1 h on ice, washed twice with ice-cold PBS to remove [3H]CGP-12177, and cells released by trypsin were transferred to scintillation vials for counting.
Efficacy Measurements for Agonist Stimulation of Adenylyl Cyclase. The methodology for the determination of efficacy (or coupling efficiency) for activation of adenylyl cyclase in membrane preparations by agonists of the β2AR has been described in detail in previous publications (Whaley et al., 1994; January et al., 1997; Clark et al., 1999; E. Qunaibi and R. Barber, submitted) and is based on the formulation that we have shown to accurately describe the kinetics of agonist activation over a 1000-fold range of β2AR levels (Whaley et al.). In this formulation, efficacy is defined as k1r/k-1, where k1 is the rate constant for activation of adenylyl cyclase, k-1 is the rate constant for inactivation, and r is the receptor level. Our efficacy is equivalent to Furchgott's parameter e as previously discussed (Clark et al., 1999).
Efficacy (k1r/k-1) is calculated by either of two equations. The first of these is k1r/k-1 = Vmax/(V100 - Vmax), where Vmax is the maximum activity for a given agonist and V100 is the maximum activity for the strongest agonist (epinephrine). The second formulation is k1r/k-1 = (Kd - EC50)/EC50, where EC50 is the concentration of agonist that gives 50% of its maximal activation of adenylyl cyclase and Kd is the dissociation constant for agonist binding in the presence of GTP (or guanosine 5′-O-(3-thio)triphosphate). For the determination of efficacy parameters (EC50, Vmax, and Kd), membrane preparations of the Hβ2AR cells expressing 2.5 pmol of Hβ2ARH/mg of protein were used. The EC50 and Vmax for agonist stimulation of adenylyl cyclase and the Kd for binding using displacement of [125I]iodocyanopindolol were determined as described previously (Williams et al., 2000). Because the receptor level (r) and k-1 are the same for all agonists in the membrane preparations, they are not included in the calculation. Choice of which equation to use was based on error analysis (E. Qunaibi and R Barber, submitted). For membrane preparations expressing high levels of receptor, as in the present study with the Hβ2ARH, where for moderate to strong agonists there is a reasonably large difference (> 5-fold) between the EC50 and the Kd, the second equation is used. Where there is a very small difference between these parameters (i.e., where the EC50 and the Kd differ by ≤5-fold), the first equation is used and is therefore the most accurate for either membranes expressing low levels of receptor or for very weak agonists such as ephedrine and dobutamine, wherein changes in the Vmax for these agonists relative to epinephrine were more pronounced. An additional problem was encountered for salmeterol, which, because of its hydrophobicity and putative exosite binding, makes measurements of EC50 and Kd difficult, as discussed previously (Clark et al., 1996; January et al., 1998). For that reason, its efficacy was also determined by Vmax/(V100 - Vmax).
Results
Specificity of GRK and PKA Site Phosphoserine-Specific Antibodies. The specificity of the monoclonal and polyclonal antibodies was characterized by their reactivity against the double-epitope tagged Hβ2ARH stably overexpressed in HEK 293 cells. The desensitization, internalization, and phosphorylation of this receptor and various substitution mutants (all double-epitope tagged, including the PKA- that lacks both PKA consensus sites) in response to various agonists were previously extensively characterized by our group (January et al., 1997; Seibold et al., 2000). HEK 293 cells expressing either the Hβ2ARH or mutant receptors were treated with 10 μM epinephrine, and the receptor was purified by passage over the Talon affinity resin followed by treatment with N Gly F to remove glycosyl residues. On immunoblots, the β2AR appears as a major band around 48 to 50 kDa and a minor band that shows variable intensity at a slightly lower molecular mass. The lower band seems to be an N-terminally truncated β2AR, because this band reacts with the C-tail antibody but not with an anti-HA antibody that is on the N terminus (see Fig. 1).
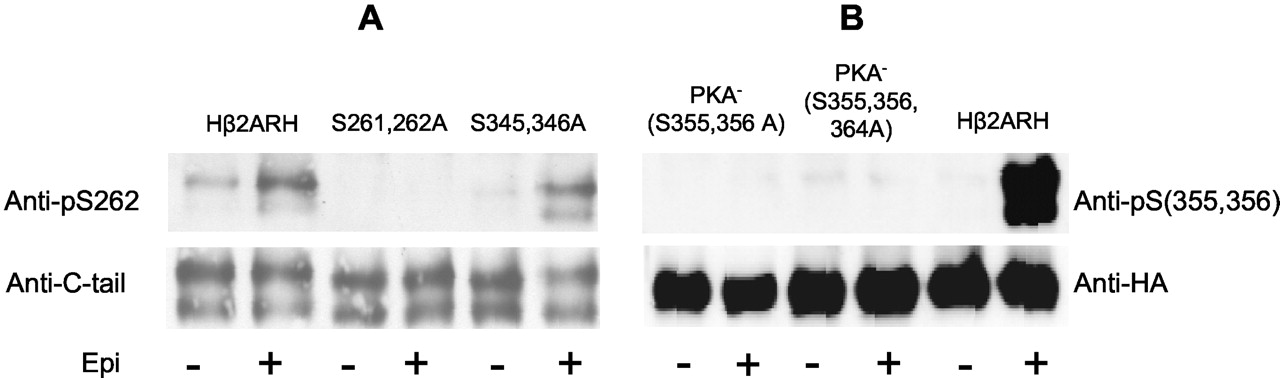
Specificity of anti-phosphoserine antibodies. HEK293 cells stably expressing either Hβ2ARH or various mutant epitope-tagged receptors in six-well plates were stimulated with 10 μM epinephrine (Epi) or carrier AT for 5 min. The β2AR was solubilized, purified over Talon affinity columns, treated with N Gly F and immunoblotted with either anti-pSer262 mAb (2G3) (A) or anti-pSer(355,356) Ab (B). Levels of β2AR were probed with either anti-C-tail antibody or anti-HA as indicated.
For the PKA site Ser262, five mAbs were found that generated a positive specific reactivity with cells treated for 5 min with 3 nM or 10 μM epinephrine. Antibody specificity for the PKA site was tested by immunoblotting the Talon-purified Hβ2ARH and the mutant Hβ2ARHs that lacked either the Ser261, Ser262, or Ser345, Ser346 PKA consensus sites after treatment of cells with either the carrier AT or 10 μM epinephrine. The characterization of the specificity of PKA site anti-pSer262 (mAb 2G3) is shown in Fig. 1A. The mutant lacking the carboxyl-tail PKA site serines (S345,346A) and the Hβ2ARH showed equivalent results, a 3- to 5-fold elevation of the signal upon epinephrine treatment. There was no reactivity of the anti-pSer262 with the substitution mutant lacking the third intracellular-loop PKA site serines (S261,S262A) with or without epinephrine stimulation. Based on these criteria, antibody 2G3 was chosen for further analysis of epinephrine-induced phosphorylation of the PKA site Ser262.
Characterization of the specificity of the polyclonal rabbit antibody raised to pSer(355,356) is shown in Fig. 1B. Screens were performed against purified receptor isolated from cells expressing Hβ2ARH or mutant receptors that were treated with 10 μM epinephrine or AT. A very strong specific signal was found in probes of the Hβ2ARH, with epinephrine stimulation generating a ≈15-fold elevation over control. There was no reactivity of the polyclonal antibody anti-pSer(355,356) with substitution mutants of the PKA- receptors lacking either both sites (S355,356A) or all three putative GRK sites (S355,356,364A).
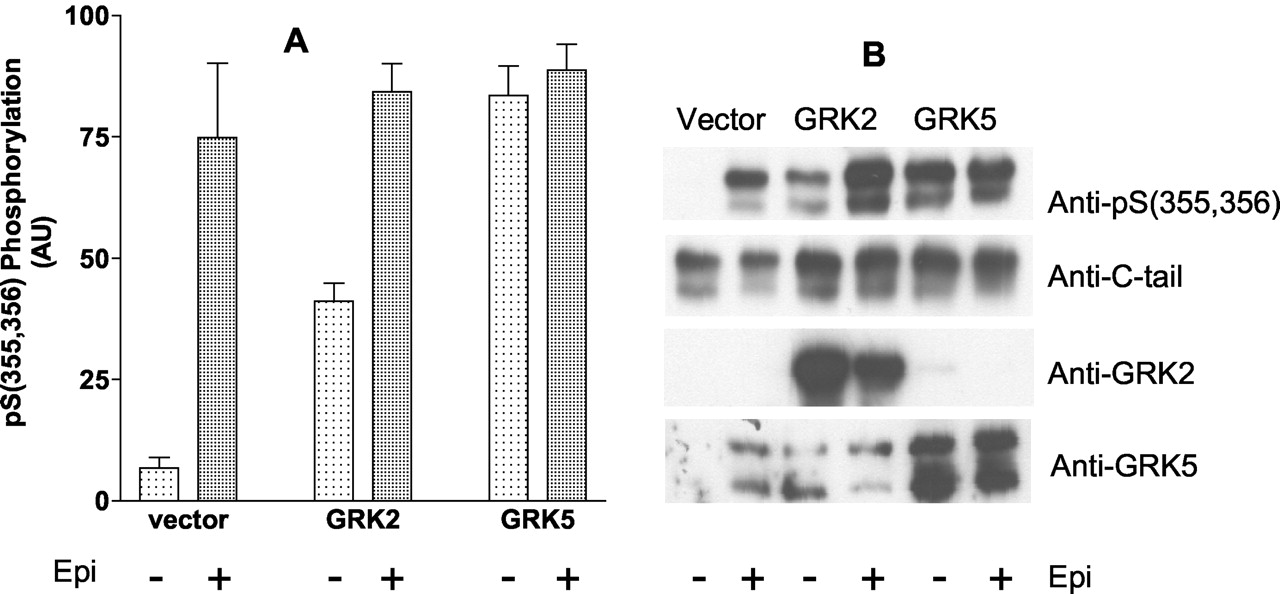
To demonstrate that the anti-pSer(355,356) polyclonal antibody was specific for GRK phosphorylation of sites Ser355, Ser356, COS-7 cells were transiently transfected with the double epitope-tagged Hβ2ARH in combination with either vector, GRK2, or GRK 5. COS-7 cells were chosen because previous work had shown that they were considerably more deficient in GRKs relative to the HEK 293 cells (Menard et al., 1997). The Talon-purified receptors were immunoblotted, and the results in Fig. 2 demonstrate that transfection with either GRK2 or GRK5 caused a dramatic increase in the basal (AT-treated) level of phosphorylation (≈5- and 12-fold, respectively). However, no significant increase in the level of phosphorylation of the Hβ2ARH was achieved with 10 μM epinephrine treatment of the cells. The Hβ2ARH cells were also transiently transfected with GRK2 and GRK5, and although the effects of GRK2 and GRK5 were not as large on basal phosphorylation (3- and 7-fold, respectively) relative to epinephrine-stimulated reactivity (20-fold), they were nonetheless considerable (data not shown). As with the COS-7 cells, there was no augmentation of the epinephrine-induced signal. These data demonstrate that anti-pSer(355,356) recognizes the product of GRK-mediated phosphorylation, although the specific GRK(s) responsible for the phosphorylation in HEK 293 cells have not been determined.

Effect of transient overexpression of GRK2 or GRK5 on basal and epinephrine-stimulated phosphorylation of Ser355, Ser356 of Hβ2ARH in COS-7 cells. COS-7 cells in six-well dishes were transiently transfected with Hβ2ARH in combination with either vector, GRK2, or GRK5 plasmids. After 48 h, cells were stimulated with either carrier (AT) or 10 μM epinephrine for 5 min. The Hβ2ARH was solubilized, purified over Talon affinity columns, and immunoblotted serially with anti-pSer(355,356) antibody, to measure phosphorylation, and anti-C-tail antibody to assess Hβ2ARH levels. Solubilized extracts were probed with anti-GRK2 antibody, and anti-GRK5 antibody to determine GRK expression. A, phosphorylation was normalized to Hβ2ARH levels and to the maximum level of stimulation in each experiment. The data shown are the means ± S.E.M. (n = 4) expressed as arbitrary units (AU). B, immunoblots from a typical experiment.
Time and Concentration Dependence of Epinephrine Stimulation of Phosphorylation of the Third Intracellular-Loop PKA Site (Ser262) after Treatment with Epinephrine. Our previous studies with a number of different cultured cell types had indicated that there was both a high-potency PKA-mediated desensitization of the third intracellular loop PKA consensus site (low concentrations of epinephrine and very low occupancy), and an occupancy-dependent desensitization mediated by GRKs that more closely followed high occupancy (Clark et al., 1999). However, as noted above, individual assessment of the phosphorylation of these sites has never been shown directly, and was based on the use of site-directed substitution mutants of these sites and/or measurement of phosphorylation by 32P-prelabeling. Figure 3, A and B, shows the time course and concentration-dependence of the phosphorylation of the PKA site in response to treatment of the Hβ2ARH cells with epinephrine concentrations in the range of 0.01 to 100 nM using the 2G3 anti-pSer262 mAb. Maximal phosphorylation (≈4- to 5-fold over AT-treated) was achieved after 5 to 10 min for all concentrations, and the EC50 for epinephrine was approximately 0.03 nM. Phosphorylation after 1 min of stimulation was not significantly different from AT for the treatments with 0.01 and 0.3 nM epinephrine (p > 0.05), but was significant for the 3.0 and 100 nM treatments (p ≤ 0.03). After 2-min treatments, the values with all epinephrine concentrations were significantly different from the control cells (p ≤ 0.02). Epinephrine at 0.01 nM caused a significant increase in PKA site phosphorylation over control cells at the 2- to 20-min stimulation time points (p ≤ 0.04). A precise value for the half-time to peak stimulation was not possible given the spread of values after 1 min stimulations, but in the aggregate was between 1 and 2 min. For the most part, maximal levels of phosphorylation stayed constant with time; the only exception was the 20-min treatment with 100 nM, which was significantly lower (p = 0.008) than the value at 5 min.
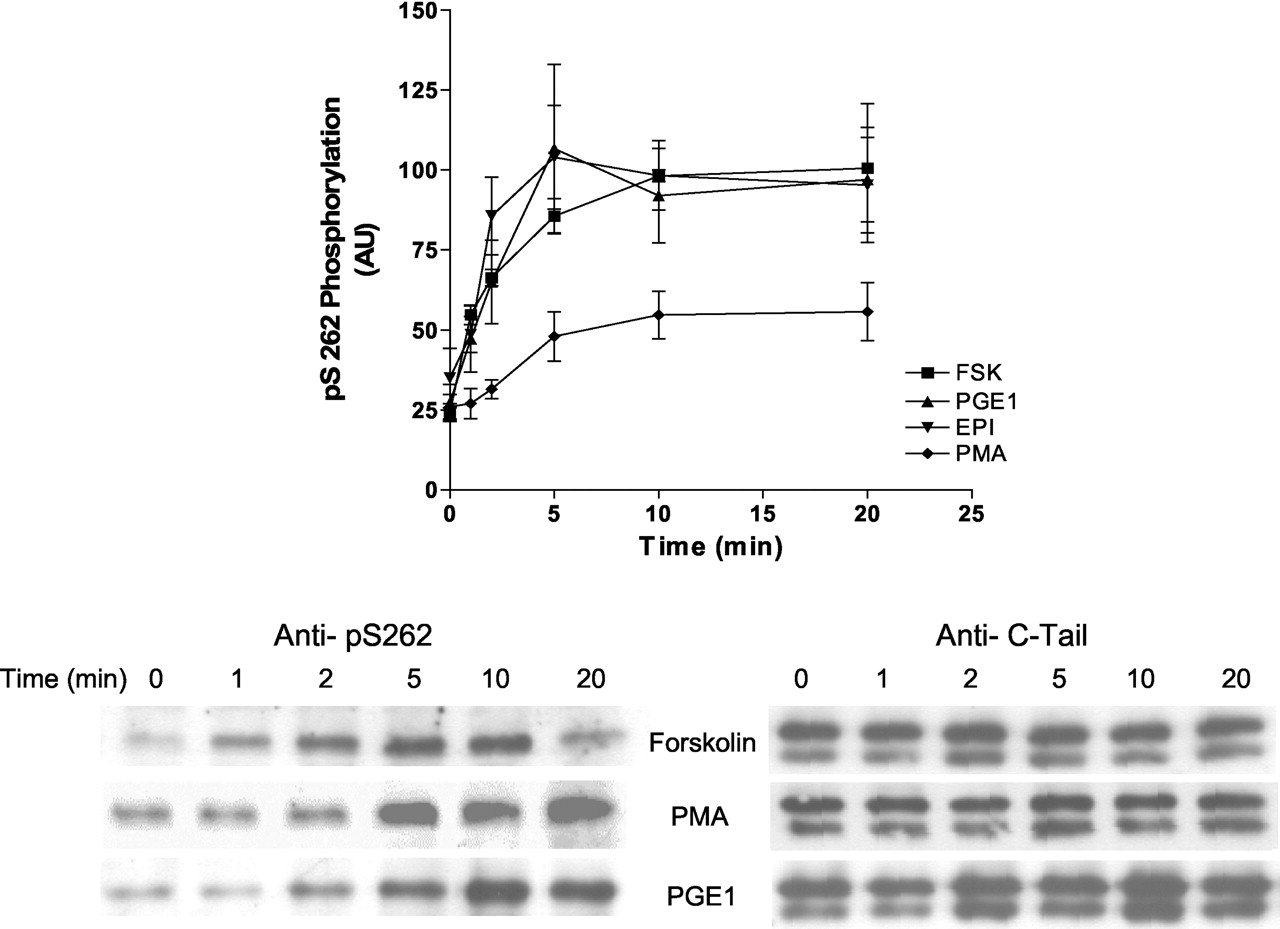
Heterologous Activation and Phosphorylation of the Ser262 PKA Consensus Site. Previous studies have shown that PKA desensitization of the β2AR was heterologous; that is, that any stimulation of cAMP and PKA could lead to a desensitization of epinephrine stimulation of adenylyl cyclase. We and others have also shown that activation of PKC with PMA led to a similar desensitization in a number of cell types (Clark et al., 1988, 1989; Kunkel et al., 1989; Johnson et al., 1990; Bouvier et al., 1991; Hausdorff et al., 1991; Yuan et al., 1994). Both the PKA- and PKC-mediated desensitizations were shown to be ablated with an S261,262A substitution mutant of the β2AR (Johnson et al., 1990; Yuan et al., 1994). To examine whether either heterologous activation of adenylyl cyclase/PKA or PKC activation stimulated a phosphorylation of Ser262, Hβ2ARH cells were treated for 1 to 20 min with either 3.0 nM epinephrine, 10 μM PGE1, 10 μM forskolin, or 500 nM PMA. The results shown in Fig. 4 demonstrate that PGE1, forskolin, and epinephrine all caused a 4-fold stimulation of phosphorylation of Ser262; maximal levels were achieved for all stimulations at 5 to 10 min and half-maximal levels by 1 to 2 min. PMA stimulated a 2-fold increase in the level of phosphorylation of Ser262 over the basal value. We also examined whether stimulation of the endogenous muscarinic receptor with carbachol caused phosphorylation of Ser262, but it was without effect (data not shown).

Heterologous phosphorylation of Ser262 after stimulation of Hβ2ARH cells with epinephrine, PGE1, forskolin, or PMA. Hβ2ARH cells in six-well dishes were stimulated with either the appropriate carrier or with 3.0 nM epinephrine, 10 μM PGE1, 10 μM forskolin, or 500 nM PMA for the indicated times. Cells were solubilized, purified on Talon affinity columns, and immunoblotted with anti-pSer262 mAb 2G3. Data were first normalized to receptor levels using the anti-C-tail Ab, and then for comparison of separate experiments further normalized to the forskolin value at 10 min. The data shown are the means ± S.E.M., where n values for forskolin, PGE1, epinephrine, and PMA were 5, 4, 3, and 3, respectively.
Time and Concentration Dependence of Epinephrine Stimulation of GRK Phosphorylation Using anti-pSer(355,356) Antibody. Hβ2ARH cells were stimulated for 0 to 30 min with a range of epinephrine concentrations from 3 nM to 10 μM, and GRK site phosphorylation was assessed using the anti-pSer(355,356) antibody as shown in Fig. 5. Maximal levels of phosphorylation were rapidly achieved (2 min) in response to stimulation with 10 μM epinephrine, but required considerably longer times for the 3, 10, and 50 nM concentrations. GRK-mediated phosphorylation was quite stable, although there seemed to be some decline in time with the higher concentrations. Stimulation of cells with 50 nM epinephrine for 30 min caused a phosphorylation that was not significantly different from the level observed after stimulation with a 200-fold higher concentration. The EC50 for phosphorylation of the receptor at 1 min was ≈200 nM, or about 4000-fold higher than for PKA site phosphorylation. In addition, we found that the EC50 was markedly reduced with time of treatment, shifting to about 20 nM after 10 to 30 min of treatment. Thus, the data show that although the initial rate of GRK site phosphorylation was proportional to agonist occupancy (Kd for epinephrine is ≈280 nM), this correlation did not hold with longer times of stimulation.

Effect of time and concentration of epinephrine stimulation on GRK site phosphorylation of Ser355,Ser356. Hβ2ARH cells in six-well dishes were stimulated with epinephrine or carrier AT for the times (A) and concentrations (B) as indicated. Hβ2ARH was solubilized, purified on Talon affinity columns, N Gly F-treated, and immunoblots were performed serially with anti-pSer(355,356) antibody to measure phosphorylation and anti-C-tail antibody to measure Hβ2ARH levels. Phosphorylation was first normalized to receptor levels, and for the comparison of experiments, the data were further normalized to the maximum epinephrine-stimulated value in each experiment. Data shown are the means ± range of two experiments. Representative immunoblots are shown for the 1-, 10-, and 30-min epinephrine treatments.
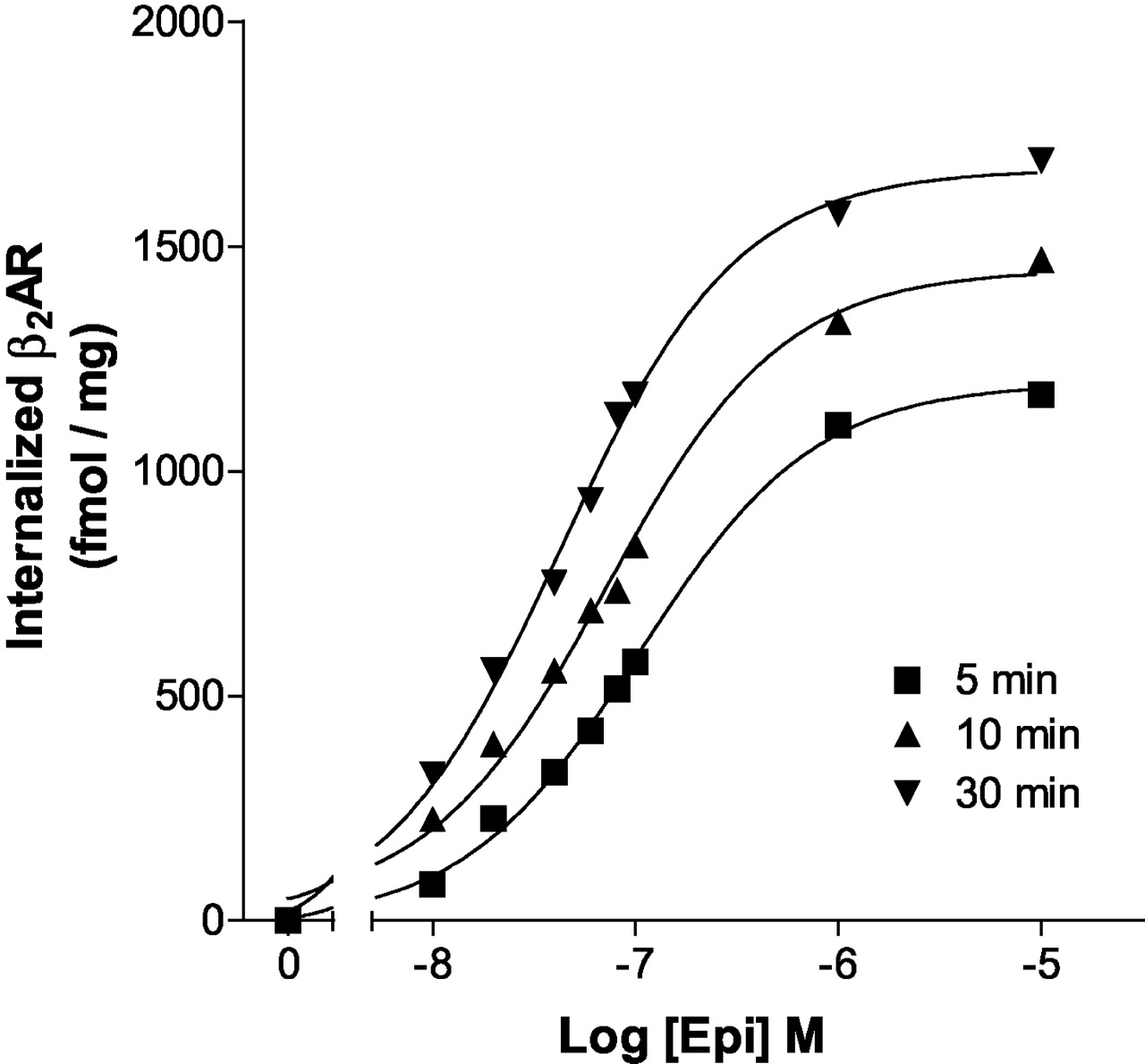
It is well established that arrestin binding follows GRK site phosphorylation and in turn triggers internalization. This suggests that internalization might show a similar dependence on time and concentration of epinephrine. To explore this possibility, we measured the EC50 for internalization of the Hβ2ARH after the loss of [3H]CGP-12177 binding (Fig. 6). As with GRK site phosphorylation, we found a shift to lower EC50 values for epinephrine with time; i.e., the EC50 for epinephrine-induced internalization after 5 min was ≈100 nM and shifted to ≈40 nM after 30 min. The EC50 for internalization at shorter time points could not be measured because the data were too variable. These data support the proposal that GRK site phosphorylation and internalization of the β2AR are tightly coupled.

Effect of time and concentration on epinephrine stimulation of internalization. Hβ2ARH cells in 12-well plates were treated with the various epinephrine concentrations for the times indicated. Cells were washed six times with ice-cold PBS and incubated with [3H]CGP-12177 at 4°C for 1 h. Surface receptor binding as fmol/mg of cell protein was quantitated as described under Materials and Methods. The data shown are femtomoles of internalized receptor calculated by subtracting the surface levels after the different treatment times from the starting surface expression (2116 fmol/mg of cell protein). This experiment is representative of two similar experiments.
GRK Site Phosphorylation in Response to Stimulation with Agonists of Varied Efficacies. In previous work, we reported that phosphorylation of the Hβ2ARH is roughly correlated with the efficacy (coupling efficiency, or intrinsic activity) of agonist activation of adenylyl cyclase. However, these studies were based on 32P-prelabeling and could not distinguish the individual contributions of GRK and PKA site phosphorylations (January et al., 1997, 1998). Using the anti-pSer(355,356), we have now determined the correlation of a range of agonists of widespread efficacies with GRK site phosphorylation. Hβ2ARH cells were stimulated for 2 min with various agonists at concentrations that were 10- to 20-fold above their Kd values. In addition, we examined the effects of an antagonist, propranolol, and an inverse agonist, ICI-118,551. GRK site phosphorylation for these drugs is shown in Fig. 7A. Fig. 7B shows the correlation of the initial levels of GRK site phosphorylation with partial agonist efficacies. Efficacy values for the agonists, determined as described under Materials and Methods, are tabulated in the inset to Fig. 7B. S.E.M. for these efficacy values was less than 5% of the mean for all but salmeterol and formoterol. With the exception of these agonists, the greater error in the comparison of agonist efficacies with GRK site phosphorylation was for the most part with the measurement of phosphorylation; initial rates (see Fig. 7C) were difficult to measure, especially for the weakest agonists. The extent of phosphorylation in response to the two strongest agonists, fenoterol and formoterol, did not differ significantly (p = 0.06 and 0.31, respectively) in comparison to epinephrine. The phosphorylation in response to agonists with efficacies in the intermediate range (zinterol, terbutaline, and albuterol) was significantly decreased relative to epinephrine (p ≤ 0.0001). Salmeterol, whose efficacy was 13% of epinephrine, stimulated GRK site phosphorylation that was 31% of epinephrine. The weakest agonists, dobutamine and ephedrine, stimulated only a slight increase in phosphorylation after the 2-min treatment that was not significantly different from basal levels. Nonetheless, as the data shown in Fig. 7C demonstrate, both ephedrine and dobutamine stimulated significant phosphorylation after extended times of treatment, and ephedrine, the weakest agonist, clearly promoted less phosphorylation than dobutamine after 20- and 30-min stimulations. CGP-12177, a weak partial agonist in the stimulation of adenylyl cyclase (Baker et al., 2002), the antagonist propranolol, and the negative agonist ICI-118,551 did not cause a significant increases in the initial levels of phosphorylation relative to basal (p > 0.21)
The correlation of efficacy with initial levels of GRK site phosphorylation suggests that all of the agonists share a similar activated state, at least within the accuracy of these measurements. This would mean that partial agonists for the most part mimic fractional occupancy of the receptor with epinephrine (Fig. 5B) without consideration of agonist-specific states, even though these specific states must be different based on thermodynamic considerations (Christopoulos and Kenakin, 2002). Based on these similarities in behavior with partial agonists and fractional occupancy of epinephrine, we expected the correlation of phosphorylation with efficacy to break down with prolonged stimulation. The results shown in Fig. 7C demonstrate that whereas albuterol (efficacy ≈25% of epinephrine) and especially salmeterol (efficacy ≈13% of epinephrine) caused a much-reduced phosphorylation over the early time points relative to epinephrine, they eventually caused phosphorylations (10–30 min stimulations) that almost equaled that of epinephrine. These data are consistent with the results shown in Fig. 5; that is, a 20- to 30-min treatment with 50 nM epinephrine caused a GRK site phosphorylation that was equal to that of 10 μM epinephrine. The time course for saturating salmeterol, whose efficacy was comparable with 50 nM epinephrine (15% occupancy), and that for dobutamine whose efficacy (4% of epinephrine) is comparable with 10 nM epinephrine (3.4% occupancy), were similar to the time courses of the respective epinephrine concentrations (Fig. 5). Ephedrine, the weakest agonist used in these studies (efficacy ≈ 2.7% of epinephrine), remarkably caused a 3- to 4-fold increase in GRK site phosphorylation after 10 min of stimulation. These results show that although initial rates of stimulation are in fact proportional to coupling efficiency, that proportionality breaks down with time of treatment.
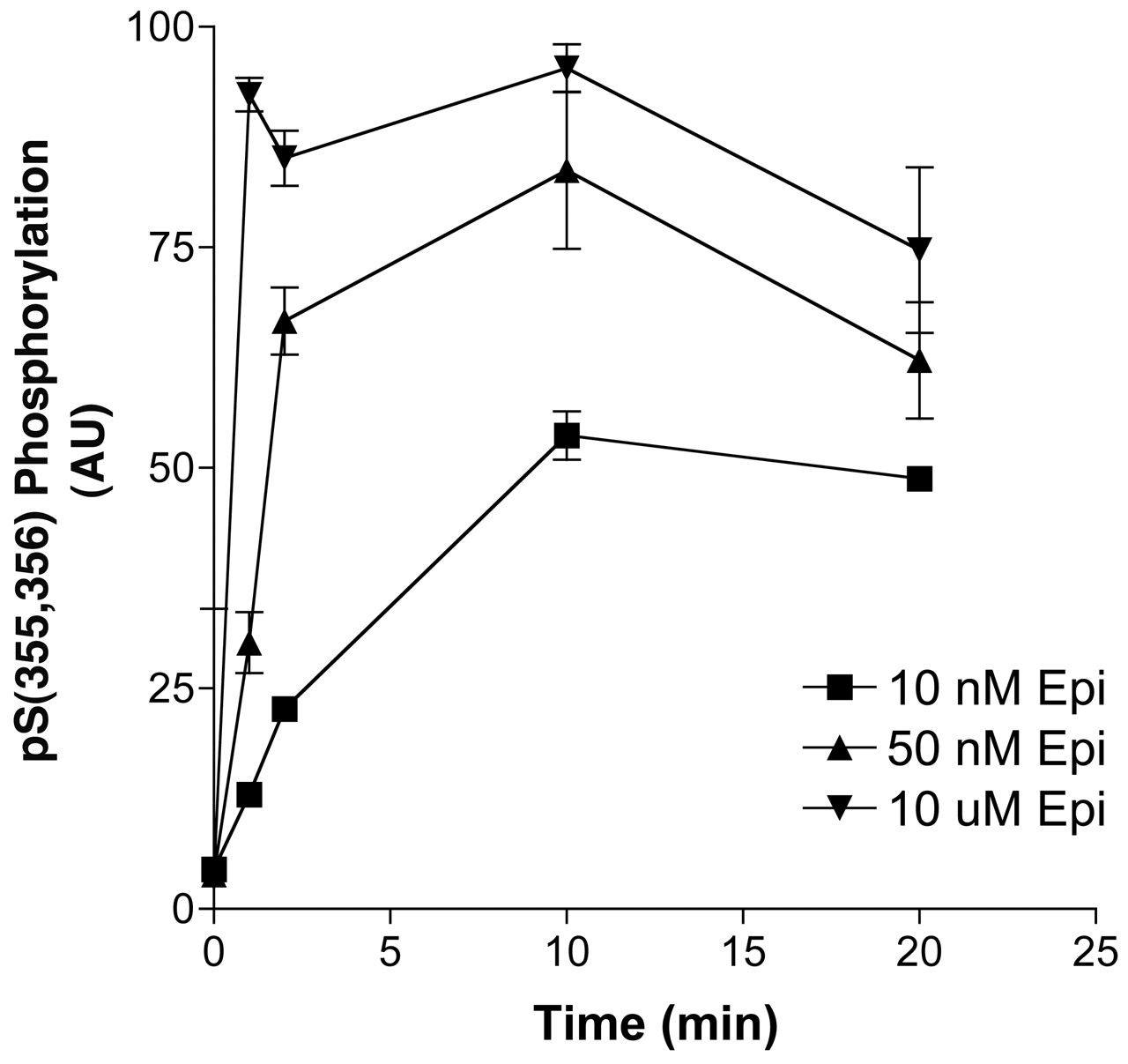
Absence of PKA Sites Does Not Alter GRK Site Phosphorylation. The possibility exists that there are hierarchical effects of carboxyl-terminal PKA site phosphorylation (S345,S346) on GRK phosphorylation (Moffett et al., 2001); however, in the absence of the direct means of assaying GRK site phosphorylation, it has been difficult to test that proposal. With the GRK site antibody, direct tests of this proposal were possible, and we performed two experiments to assess hierarchy. First, we measured the time course of the GRK site phosphorylation of the mutant receptor, PKA-, which lacks both PKA consensus sites, with anti-pSer(355,356) antibody. The data shown in Fig. 8 demonstrate that there was no apparent effect of the lack of these sites on the time and concentration dependence of epinephrine stimulation of GRK site phosphorylation relative to epinephrine stimulation of the wild-type Hβ2ARH. Second, Hβ2ARH cells were pretreated with forskolin for 30 min, and GRK site phosphorylation measured after 50 nM or 10 μM epinephrine stimulation. No significant effects of forskolin pretreatment were observed (data not shown). These results are consistent with our previous observations that the GRK-mediated desensitization proceeds unimpaired in the PKA- and that the rate and extent of GRK desensitization and internalization are unaltered relative to the WTβ2AR (Seibold et al., 2000).

Time course of GRK site phosphorylation after stimulation of PKA- cells with epinephrine. PKA- cells expressing a mutated Hβ2ARH lacking the two PKA consensus sites (S261,262A and S345,346A) grown in six-well dishes were stimulated for the indicated times with or 10 nM, 50 nM, or 10 μM epinephrine. The cells were solubilized and the β2AR was purified over Talon affinity columns, treated with N Gly F, and immunoblotted sequentially with anti-pSer(355,356) antibody and the anti-C-tail antibody. Phosphorylation data were first normalized to receptor levels determined with the anti-C-tail antibody and then to the maximal epinephrine stimulation in each experiment. Data shown are the means ± range (n = 2).
Kinetics of GRK Site Phosphorylation in A431 Cells Expressing Only Endogenous Receptor. Because all of the above studies were conducted with HEK 293 cells overexpressing the Hβ2ARH, we examined GRK and PKA site phosphorylation in A431 cells expressing only endogenous levels of the human β2AR (∼500 fmol/mg membrane protein). As the results shown in Fig. 9 demonstrate, 10 μM epinephrine was able to rapidly stimulate GRK site phosphorylation of the WTβ2AR in A431 cells. Although activation of GRK site phosphorylation by 5 nM epinephrine was slightly increased at 2 to 10 min, it seemed to be significant only at 30 min. The kinetics of the responses to 10 μM epinephrine over the various times of treatment were reasonably similar to those observed with the Hβ2ARH. We also found a significant 4- to 5-fold stimulation of PKA site phosphorylation after a 10-min stimulation with 10 nM and 1.0 μM epinephrine, although this required purification of the β2AR on C-tail antibody affinity resins (data not shown).

Time and concentration dependence of epinephrine stimulation of GRK site phosphorylation in A431 cells. A431 cells grown in six-well dishes were stimulated for various times with either carrier, 5 nM epinephrine, or 10 μM epinephrine as indicated. The cells were solubilized in 1 ml of solubilization buffer, treated with N Gly F, and 20 μl of 100-μl samples was subjected to SDS-polyacrylamide gel electrophoresis as described under Materials and Methods. Gels were transferred and incubated with anti-pSer(355,356) polyclonal antibody and quantitated after normalization to receptor levels assessed with the C-tail antibody. Data were further normalized to the maximal epinephrine value at 5 min and the values shown are the means ± range (n = 2). Immunoblots of representative data are shown in the inset.
Discussion
Previous studies of the phosphorylation of the β2AR were limited by the inherent difficulties and ambiguities in distinguishing the kinetics and concentration dependence of agonist-induced PKA and GRK site phosphorylations based on 32P labeling and mutagenesis. To surmount these problems, we explored the possibility of generating phospho-site-specific antibodies. Our results with these reagents have demonstrated the phospho-acceptor roles of Ser262 in the PKA-mediated desensitization, provided further evidence for the role of Ser355 and Ser356 in GRK-mediated desensitization, and allowed us to define the kinetics and concentration dependence of their phosphorylations. The most striking result was the remarkable difference in the concentration dependence of the epinephrine-induced phosphorylation of the PKA site relative to the phosphorylation of the putative GRK sites Ser355 and Ser356, revealing a difference in EC50 of more than 400- to 4000-fold, depending on whether the initial levels of GRK phosphorylation or the 30-min accumulations were compared. Our results also demonstrate that there is no obvious hierarchy of phosphorylation of these sites. That is, PKA site phosphorylation of Ser262 was not impaired by concentrations of epinephrine that drive GRK site phosphorylation, demonstrating that the desensitization caused by higher concentrations reflects the combined actions of the two protein kinases as well as the contribution from internalization. Furthermore, we found that the lack of PKA consensus sites did not alter the phosphorylation of the GRK sites. This conclusion is consistent with our previous studies of mutant receptors, such as the PKA- (Seibold et al., 2000) and the S49 lymphoma cell mutants cyc- and kin- lacking either Gs or PKA (Green and Clark, 1981). From these results, it can be extrapolated that the highly amplified PKA-mediated phosphorylation reflects the primary mechanism of desensitization for β2ARs exposed to blood levels of catecholamines that rarely exceed 1 to 2 nM, whereas the occupancy-dependent GRK site mechanism will become more pronounced at the higher concentration range that is the likely scenario at sympathetic neuroeffector synapses in the central nervous system or after treatment with β2AR-agonists. We have previously presented evidence based on the study of mutants of the PKA and GRK sites (January et al., 1997, 1998; Seibold et al., 1998, 2000) that the net desensitization at high concentrations of epinephrine for the Hβ2ARH is caused by approximately additive effects of the PKA site phosphorylation (2- to 3-fold decrease in potency of epinephrine activation of adenylyl cyclase), and the GRK site phosphorylation/internalization (3- to 4-fold decrease in potency).
The downshift in the EC50 for epinephrine-stimulated phosphorylation of the GRK sites Ser355 and Ser356 was striking and not fully understood. Our observation of a similar downshift in the EC50 for epinephrine stimulation of internalization is further evidence for the tight coupling and likely dependence of internalization on GRK site phosphorylation. It is well accepted that the GRK site phosphorylations and arrestin binding either greatly promote or are required for internalization (Krupnick and Benovic, 1998). A possible explanation for the observation that GRK phosphorylation with low occupancy (50 nM epinephrine) catches up in time with high occupancy (10 μM) is the differential actions of phosphatases on the GRK sites depending on the cellular location of the β2AR. Previous studies have suggested that dephosphorylation of the GRK sites requires internalization (Krupnick and Benovic, 1998). Figure 6 shows that with 50 nM epinephrine stimulation, the fraction of internalized receptor is considerably less than with 10 μM stimulation after either 5 or 30 min of stimulation. If internalization promotes dephosphorylation of the GRK sites there will be a higher rate of dephosphorylation in cells stimulated with 10 μM epinephrine. Preliminary studies of the washout of phosphorylation of the GRK sites after propranolol treatment (data not shown) indicate that there is a significant (≈10 min) delay of dephosphorylation of the GRK sites when propranolol is added after just 2 min of stimulation with 1 μM epinephrine (20–25% of the β2AR internalized), whereas there is no delay in dephosphorylation after a 20-min stimulation when 70 to 75% of the receptor is internalized.
In this study, we demonstrate that transient overexpression of Hβ2ARH in combination with either GRK2 or GRK5 in COS-7 cells caused an increase in GRK site phosphorylation without agonist stimulation, but had no significant effect on epinephrine stimulation. A qualitatively similar augmentation was seen in the Hβ2ARH cells transfected with the GRKs. In both cells, GRK5 was more effective than GRK2. Our results extend the previous observation that GRK2 overexpression augmented internalization in COS cells (Menard et al., 1997). It is possible that the overexpression of the receptor and GRKs induces a stable constitutive phosphorylation caused by increased levels of the activated state of the receptor (R*), greatly increasing the rate of GRK-mediated phosphorylation, and saturation of phosphatase activity. Although this may be the most parsimonious interpretation, it is also possible that transient receptor and GRK overexpression disrupt the normal subcellular localization and/or other controls of GRK activity. Aside from showing that the anti-pSer(355,356) antibody recognizes the putative GRK sites in the β2AR, these experiments raise the interesting possibility that physiologically relevant processes that increase GRK levels or activity may lead to agonist-independent desensitization of GPCRs. The lack of effect of either GRK2 or GRK5 on epinephrine stimulation probably reflects that maximal epinephrine stimulation fully saturates the putative GRK sites in both HEK 293 and COS-7 transfected cells. This is consistent with the hyperbolic nature of the relationship between GRK phosphorylation and efficacy (Fig. 7B). However, further studies will be required to determine the specific GRKs involved.
Prior studies had indicated that phosphorylation of the third intracellular loop PKA site was heterologous. That is, agonist occupancy was not required; rather any non-β2AR agonist or drug activation of PKA could result in desensitization of epinephrine stimulation (Clark et al., 1988, 1989; Kunkel et al., 1989; Hausdorff et al., 1991; Yuan et al., 1994). However, these conclusions were based on the measurement of the desensitization of adenylyl cyclase, and localization of the β2AR sites involved in heterologous desensitization based on site-directed mutagenesis. In this study, we show that, as expected, PGE1 and forskolin stimulations caused phosphorylation of Ser262 and that the time course and extent of phosphorylation were similar to those for epinephrine stimulation. PMA stimulation also generated some phosphorylation, although it was reduced by about 50% relative to epinephrine activation. This result is consistent with previous studies that demonstrated that PKC-mediated desensitization seemed to require both Ser261 and Ser262 (Johnson et al., 1990; Yuan et al., 1994).
Many epinephrine analogs are used in clinical medicine for the treatment of asthma and cardiovascular disease. The discovery of analogs of epinephrine that both produce strong activations of adenylyl cyclase and show relatively reduced desensitization would be of considerable use. It is clear that agonist-specific states are a thermodynamic necessity, and there is evidence for agonist-specific states of the β2AR (Gether et al., 1995; Krumins and Barber, 1997), as well as for other GPCRs (Berg et al., 1998; Clarke and Bond, 1998; Christopoulos and Kenakin, 2002). There have been reports of agonist stimulations of GPCR desensitization and down-regulation that deviate from predictions based on efficacy (Clark et al., 1999; Borgland et al., 2003). However, in our study of frequently used drugs, there was a strong correlation of efficacy with initial levels of GRK site phosphorylation, and we did not find a significant dissociation of efficacy and GRK site phosphorylation. The sensitivity of the GRK site antibody has allowed us to measure phosphorylation for even the weakest agonists, such as ephedrine and dobutamine. In our prior studies of the correlation of agonist efficacy with β2AR phosphorylation in the Hβ2ARH cells based on 32P-prelabeling, this sensitivity was not possible, nor could we distinguish whether the loss of phosphorylation with partial agonists was attributable to reduced PKA or GRK site phosphorylation.
In conclusion, the use of phospho-specific antibodies to the human β2AR phosphorylation sites as well as to many other GPCRs will be very useful in future studies. It will be of considerable interest and clinical importance to determine how these differences in the kinetics of protein kinase-mediated phosphorylations of the β2AR and other GPCRs in specific tissues affect their function in vivo. Our studies of the endogenous β2AR in human epidermoid carcinoma A431 cells, demonstrate that the antibodies to GRK sites Ser355 and Ser356 and PKA site Ser262 are probably sufficient for the study of endogenous receptors. Our studies indicate that these antibodies will be of use for the following: 1) directly discerning the specific in vivo actions of the GRK family of protein kinases; 2) evaluating structural requirements of the GRKs; 3) studying GRK regulation by other protein kinases such as Ca2+/calmodulin, mitogen-activated protein kinase, PKC, and Src (Krupnick and Benovic, 1998; Pronin et al., 1998; Fan et al., 2001; Kohout and Lefkowitz, 2003); 4) determining possible insulin counterregulation of the β2AR through activation of phosphatidyl inositol 3-kinase and subsequent Akt-mediated phosphorylation of the β2AR on PKA consensus site residues Ser345,Ser346 (Doronin et al., 2002); 5) establishing the role of phosphatases in reversal of GRK- and PKA-mediated effects in cells (Pitcher et al., 1995); and 6) screening inhibitors (Benovic et al., 1990; Penn et al., 1999), all without resorting to cell-free assays.
Prior characterizations of GRK2 and GRK5 using in vitro assays and knockouts have demonstrated that there is likely to be substantial specificity of their actions on the β2AR as well as many other GPCRs (Kunapuli et al., 1994; Krupnick and Benovic, 1998; Pronin et al., 1998; Kohout and Lefkowitz, 2003). In this study with transient transfection of an excess of GRKs into both COS-7 and HEK 293 cells, we found that GRK5 caused a greater increase in basal GRK site phosphorylation relative to GRK2. It is possible that we will ultimately find subtle or not so subtle physiological regulation of, for example, GRK5 versus GRK2 caused by their preferred phosphorylation of specific sites. It was shown in cell-free assays using artificial peptides as substrates that GRK5 may favor phosphorylation of sites not dependent on acidic residues such as Ser355 and Ser356, whereas GRK2 may favor phosphorylation of sites with proximal acidic residues (Kunapuli et al., 1994; Pronin et al., 1998). Use of knockouts, dominant-negative constructs, or interfering mRNA coupled with measurement of GRK localization and β2AR GRK site phosphorylation in future studies will help to resolve questions of the role of the GRKs in the regulation of β2AR phosphorylation in varied tissue and cell types, with the caveat that redundancy of the GRKs and other problems with knockdowns or knockouts must first be surmounted (Clark and Rich, 2003).
Footnotes
-
A detailed description of the measurement of coupling efficiencies for agonist activation of adenylyl cyclase in membranes from Hβ2ARH cells has been discussed by Eyad Qunaibi and Dr. Roger Barber, University of Texas Houston, Medical School, manuscript submitted.
-
ABBREVIATIONS: β2AR, β2-adrenergic receptor; GRK, G protein-coupled receptor kinase; PKA, cyclic AMP-dependent protein kinase; PKA-, double epitope-tagged β2AR with serines in both PKA consensus site substituted (S261,262A and S345,346A); Epi, epinephrine; PG, prostaglandin; HEK, human embryonic kidney; PMA, phorbol 12-myristate 13-acetate; WT, wild-type; AT, ascorbate/thiourea; DBM, dodecyl-β-maltoside; N Gly F, N-glycosidase F; mAb, monoclonal antibody; HA, hemagglutinin antigen; PKC, protein kinase C; Hβ2ARH, double epitope-tagged (HA tag N-terminal and His6 tag C-terminal) wild-type β2AR; CGP-12177, 4-[3-[(1,1-dimethylethyl)amino]2-hydroxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one; ICI-118,551, (±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol.
- Received June 5, 2003.
- Accepted October 3, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}