


Abstract
Heat shock proteins represent an emerging model for the coordinated, multistep regulation of apoptotic signaling events. Although certain aspects of the biochemistry associated with heat shock protein cytoprotective effects are known, little information is found describing the regulation of heat shock protein responses to harmful stimuli. During screening for noncanonical β adrenergic receptor signaling pathways in human urothelial cells, using mass spectroscopy techniques, an agonist-dependent interaction with β-arrestin and the 27-kDa heat shock protein was observed in vitro. Formation of this β-arrestin/Hsp27 complex in response to the selective β adrenergic receptor agonist isoproterenol, was subsequently confirmed in situ by immunofluorescent colocalization studies. Radioligand binding techniques characterized a homogeneous population of the β2 adrenergic receptor subtype expressed on these cells. Using terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling, immunoblot analysis and quantitation of caspase-3 activity to detect apoptosis, preincubation of these cells with isoproterenol was found to be sufficient for protection against programmed cell death initiated by staurosporine. RNA interference strategies confirmed the necessity for Hsp27 as well as both β-arrestin isoforms to confer this cytoprotective consequence of β adrenergic receptor activation in this cell model. As a result, these studies represent the first description of an agonist-dependent relationship between a small heat shock protein and β-arrestin to form a previously unknown antiapoptotic “signalosome.”
Programmed cell death or apoptosis is a natural mechanism by which proliferating tissue can maintain proper size and function. In addition, metabolic stresses can also initiate signals that mediate cell death or alternatively activate survival pathways that allow the cell to tolerate or recover from the same imposed damaging stimuli. This tight regulation of the pro- and antiapoptotic response ensures that neither aberrant cell survival nor inappropriate cell death occurs. Deviation from this strict control is thought to underlie the pathophysiological state for many human disorders. Considerable information is known about signaling mechanisms that initiate programmed cell death. However, the hallmark of apoptosis is the subsequent activation of cysteine proteases called caspases that mediate the biology of cell death (Thornberry and Lazebnik, 1998). Unfortunately, little information is known concerning the regulation of caspase activation.
The heat shock protein (HSP) subfamilies HSP90, HSP70, and HSP27 have been implicated for having an antiapoptotic role in response to various proapoptotic stimuli (Beere, 2005). Their ability to fold, transport, and form complex protein structures make these HSPs well suited to protect stressed cells by recognizing unstructured regions of proteins and exposed hydrophobic stretches of amino acids. In addition, HSPs also interact with several intracellular signaling molecules providing the cell with an ability to know whether to grow, divide, differentiate, or die. Small HSPs such as HSP27 mediate their antiapoptotic effects in an ATP-independent manner, which acutely protects stressed cells from damage when ATP levels are normally low (Parcellier et al., 2003). For example, one HSP27 regulatory effect on apoptosis is suggested to involve the activation of protein kinase B (Akt) (Rane et al., 2003). In addition, HSP27 has been shown to modulate cell death by complexing with cytochrome c, preventing the apoptotic protease activating factor-1 (Apaf-1) from interacting with procaspase-9 (Bruey et al., 2000). Although the biochemical interactions for many of these HSP subfamilies have been studied in detail, the regulatory mechanisms that generate these antiapoptotic signaling complexes remains obscure.
β-Adrenergic receptors (ARs) represent a larger family of seven-transmembrane receptors (7TMRs) that have been used as a model system to study agonist-dependent receptor signaling mechanisms (Pierce et al., 2002). In general, agonist stimulation of βARs leads to activation of the cAMP second messenger system that commonly requires selective receptor-Gαs protein interactions. G protein-independent signaling pathways related to βAR activation have been identified coupled through β-arrestin, a cytosolic protein historically associated with receptor desensitization (Luttrell et al., 1999). Consequently, similar noncanonical β-arrestin signaling pathways have been documented, not only for other 7TMRs but also for tyrosine kinase receptors (Lefkowitz and Shenoy, 2005). Some of the effects mediated through these atypical pathways include regulation of nuclear signaling, innate immune activation, and apoptosis.
Our laboratory reported the βAR-mediated transduction of pro-inflammatory signals in cultured human urothelial cells through a protein kinase A-independent signaling mechanism (Harmon et al., 2005). Therefore, we were subsequently interested in further investigating these alternative βAR signaling pathways (i.e., β-arrestin complexes). While examining potential noncanonical βAR signaling pathways, we initially identified, with the use of mass spectrometric analysis, a novel agonist-dependent interaction between β-arrestin and HSP27. Formation of this β-arrestin/HSP27 complex was hypothesized to be associated with protection against an apoptotic challenge and ultimately was found, using RNA interference (RNAi) strategies, to be necessary for this cytoprotective effect. These results are the first to describe an agonist-dependent relationship between a small HSP and β-arrestin to form a previously unknown antiapoptotic signaling complex.
Materials and Methods
Cell Culture. The immortalized human urothelial cell line (UROtsa) was a gift from Donald Sens (University of North Dakota, Grand Forks, ND) and was propagated as reported previously (Harmon et al., 2005). In brief, undifferentiated UROtsa were grown to confluence in serum-containing Dulbecco's modified Eagle's medium (DMEM) under standard cell culture conditions (37°C plus 5% CO2). Confluent UROtsa cells were washed in serum-free DMEM and preincubated for 1 h before the addition of the βAR agonist isoproterenol (ISO). Unless noted otherwise, ISO (Sigma-Aldrich, St. Louis, MO) was added to cells at a final concentration of 100 nM. At timed intervals after ISO addition (0, 2, and 5 min), the cross-linking reagent dithiobis succinimidyl proprionate (Sigma-Aldrich, St. Louis, MO) was added at a concentration of 2 mM. After 30 min of rocking at room temperature with dithiobis succinimidyl proprionate, cells were washed 2 times with Hanks' balanced salt solution and lysed with Mammalian Protein Extraction Reagent (Pierce, Rockford, IL). Protein concentrations were determined by standard Bradford analysis (Bradford, 1976).
Membrane Preparation for Receptor Binding. A crude UROtsa cell membrane preparation was prepared as described previously (Harmon et al., 2005). In brief, membranes were prepared by scraping and transferring cells to a 50-ml conical tube and twice washing by centrifugation at 500g using ice-cold Hanks' balanced salt solution. The intact cell pellet was resuspended in 10 ml of 0.25 M sucrose containing 10 μg/ml bacitracin, 10 μg/ml benzamidine, 10 μg/ml leupeptin, and 20 μg/ml phenylmethylsulfonyl fluoride. The cells were disrupted by freezing followed by Dounce homogenization of the thawed suspension using 25 strokes from a loose-fitting (B) pestle. This mixture was then centrifuged at 2100g for 15 min at 4°C. Buffer A (20 mM HEPES, pH 7.5, 1.4 mM EGTA, and 12.5 mM MgCl2) was added to the supernatant and centrifuged again at 30,000g for 15 min at 4°C. The resultant pellet was kept, resuspended in buffer A, and then centrifuged once more at 30,000g for 15 min at 4°C. The final crude membrane pellet was resuspended in buffer A containing 10% glycerol and stored in aliquots at -80°C until used for radioligand binding. Protein concentrations were measured using the method of Bradford (1976).
Radioligand Binding. The radioligand binding protocol used for this study was performed as described previously (Harmon et al., 2005). In brief, the density of expressed βARs on UROtsa cells was determined by saturation binding experiments using the nonselective βAR antagonist (-)3-[125I]iodocyanopindolol ([125I]CYP) as the radiolabel (PerkinElmer Life and Analytical Sciences, Waltham, MA). Crude UROtsa cell membranes were allowed to equilibrate at 37°C with increasing concentrations of [125I]CYP (10-500 pM) in a 0.25-ml total volume of buffer A using 10 μM propranolol to determine nonspecific binding. Binding was stopped by filtering the membranes though Whatman GF/C glass fiber filters, followed by five 5-ml washes with ice-cold buffer A to remove any unbound drug. Amounts of total and nonspecific radiolabel bound to cell membranes were calculated from radioactive counts remaining on the glass fiber filters. From the plotted saturation hyperbola, βAR density (Bmax) and affinity value (Kd) of [125I]CYP for UROtsa binding sites were calculated using iterative nonlinear regression analysis (Prism 5; GraphPad Software, San Diego, CA). Competition binding studies using increasing concentrations of the unlabeled AR ligands atenolol, CGP 20712A, ICI 118,551, and SR 59230A (Tocris Bioscience, Ellisville, MO) were performed in the same buffer used in saturation binding experiments. Iterative nonlinear regression analysis (GraphPad Prism) was again used to determine IC50 concentrations from which affinity values (Ki) of competing AR ligands for βARs expressed on UROtsa cells were calculated using the method of Cheng and Prusoff (1973).
Immunoprecipitation. A polyclonal antibody to pan-arrestin (AbCam, Cambridge, UK) was coupled to the AminoLink Plus coupling gel as part of the Seize Primary Mammalian Immunoprecipitation Kit (Pierce) according to the kit instructions. In brief, 400 μg of antibody was coupled to 200 μl of the AminoLink Plus gel. The antibody coupled gel was incubated with cross-linked UROtsa cell lysates in Mammalian Protein Extraction Reagent at 4°C for 16 h. After the incubation, the gel was washed three times with 0.4 ml of immunoprecipitation buffer (0.025 M Tris and 0.15 M NaCl, pH 7.2) and then the bound protein complexes were eluted in three fractions using ImmunoPure Elution buffer according to manufacturer's instructions. These three fractions were combined, and 30 μl of combined eluant with 2 μl of 1 M reducing agent dithiothreitol were run on Tris-HCl Ready Gels (Bio-Rad Laboratories, Hercules, CA) by standard SDS polyacrylamide gel electrophoresis (PAGE) methods (Harmon et al., 2005).
Mass Spectrometry. UROtsa cell lysates were prepared as detailed under Cell Culture and Immunopreciptitation. Immunoprecipitated (IP) lysates were then run on 4-to-20% gradient Ready Gels (Bio-Rad Laboratories) by standard reducing SDS-PAGE methods. Protein bands in the SDS-PAGE gel were excised and subjected to in-gel tryptic digestion as described by Miyagi et al. (2002), and the digest analyzed by liquid chromatography/tandem mass spectrometry (LC/MS/MS) techniques (Rao et al., 2005). Proteins were identified by comparing all of the experimental ion spectra with the National Center for Biotechnology Information human protein database using Mascot database search software (Matrix Science, London, UK). S-Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionine (methionine sulfoxide) was set as a variable modification in the database search. Mass tolerances for protein identification on precursor and product ions were both set to 0.2 Da. Strict trypsin specificity was applied, but allowed for one missed cleavage. A minimum Mascot search score of 15 was used as the cutoff for a positive identification.
Western Hybridization. IP or whole-cell lysates were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). Protein expression was measured by immunoblotting overnight at 4°C with diluted antibodies that recognize HSP27 (1:2000), caspase-3 (1:1000; both from Cell Signaling Technology, Danvers, MA, pan-arrestin (1:300; AbCam) or actin (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA). In some instances, β-arrestin isoform expression was measured using a 1:2500 dilution of the polyclonal antibody A1CT (Attramadal et al., 1992). After three washes in 20 mM Tris, pH 7.5, containing 140 mM NaCl and 0.05% Tween 20, the membranes were incubated at room temperature for 90 min with 1:5000 diluted horseradish peroxidase-conjugated secondary antibody (Jackson Immunoresearch Laboratories, West Grove, PA). After repeated washing, bound antibody was visualized using chemiluminescence (Pierce, Rockford, IL). Identified protein bands were photographed and quantified by standard densitometric analysis, using LabWorks Imaging Software (UVP, Inc., Upland, CA).
DNA Transfection, Immunocytochemistry, and Confocal Microscopy of HEK293 Cells. HEK293 cells were transfected with a hemagglutinin (HA)-tagged β2AR construct as described previously (Luttrell et al., 1999). In brief, HEK293 cells were seeded onto 25-mm coverslips and grown to 50% confluence. A pcDNA3.1 HA-tagged β2AR construct (Missouri University of Science and Technology cDNA Resource Center, Rolla, MO) was transfected using Gene-Jammer according to the manufacturer's instructions (Stratagene, La Jolla, CA). Forty-eight hours after gene transfection, HA-tagged β2AR were clustered at the HEK293 cell membrane as described previously (Luttrell et al., 1999). In brief, HEK293 cells were incubated with mouse, rabbit, or goat anti-HA antibodies (AbCam) diluted at 1:500 in serum-free DMEM for 1 h at 37°C. Cells were then incubated with secondary antibodies diluted in serum-free media for 1 h at 37°C to label clustered receptors. Secondary antibodies included fluorescein isothiocyanate-conjugated antimouse at 1:200, TexasRed-conjugated antirabbit at 1:100 (Jackson ImmunoResearch Laboratories, West Grove, PA), or horseradish peroxidase-conjugated anti-Goat at 1:100 (Santa Cruz Biotechnology).
After a 5-min subsequent incubation with or without 10 μM ISO, cells were fixed with 4% paraformaldehyde (USB, Cleveland, OH) for 10 min at room temperature. Fixed cells were washed in phosphate-buffered saline (PBS), permeabilized with 30 μM digitonin (Sigma-Aldrich, St. Louis, MO) for 10 min, then washed again in PBS. After a 30-min blocking incubation with 5% normal goat serum and 0.1% bovine serum albumin in PBS, cells were incubated with primary antibodies in a 1% normal goat serum solution for 1 h at 37°C. Rabbit anti-pan-arrestin (AbCam) and mouse anti-HSP27 (Cell Signaling Technology) were both used at a 1:50 dilution. Fluorescent secondary antibodies were incubated on the cells for 1 h at 37°C at a 1:100 dilution in 0.1% bovine serum albumin/PBS. Labeled coverslips were washed in PBS and mounted in VectaShield (Vector Laboratories, Burlingame, CA). Confocal microscopy was performed using a Zeiss 510 META LSCM microscope with a Zeiss confocal 63× oil immersion lens (Carl Zeiss Inc., Oberkochen, Germany). Antibody specificity was confirmed by examination of single-labeled, secondary only, and converse secondary hybridization slides.
Small Interfering RNA Transfection of UROtsa Cells. To reduce the levels of β-arrestin in UROtsa cells, specific small interfering RNA (siRNA) constructs were transfected as described previously (Ahn et al., 2003). Prevalidated siRNA sequences shown to target β-arrestin 1 and 2 isoforms were obtained along with nonsilencing siRNA duplexes (QIAGEN, Valencia, CA). The siRNA sequences targeting β-arrestin 1 (NM_020251) and β-arrestin 2 (NM_004313) are 5′-AAAGCCUUCUGCGCGGAGAAU-3′ and 5′-AAGGACCGCAAAGUGUUUGUG-3′ and correspond to the positions 439 to 459 and 201 to 221 relative to the start codon, respectively. The nonsilencing siRNA sequence (5′-UUCUCCGAACGUGUCACGU-3′) has no homology to any known mammalian gene and has been validated using genechip microarrays (Affymetrix, Santa Clara, CA). When UROtsa cells reached approximately 50% confluence in 100-mm plates, cells were transfected using HiPerFect Transfection Reagent according to manufacturer's instructions (QIAGEN). In brief, 50 μl of HiPerFect Reagent with or without concentrated nonsilencing or β-arrestin 1 and/or β-arrestin 2 siRNA solutions was added to 200 μl of DMEM. After incubating 10 min at room temperature, the mixture was added drop-wise to cells for a final individual siRNA concentration of 100 nM. After an additional 48-h incubation at 37°C in 5% CO2, cells were divided onto glass coverslips in six-well plates. After an additional 24-h incubation at 37°C in 5% CO2, cells were treated as described under Apoptosis Induction and Detection.
HSP27 levels were also reduced via a siRNA-mediated gene knock-down strategy (Park et al., 2003). The siRNA sequence (5′-ACGGUCAAGACCAAGGAUG-3′) targeting HSP27 (NM_001540) corresponded to positions 435 to 453 relative to the start codon. Transfection was done according to instructions of SignalSilence HSP27 siRNA Kit (Cell Signaling Technology). In brief, UROtsa cells were transfected with siRNA duplexes as described previously, with slight changes in the transfection reagent used, as per manufacturer's instructions. This change included using 10 μl of SignalSilence transfection reagent incubated in 500 μl of DMEM for 5 min at room temperature, followed by addition of nonsilencing or HSP27 siRNA. After an additional 5-min room temperature incubation, the entire transfection mixture was added to UROtsa cells in 100-mm plates for a final siRNA concentration of 100 nM. After 48 h incubation at 37°C in 5% CO2, cells were plated onto glass coverslips in six-well plates and incubated (37°C in 5% CO2) for an additional 24 h, then treated as described under Apoptosis Induction and Detection.
Apoptosis Induction and Detection. UROtsa cells were grown to confluence on glass coverslips for cytoprotection analysis. After a 30-min incubation (37°C in 5% CO2) in serum-free media, confluent UROtsa cells were treated with or without 100 nM ISO for 5 min. To induce apoptosis, staurosporine (STS; Sigma-Aldrich) was added at a concentration of 1 μM. Negative control samples were incubated in serum-free media containing either ISO alone or no additives. After 4.5 h of incubation at 37°C in 5% CO2, cells were fixed in 10% buffered formalin for 30 min and permeabilized with a 0.1% Triton X-100, 0.1% sodium citrate solution for 2 min. Apoptotic cells were then detected using the In Situ Fluorescein Cell Death Detection Kit (Roche, Indianapolis, IN) according to manufacturer's instructions. In brief, permeabilized cells were washed in PBS and were then incubated with terminal deoxynucleotidyl transferase biotin-dUTP nick-end labeling (TUNEL) enzyme solution for 60 min at 37°C. Cells were then washed in PBS and mounted in VectaShield containing the nuclear stain propidium iodide (PI). Slides were analyzed and photographed under standard fluorescent imaging techniques. Five microscope fields with an average of 64 ± 2 cells/field/condition were photographed at 40× magnification per slide. In addition, two slides were prepared for each experimental condition. Apoptotic cells were quantitated as a percentage of TUNEL-positive (green) nuclei based on the total number of PI (red) labeled nuclei per microscope field. Percentages reported represent the mean of all analyzed fields ± S.E. for n separate experimental days.
Caspase-3 Activity Assay. UROtsa cells were seeded in 96-well plates at a density of 12,500 cells/well and allowed to grow for 24 h. UROtsa cells were subsequently treated as detailed under Apoptosis Induction and Detection. After a 4.5-h incubation (37°C in 5% CO2) the caspase-3 activity was determined according to the manufacturer's instruction (Promega, Madison, WI). In brief, an equal volume (100 μl) of cell lysis/activity buffer containing the profluorescent substrate bis-N-benzyloxycarbonyl-l-aspartyl-l-glutamyl-l-valyl-l-aspartic acid amide, was added to the treated cells and incubated at room temperature for 14 h with constant shaking (300 rpm). The fluorescence of each well was then measured using an excitation wavelength of 485 nm and an emission wavelength of 528 nm. Data are presented as relative fluorescent units (RFU) after background is subtracted.
Statistical Analysis. For each individual binding experiment, the fitted iterative nonlinear regression curve that best represented the data were determined using a partial f test (Prism 5). All values are reported as the mean ± S.E. for n experiments. Significance between experimental groups was tested using an unpaired two-tailed Student's t test or, when appropriate, a one-way ANOVA with a post-hoc Newman-Keuls multiple comparison test (Prism 5). Calculated p values less than 0.05 were used as the level of significance.
Results
To discover novel proteins that conditionally bind to β-arrestin after βAR activation, UROtsa cells were stimulated with 100 nM ISO for 0, 2,, and 5 min. Afterward, cell lysates were cross-linked then immunoprecipitated with a pan-arrestin polyclonal antibody and the eluted protein complexes were separated by SDS-PAGE. Resolved proteins were visualized by SyproRuby staining and those bands whose intensities were significantly increased in the IP protein fractions of ISO treated cells were excised and digested by trypsin (data not shown). The resulting peptides were analyzed by LC/MS/MS, and the data were subjected to human database analysis for protein identification (Table 1). The database search included the identification of HSP27 by assigning three peptides to the protein (30% sequence coverage). No other statistically significant matches were recognized for these three peptides.
Human database analysis of statistically significant matches for resolved β-arrestin IP proteins from ISO treated UROtsa cells
Accession numbers refer to “GenInfo Identifier” sequence identification number from NCBI.
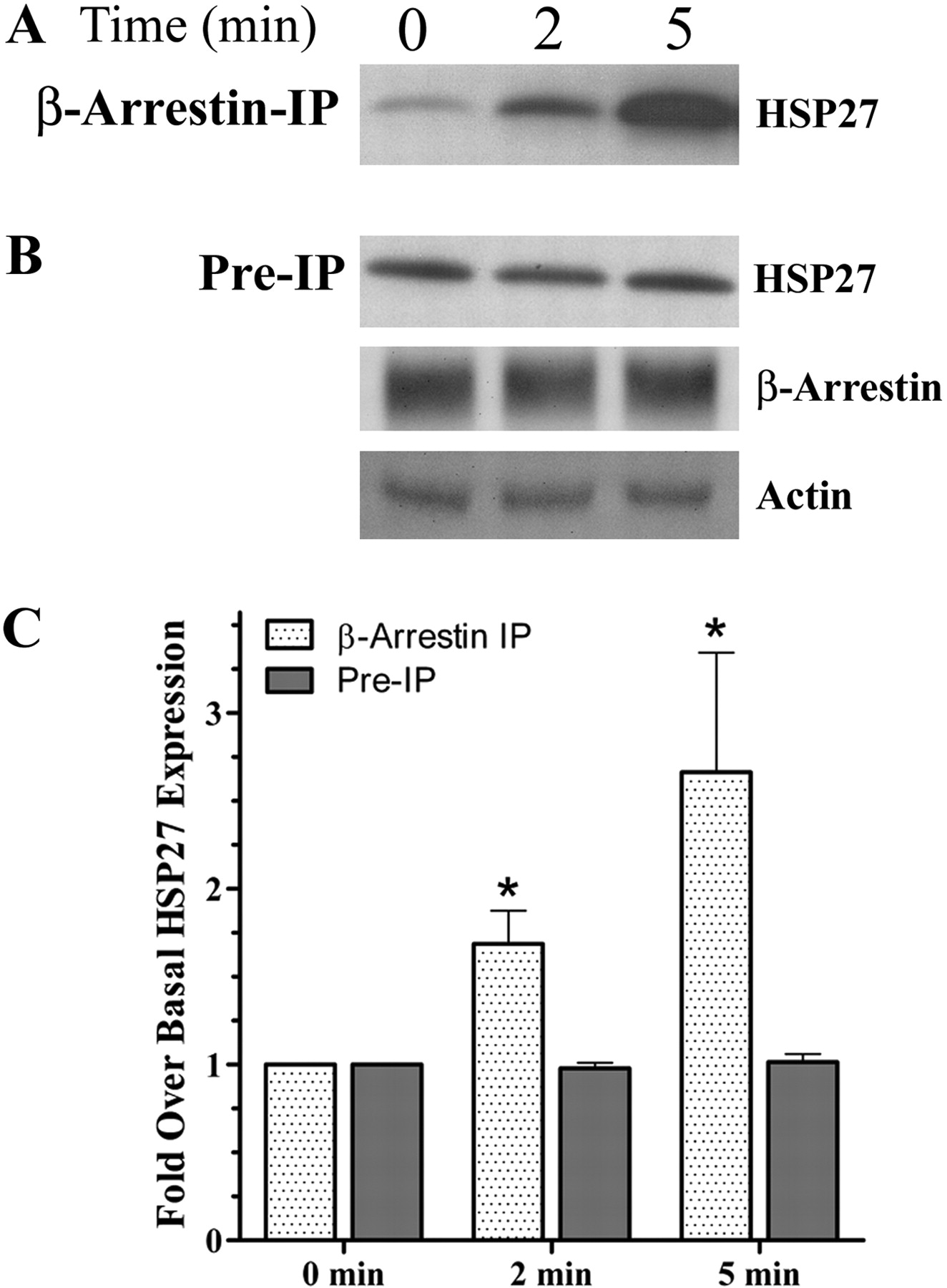
Although mass spectrometry revealed an ISO-induced interaction of HSP27 with β-arrestin, it was important to confirm these findings using Western analysis, especially because a limited number of bands were analyzed by LC/MS/MS. Therefore, β-arrestin IP protein complexes were again separated by SDS-PAGE. Probing the membrane with an anti-HSP27 antibody revealed significant increases in the binding of HSP27 with β-arrestin after ISO addition (Fig. 1A). Although a small amount of HSP27 was found complexed with β-arrestin after a 30-min preincubation in serum free-media, within 2 min after ISO addition, an increased amount of HSP27 was found associated with β-arrestin and by 5 min, the amount of coIP HSP27 was drastically increased over basal levels (n = 3). Figure 1C illustrates the densitometric quantification, which revealed 1.7 ± 0.2- and 2.7 ± 0.7-fold increases after 2 and 5 min, respectively, that was significant from basal (p < 0.05). These observations suggest that βAR stimulation resulted in an increased association of HSP27 with β-arrestin.
To confirm that these effects were not the consequence of a global increase in HSP27 levels throughout the cell, we analyzed UROtsa whole-cell lysates (i.e., preIP) 0, 2, and 5 min after ISO addition. Probing these whole cell lysates with anti-HSP27 antibody showed an unvarying level of HSP27 over the same time course (n = 3; Fig. 1B). In addition, equivalent amounts of β-arrestin were loaded onto the coupling gel before IP. Moreover, equal amounts of total protein lysates were used from each treatment for the IP protocol as indicated by similar actin band intensities. Finally, densimetric analysis of these band intensities confirmed no significant changes in HSP27 preIP levels, 2 and 5 min after ISO addition (Fig. 1C). These results demonstrate a specific subcellular association of HSP27 with β-arrestin after stimulation of βAR with ISO.
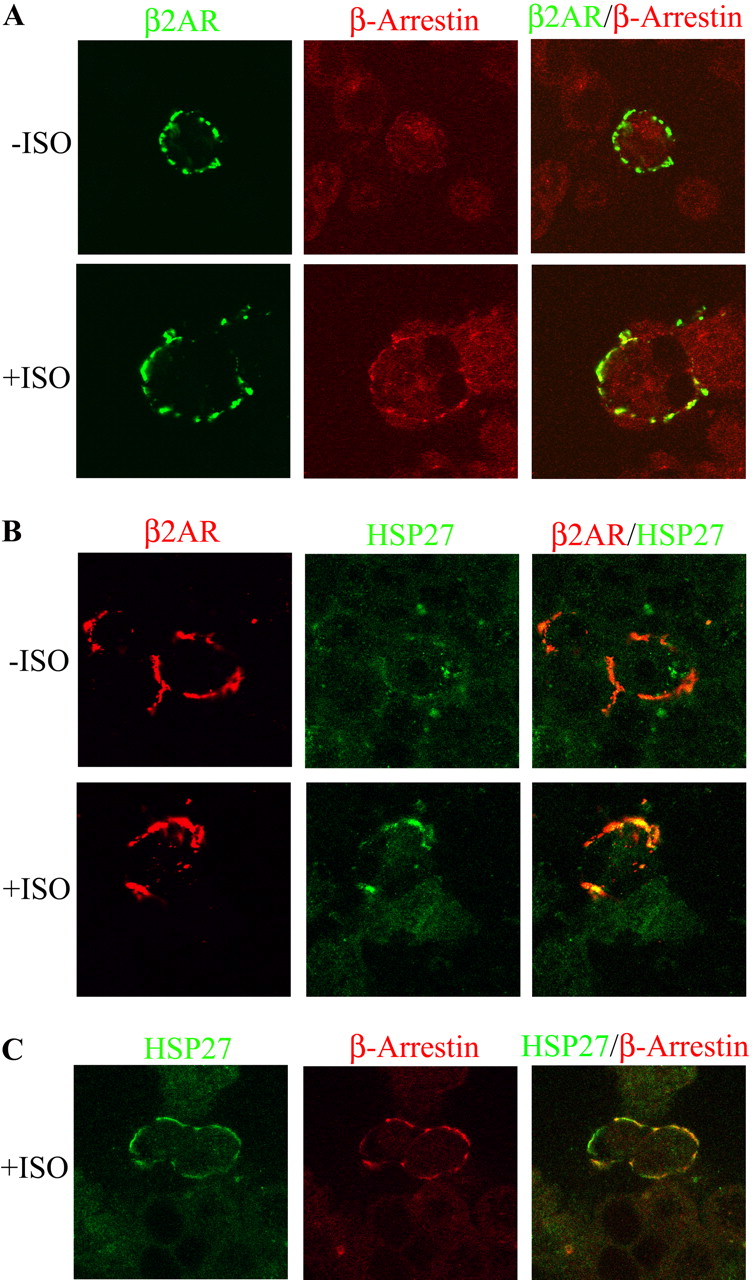
Although in vitro analysis demonstrated an increased agonist-dependent interaction of HSP27 with β-arrestin after βAR stimulation, it was necessary to show whether these proteins associated in living cells. Therefore, to visualize formation of AR signaling complexes, HEK293 cells were transfected with an N-terminally HA-tagged β2AR construct. The expressed receptors were labeled with antibodies against the HA-epitope and were subsequently clustered at the cell membrane surface with an appropriate conjugated fluorescent secondary antibody (fluorescein isothiocyanate or TexasRed). After 5-min incubation with or without 10 μM ISO, these cells were fixed, and the subcellular localization of β-arrestin or HSP27 was observed using confocal immunofluorescence microscopy. In nonstimulated cells, endogenous β-arrestin was found throughout the cellular cytoplasm with no distinct aggregates corresponding to the distribution of β2AR clusters. However, after a 5-min incubation with ISO, a portion of β-arrestin was found associated with labeled β2ARs (n = 3; Fig. 2A). This result is similar to observations by others that describe an agonist-dependent translocation of β-arrestin to βARs (Luttrell et al., 1999). In the same manner, HSP27 labeling showed diffuse staining throughout the cytoplasm in the absence of ISO that did not match up with β2AR punctate staining. However, a 5-min stimulation with ISO induced a dramatic orientation of HSP27 to the cell membrane that was perfectly colocalized with aggregated β2ARs at the extracellular surface (n = 3; Fig. 2B). This observation demonstrates a recruitment of HSP27 to activated βARs possibly through association with β-arrestin.

βAR activation increases association of β-arrestin with HSP27 in vitro. A, UROtsa cells stimulated with 100 nM ISO were IP using a pan-arrestin antibody, resolved by SDS-PAGE, and immunoblotted with an anti-HSP27 antibody. The representative blot (n = 3) shows a time-dependent increase in HSP27 immunoreactivity (27 kDa) associated with β-arrestin IP lysates. B, ISO-stimulated whole-cell lysates resolved by SDS-PAGE and immunoblotted with an anti-HSP27 (27 kDa), pan-arrestin (49 or 47 kDa), and actin (42 kDa) antibodies. The representative blot shows no change over time in the cytosolic levels of these proteins after ISO stimulation. C, densitometric quantification of immunoblot digital images illustrate the significant (p < 0.05) time-dependent increase (1.7 ± 0.2 and 2.7 ± 0.7-fold after 2 and 5 min, respectively) of HSP27 associated with β-arrestin IP UROtsa cell lysates with no change in preIP levels compared with basal (n = 3).

In situ analysis of HSP27 interaction with a βAR signaling complex transiently transfected in HEK293 cells. Subcellular localization of β-arrestin or HSP27 with immunoclustered HA-tagged β2AR subtypes after 5 min with or without 10 μM ISO was observed using confocal immunofluorescence microscopy. A, as described previously (Luttrell et al., 1999), endogenous β-arrestin in nonstimulated cells were diffusely labeled throughout the cytoplasm. However, after ISO incubation, a portion of the β-arrestin staining was confined with clustered β2ARs (n = 3). B, HSP27 fluorescent labeling was diffuse throughout the cytoplasm in the absence of ISO, which did not match up with clustered β2AR staining. Conversely, ISO stimulation induced a dramatic orientation of HSP27 to the cell membrane, which colocalized with aggregated β2ARs (n = 3). C, transiently expressed β2AR subtypes were clustered using nonfluorescent antibodies before the addition of ISO. Endogenous β-arrestin and HSP27 were subsequently labeled with fluorescent antibodies and found to be colocalized in the same agonist-dependent complex (n = 3).
To validate that β-arrestin and HSP27 proteins could be found in the same membrane complex, HA-tagged β2ARs were clustered using nonfluorescent antibodies before the addition of ISO. Endogenous β-arrestin and HSP27 proteins were then fluorescently labeled and found to be colocalized in an agonist-dependent manner (n = 3; Fig. 2C). This result supports our in vitro analysis using UROtsa cell lysates and suggests that HSP27 and β-arrestin are found together in the same receptor agonist induced complex after βAR activation, which is a relationship currently not described in the literature.
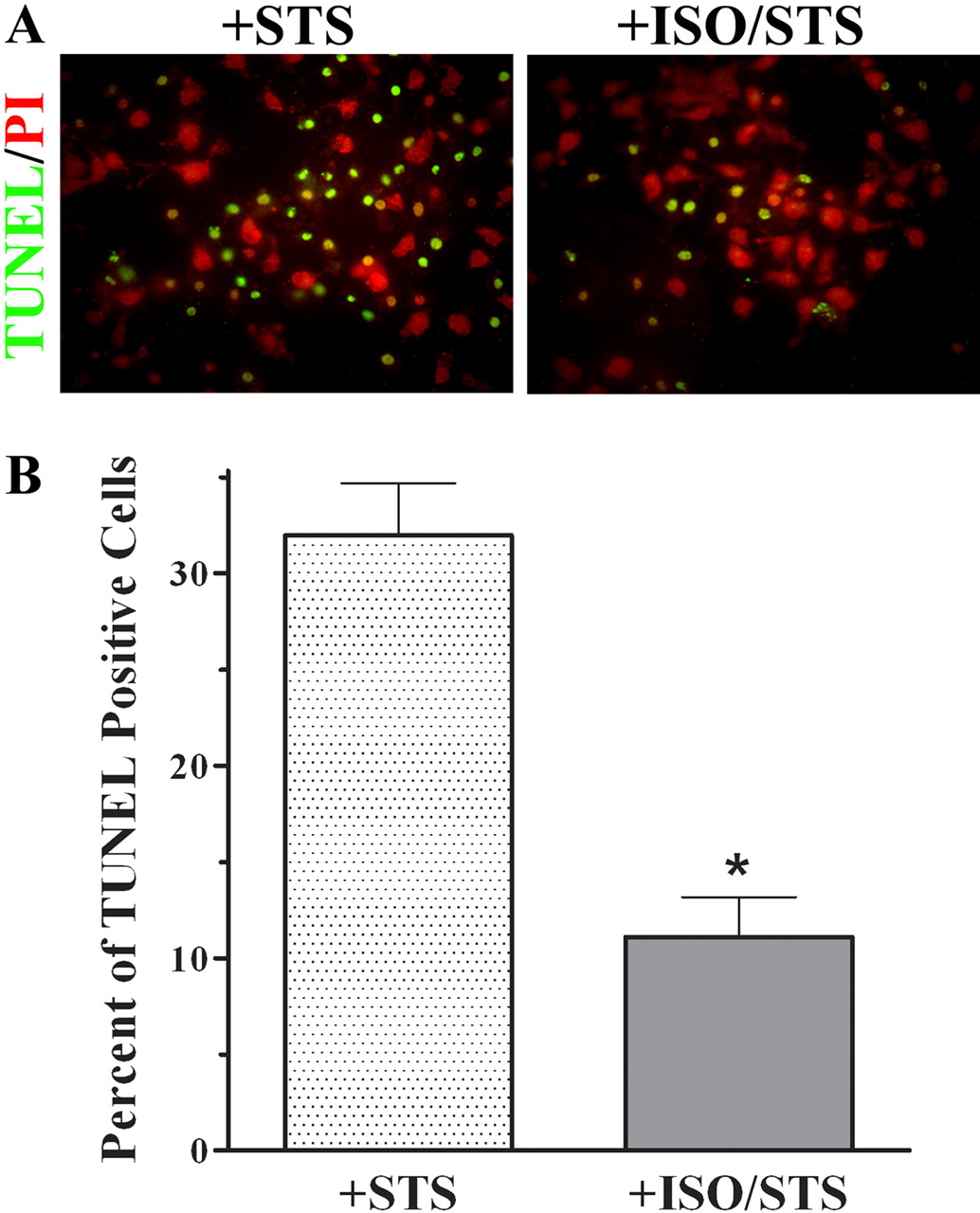
Because HSP27 has a well characterized cytoprotective function, we investigated whether ISO recruitment of this β-arrestin/HSP27 complex is associated with an increased resistance to apoptosis. A 4.5-h incubation with 1 μM STS, a proapoptotic drug, increased the number of TUNEL-positive UROtsa cells to 32 ± 3% of total PI-stained cells (n = 3; Fig. 3). A 5-min preincubation with 100 nM ISO before treatment with STS significantly reduced (p < 0.05) the number of TUNEL-positive cells to 11 ± 2% of total (n = 3). Less than 1% of total PI stained UROtsa cells pretreated with ISO only or media alone were TUNEL-positive after a 4.5-h incubation (n = 3, data not shown).
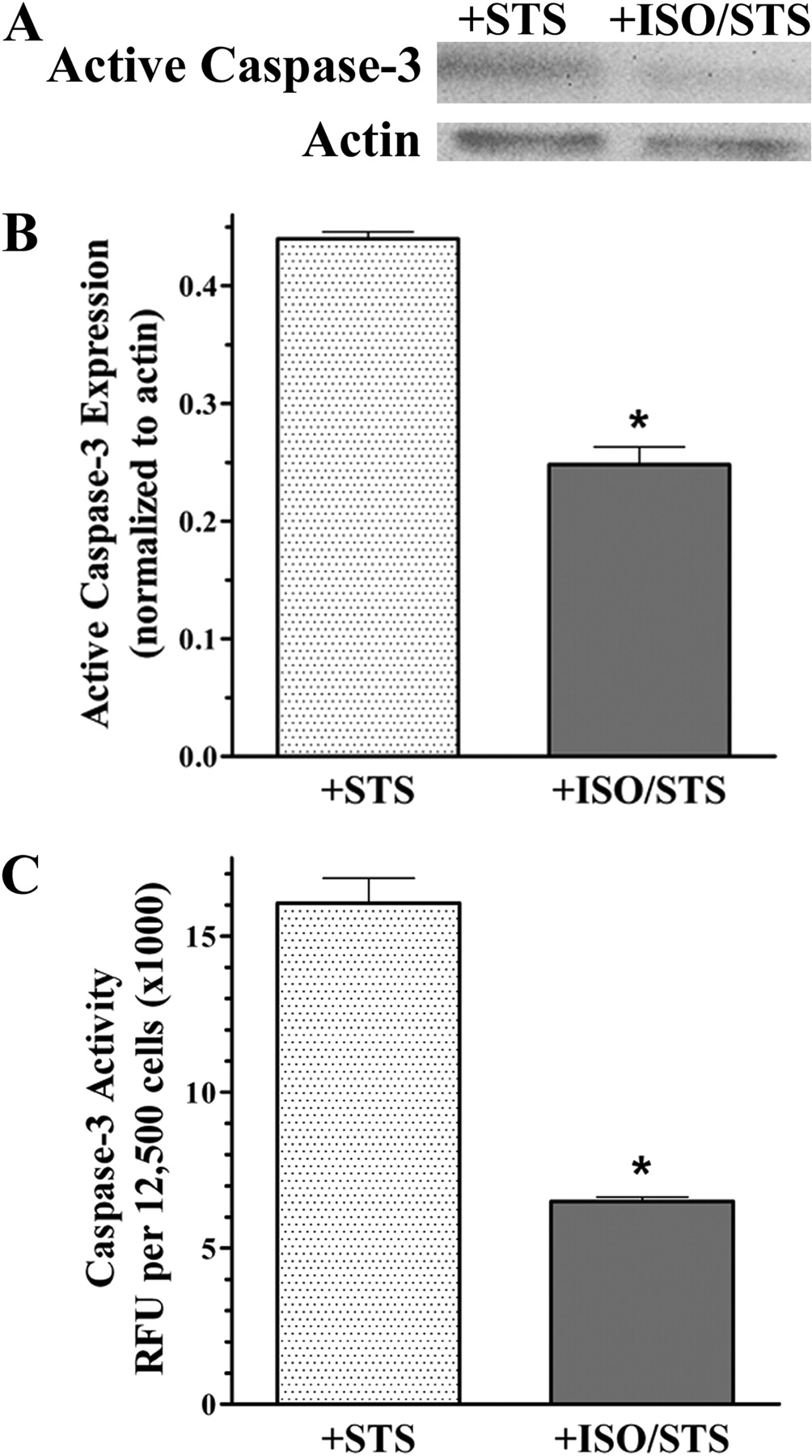
The apoptotic nuclear morphology detected by TUNEL is a direct result of active caspase-3 cleaving an inhibitor, which regulates the nuclease responsible for DNA fragmentation (Enari et al., 1998). Therefore, to support our TUNEL results, we examined amount and activity of active caspase-3 generated in UROtsa cells using the same treatment described previously to induce apoptosis with and without ISO pretreatment. In control cell lysates, the 17-kDa cleaved fragment of active caspase-3 was not detected (n = 3; results not shown). Conversely, resolved lysates from STS-only treated cells showed a substantial increase in the expression of this 17-kDa protein band, complementing the TUNEL results illustrated earlier (Fig. 4A). However, there was a decreased amount of active caspase-3 generated in UROtsa cells pretreated with ISO (Fig. 4A). Densitometric quantification normalized to actin illustrates the approximate 2-fold significant (p < 0.05) decrease in expression of active caspase-3 from cell lysates that had been pretreated with ISO (0.25 ± 0.02) compared with STS only (0.44 ± 0.01; n = 3; Fig. 4B). This diminished generation of active caspase-3 in ISO pretreated cells compared with STS treatment alone correlated to a decrease in RFU/12,500 cells when using a specific profluorescent substrate to measure caspase-3 enzymatic activity (Fig. 4C). In this study there was a significant (p < 0.05) 2.5-fold decrease in caspase-3 activity in cells pretreated with ISO (6485 ± 88 RFU) compared with cells treated with STS only (16,078 ± 592 RFU; n = 3; Fig. 4C). Together, these caspase-3 and TUNEL results demonstrate that βAR-activated subcellular recruitment of HSP27 with β-arrestin is associated with an increased resistance to an apoptotic challenge in cultured urothelial cells.

βAR activation provides protection from an apoptotic challenge. A, representative images (n = 3) of fixed TUNEL positive UROtsa cells costained with PI that had been exposed to 1 μM STS for 4.5 h after pretreatment with or without 100 nM ISO for 5 min. B, percentage of TUNEL positive cells normalized to total PI-stained cells calculated from five microscopic fields for each treatment performed in duplicate. STS treatment alone increased the number of TUNEL-positive cells to 32 ± 3% of total, whereas ISO preincubation before STS treatment significantly reduced (p < 0.05) the number of TUNEL positive cells to 11 ± 2% of total (n = 3).

Decreased caspase-3 generation and function after βAR activation. A, representative blot (n = 3) of resolved UROtsa cell lysates that had been exposed to 1 μM STS for 4.5 h after pretreatment with or without 100 nM ISO for 5 min. There was a decreased immunoreactivity of the large cleaved fragment representing active caspase-3 (17 kDa) in ISO-pretreated cells with no changes in the actin immunoreactivity (42 kDa) compared with STS only. B, densitometric quantification of the immunoblot digital images illustrates a significant (p < 0.05) decrease in the generation of active caspase-3 normalized to actin for cells pretreated with ISO (0.25 ± 0.02) compared with cells treated with STS (0.44 ± 0.01; n = 3). C, quantitation of caspase-3 function using the profluorescent substrate bis-N-benzyloxycarbonyl-l-aspartyl-l-glutamyl-l-valyl-l-aspartic acid demonstrated a significant (p < 0.05) decrease in enzymatic activity normalized to 12,500 cells from lysates pretreated with ISO (6485 ± 88 RFU) compared with treatment with STS only (16078 ± 592 RFU; n = 3).
In cardiac myocytes, βAR-mediated effects on programmed cell death depend on which βAR subtype is being activated (for review, in Zheng et al., 2005). In this review, stimulation of the β1AR subtype is suggested to be associated with promoting apoptosis, whereas β2AR activation is linked to inhibition programmed cell death. However, it is currently unknown in our system if ISO-mediated recruitment of β-arrestin/HSP27 is responsible for inhibiting a pro-apoptotic pathway (β1AR) or initiating an antiapoptotic signal (β2AR). Therefore, radioligand binding studies were used to quantify the amount and characteristics of the βAR subtypes expressed on UROtsa cell membranes. Increasing amounts of the iodinated nonselective βAR antagonist [125I]CYP specifically labeled a saturable, homogeneous, high-affinity binding site on these cell membranes (data not shown). The number of binding sites was determined to be 583 ± 73 fmol/mg protein (n = 5) and the equilibrium dissociation constant of [125I]CYP for these binding sites was 45 ± 18 pM (n = 5), which is similar to the calculated affinity value of this radiochemical when it was used by others to identify β-AR binding sites on cell membranes (Hoffmann et al., 2004). Competitive binding assays were also performed using a variety of AR antagonists to establish the characteristics of these specific [125I]CYP binding sites expressed on UROtsa membranes. From these experiments, competition curves were generated and used to calculate affinity values for selective βAR antagonists (Table 2). When the selective βAR antagonist, propranolol was used to compete for specific [125I]CYP binding sites the data were fit to both one- and two-site binding models. Using a partial f test, the two-site model was not found to be significantly better. Therefore, the propranolol competition curve was modeled to a one-site fit with a calculated equilibrium dissociation constant (Ki) of 1.5 ± 0.7 nM (n = 3) for these UROtsa cell binding sites. This value is similar to the calculated affinity of propranolol when used to identify β1 and β2AR subtypes in other membrane preparations (Smith and Teitler, 1999) and suggest that the β3AR subtype is not expressed in our system. Additional subtype-selective βAR antagonists were also used to establish specific βAR subtype expression on UROtsa cell membranes. Again, a one-site model fit best when the selective β1AR antagonists, atenolol, and CGP 20712A, were used for competitive displacement of specific [125I]CYP binding sites on these membranes. From these curves, single, high-value, equilibrium dissociation constants of 6668 ± 1068 nM and 5943 ± 2462 nM were calculated for CGP 20712A (n = 3) and atenolol (n = 3), respectively, which correspond to affinity values previously reported for the β2AR subtype in other systems (Smith and Teitler, 1999; Yamanishi et al., 2002). However, use of CGP 20712A and atenolol can only effectively discern expression of β1 versus the β2 or β3AR subtypes. Therefore, the β3-selective AR antagonist SR 59230A was included in the pharmacological profile of ligands used for competitive binding assays to support the notion for the lack of β3AR binding sites expressed on UROtsa cells (Table 2). Again, a one-site competition curve was best modeled to the SR 59230A data with a calculated Ki of 88 ± 41 nM (n = 3) for these cell membrane binding sites. This Ki does not correspond to previous reports, where SR 59230A was used to identify β3ARs in other preparations (Candelore et al., 1999) supporting our propranolol results, which suggests a lack of β3AR subtype expression on UROtsa cells. However, this lower SR 59230A affinity is comparable with calculated values of this ligand for endogenous β2AR binding sites expressed on cell membranes prepared from tissue (Yamanishi et al., 2002). This result, including affinity values calculated for CGP 20712A and atenolol, suggests that neither the β1 nor the β3 subtype, but instead the β2AR subtype, is expressed on UROtsa cell membranes. However, to confirm a homogenous population of β2AR is present in our system, the selective β2AR antagonist ICI 118,551 was used to compete for specific [125I]CYP binding sites. Not unexpectedly, a one-site competition curve again fit best to the data with a calculated low Ki value of 1.0 ± 1.0 nM (n = 3) for these binding sites, corresponding to previous affinity estimates identifying the β2AR subtype transiently expressed in CHO cells (Hoffmann et al., 2004). This pharmacological profile of selective βAR antagonist affinity values ([125I]CYP < ICI 118,551 = propranolol < SR 59230A < atenolol ≤ CGP 20712A) compared with equilibrium dissociation constants for all three βAR subtypes calculated in other systems indicates that a homogenous population of β2ARs, previously linked to antiapoptotic signaling mechanisms, is expressed on UROtsa cell membranes (Table 2).
Calculated Ki values of selective βAR antagonists for specific 125I-CYP binding sites
To corroborate whether these cytoprotective effects of βAR stimulation are mediated through HSP27, a RNAi strategy was employed. Previously characterized siRNA duplexes were used in this approach to successfully decrease UROtsa cellular levels of HSP27 before treatment with STS and/or ISO (Park et al., 2003). Nontransfected cells as well as those transfected with nonsilencing siRNA duplexes showed a similar percentage of TUNEL-positive cells after a 4.5-h STS incubation (46 ± 3 and 42 ± 4% of total cells, respectively; n = 3; Fig. 5A). As described previously, a 5-min preincubation with 100 nM ISO significantly reduced (p < 0.05) the percentage of TUNEL-positive UROtsa cells in both nontransfected and cells transfected with nonsilencing siRNA (23 ± 2 and 20 ± 3% of total, respectively; n = 3; Fig. 5A). However, in UROtsa cells transfected with HSP27 siRNA, a 5-min pretreatment with ISO did not have the same cytoprotective effect after a 4.5-h STS treatment as observed in controls. There was no difference in the number of TUNEL-positive HSP27 knockdown cells with (45 ± 3% of total) or without (41 ± 5% of total) ISO pretreatment (n = 3; Fig. 5A). Western hybridization techniques were also employed to authenticate the specificity of siRNA duplexes used to decrease HSP27 cellular levels in our system (Fig. 5B). There was a qualitative decrease in the amount of HSP27 expression in cell lysates that had been transfected with HSP27 specific siRNA duplexes compared with nontransfected as well as nonsilencing transfected cells (n = 3). Conversely, there were no expression level changes for either β-arrestin isoforms or actin in all siRNA treatments used, indicating a specificity of our RNAi protocol for HSP27 (n = 3). Furthermore, these observations indicate that HSP27 is a necessary signaling component for the cytoprotective effects after βAR activation in UROtsa cells.

HSP27 RNAi techniques reverse βAR mediated cytoprotection. A, percentage of UROtsa cells that were TUNEL-positive after being transiently transfected with nonsilencing or HSP27 siRNA duplexes then subsequently exposed to 1 μM STS for 4.5 h after pretreatment with or without 100 nM ISO for 5 min. Actual numbers of TUNEL-positive cells were normalized to total PI-stained cells, calculated from five microscopic fields for each treatment performed in duplicate. STS alone caused similar increases in the number of TUNEL-positive cells for control, nonsilencing, and HSP27 siRNA treatments (46 ± 3, 42 ± 4, and 41 ± 5% of total, respectively; n = 3). ISO preincubation before STS treatment significantly reduced (p < 0.05) the number of TUNEL-positive cells for control and nonsilencing siRNA transfections to 23 ± 2 and 20 ± 3% of total (n = 3). There was no difference in the percentage of TUNEL-positive HSP27 knockdown cells preincubated with ISO (45 ± 3%) compared with STS treatment alone (n = 3). B, representative blot (n = 3) of resolved UROtsa cell lysates transiently transfected with nonsilencing or HSP27 siRNA duplexes. There was a decreased HSP27 immunoreactivity (27 kDa) in cells transfected with HSP27 siRNA duplexes compared with nontransfected cells or cells transfected with nonsilencing siRNA. Conversely, there were no changes among all RNAi treatments in the protein expression of either β-arrestin isoform (49 or 47 kDa) or actin (42 kDa).
Our studies demonstrate a βAR agonist-dependent colocalization of HSP27 with β-arrestin. However, to establish that β-arrestin is also an essential component for the cytoprotective effect of HSP27 after ISO preincubation, RNAi techniques using previously characterized siRNA sequences were again employed to successfully decrease UROtsa cell levels of β-arrestin isoforms 1 and 2 before treatment with STS and/or ISO (Ahn et al., 2003). As described previously, nontransfected cells as well as those transfected with nonsilencing siRNA showed similar percentages of TUNEL-positive cells after a 4.5-h STS incubation (42 ± 3 and 46 ± 4% of total cells, respectively; n = 4; Fig. 6A). Again, a 5-min preincubation with 100 nM ISO significantly reduced (p < 0.05) the percentage of TUNEL-positive UROtsa cells in both nontransfected cells and cells transfected with nonsilencing RNA (17 ± 2 and 19 ± 2% of total, respectively; n = 4; Fig. 6A). Once more, in cells transfected with β-arrestin 1/2 siRNA, a 5-min pretreatment with ISO did not have the same cytoprotective effect after a 4.5-h STS treatment as observed in controls. As in the HSP27 siRNA transfected cells, there was no difference in the number of TUNEL-positive β-arrestin knockdown cells with (44 ± 5% of total) or without (41 ± 4% of total) ISO pretreatment (n = 4; Fig. 6A). Moreover, treatment with ISO or DMEM alone did not induce apoptosis in any of the β-arrestin knockdown cells observed (n = 4, data not shown). Western hybridization techniques were also employed to authenticate the specificity of siRNA duplexes used to decrease β-arrestin 1 and 2 cellular levels in our model (Fig. 6B). There was a qualitative decrease in the expression of both β-arrestin isoforms from cell lysates that had been transfected with β-arrestin-specific siRNA duplexes compared with nontransfected as well as nonsilencing transfected cells (n = 4). Conversely, there were no expression level changes for the actin protein, which was also used as a loading control (n = 4). This observation indicates that β-arrestin is a fundamental component for the cytoprotective effects of HSP27 after βAR activation in these cells.
To better understand the cellular mechanisms underlying this antiapoptotic response, the same RNAi technique was employed, except that individual siRNA duplexes corresponding to specific β-arrestin isoforms were used for the transient transfection of UROtsa cells. In addition, to corroborate results of the previous pan-arrestin siRNA transfections, which used TUNEL as a measurement of apoptosis, the functional response of cells transfected with individual β-arrestin siRNA duplexes were assessed by the amount of active caspase-3 immunoreactivity using immunoblot analysis. UROtsa cells transfected with siRNA duplexes specific for β-arrestin 1 transcripts showed a qualitative increase in the immunoreactivity of the large 17-kDa active caspase-3 fragment after a 4.5-h STS challenge compared with control, which correlates well with previous TUNEL analysis demonstrating an induction of apoptosis (n = 3, Fig. 6C). However, there were no differences in the amount of active caspase-3 generated in cells preincubated with 100 nM ISO before an STS challenge compared with β-arrestin 1 siRNA-transfected cells treated with STS alone. Equal amounts of resolved total protein lysates from each β-arrestin 1 siRNA treatment is indicated by similar actin immunoreactive band intensities. Moreover, semiquantitative densimetric analysis of active caspase-3 immunoblot intensities generated by STS confirmed no significant changes with (0.43 ± 0.01) or without (0.44 ± 0.01) ISO pretreatment when normalized to actin. Likewise, STS-stimulated UROtsa cells transfected with specific β-arrestin 2 siRNA duplexes also demonstrated no qualitative differences in active caspase-3 immunoreactivity with or without ISO pretreatment (n = 3, Fig. 6C). Once more, densimetric analysis of these active caspase-3 band intensities established no significant changes with (0.44 ± 0.01) or without (0.44 ± 0.01) ISO pretreatment when normalized to actin. Finally, immunoblot techniques were used to validate the effectiveness of each siRNA duplex used individually to decrease protein levels of specific β-arrestin isoforms in our system (Fig. 6D). There was a qualitative decrease in the amount of β-arrestin 1 or β-arrestin 2 expression from cell lysates transfected with isoform specific siRNA duplexes with no changes in expression of the correlate β-arrestin isoform compared with nontransfected cells (n = 3). Conversely, there were no expression level changes for either HSP27 or actin for all specific β-arrestin siRNA treatments used, indicating a selectivity of our RNAi protocol for each β-arrestin isoform (n = 3). Furthermore, no changes in the levels of active caspase-3 immunoreactivity in cells individually treated with specific β-arrestin isoform siRNA confirm our pan-arrestin RNAi results when TUNEL analysis was used as a measure of apoptosis. Finally, these observations indicate that both β-arrestin isoforms are necessary signaling components for the cytoprotective effects after βAR activation in UROtsa cells, possibly serving as scaffolding proteins linking extracellular signals through the activated receptor to a physiological effector.

β-Arrestin is a necessary component for βAR mediated cytoprotection. A, percentage of UROtsa cells that were TUNEL-positive after being transiently transfected with nonsilencing or pan-arrestin (β-arrestin 1/2) siRNA duplexes then subsequently exposed to 1 μM STS for 4.5 h after pretreatment with or without 100 nM ISO for 5 min. Actual numbers of TUNEL-positive cells were normalized to total PI-stained cells, calculated from five microscopic fields for each treatment performed in duplicate. STS alone caused similar increases in the number of TUNEL-positive cells for control, nonsilencing, and pan-arrestin siRNA treatments (42 ± 3, 46 ± 4, and 41 ± 4% of total, respectively; n = 4). ISO preincubation before STS treatment significantly reduced (p < 0.05) the number of TUNEL-positive cells for control and nonsilencing siRNA transfection to 17 ± 2 and 19 ± 2% of total (n = 4). There was no difference in the percentage of TUNEL-positive β-arrestin knockdown cells preincubated with ISO (44 ± 5% of total) compared with STS treatment alone (n = 4). B, representative blot (n = 4) of resolved UROtsa cell lysates transiently transfected with nonsilencing or pan-arrestin siRNA duplexes. There was a decreased immunoreactivity for both β-arrestin isoforms (49 or 47 kDa) in cells transfected with pan-arrestin siRNA duplexes compared with nontransfected cells or cells transfected with nonsilencing siRNA. Actin immunoreactivity (42 kDa) is shown as loading control. C, representative blot (n = 3) of active caspase-3 immunoreactivity (17 kDa) from resolved UROtsa cell lysates transiently transfected with β-arrestin 1 or β-arrestin 2 siRNA duplexes, subsequently exposed to 1 μM STS for 4.5 h after pretreatment with or without 100 nM ISO for 5 min. There was no change in active caspase-3 (17 kDa) immunoreactivity from ISO-pretreated cells compared with STS only in both sets of transiently transfected groups. Actin immunoreactivity (42 kDa) is shown as loading control. D, representative blot (n = 3) of resolved UROtsa cell lysates transiently transfected with nonsilencing, β-arrestin 1 or β-arrestin 2 siRNA duplexes. There was a selective decreased immunoreactivity in β-arrestin 1 (49 kDa) or β-arrestin 2 (47 kDa) transfected cells with no change in the corresponding isoform or HSP27 immunoreactivity (27 kDa) compared with nontransfected lysates. Actin immunoreactivity (42 kDa) is shown as loading control.
Discussion
The ability of βAR activation to control the cytoprotective effects of HSP27 through association with β-arrestin, as demonstrated in this study using Western analysis, immunofluorescence microscopy, and RNAi techniques, represents a previously unidentified mechanism for regulating apoptosis by means of 7TMR stimulation. Our data also reveal another key cytosolic protein associated with β-arrestin through a receptor agonist-dependent manner, linking 7TMR activation to an additional diverse signaling pathway. Previous reports that examine βAR-mediated signaling in isolated cardiac myocytes have demonstrated contrasting effects on programmed cell death dependent upon which βAR subtype was being activated. Sustained β1AR subtype stimulation is thought to be associated with promoting apoptosis through a Ca2+/calmodulin-dependent protein kinase II activation that is independent of protein kinase A (Zheng et al., 2005). On the other hand, long-term administration of selective β2AR agonists reduced the number of TUNEL-positive cardiac myocytes compared with control in an animal model of dilated cardiomyopathy (Ahmet et al., 2004). The mechanism of this β2AR effect is postulated to be mediated through activation of phosphoinositide 3-kinase (PI3K), which regulates the phosphorylation state of the powerful antiapoptotic factor Akt (Zhu et al., 2001). Noncanonical 7TMR signaling pathways such as these are believed to involve formation and activation of multicomponent signaling complexes (i.e., signalosomes) for which β-arrestin can serve as an adaptor, scaffold, or transducer. The association of β-arrestin has been shown for both β1- and β2AR subtypes as a consequence of receptor agonist binding and subsequent receptor phosphorylation by G protein-coupled receptor kinases (Lefkowitz and Shenoy, 2005). Moreover, β-arrestin complexes have been demonstrated to prevent programmed cell death in other receptor systems by either inhibiting proapoptotic receptor signaling (Revankar et al., 2004) or by activating antiapoptotic pathways like PI3K/Akt (Povsic et al., 2003). In our model, radioligand binding studies demonstrated that a homogenous population of β2ARs is expressed on membranes of these urothelial cells. Therefore, activation of this β2AR population is thought to initiate formation of a previously unidentified β-arrestin-dependent signalosome that recruits, compartmentalizes or transduces HSP27, resulting in protection from insults that trigger programmed cell death.
To better understand the necessity for which β-arrestin isoform was important for this antiapoptotic effect, selective RNAi protocols were used to prevent translation of β-arrestin 1 or 2 individually before initiating programmed cell death. When either group of cells, individually transfected with siRNA sequences for β-arrestin 1 or 2, were pretreated with ISO then incubated with STS there was no difference in the apoptotic response compared with STS treatment alone. These results uncover in our model, a requirement for both β-arrestin isoforms mediating a protective βAR mechanism in opposition to a proapoptotic challenge. Heterodimerization of β-arrestin isoforms as a prerequisite for protection against cell death could be a potential explanation for this observation. Recent studies have demonstrated that β-arrestin heterodimers can form and translocate to activated 7TMRs in living cells using expression levels appropriate to what is found in native tissue (Storez et al., 2005). Furthermore, β-arrestin dimerization has been shown to regulate not only its subcellular localization (Milano et al., 2006) but also β2AR-mitogen-activated protein kinase (MAPK) signaling (Xu et al., 2008) as well as cell death and proliferation (Boularan et al., 2007).
Alternatively, regulating independent yet essential signaling pathways that modulate the apoptotic cascade could be speculated for the necessity of both β-arrestin isoforms in our model. For example, β2AR-mediated p38 MAPK activation kinetics display a differential phosphorylation profile when either β-arrestin 1 or 2 expression levels were independently reduced using RNAi techniques, suggesting that each β-arrestin isoform can moderate autonomous signaling pathways (Gong et al., 2008). It is noteworthy that the early phosphorylation event of p38 MAPK described in this investigation was β-arrestin 1-dependent and facilitated F-actin rearrangement that was accompanied by phosphorylation of HSP27. More significant, however, is a description of a βAR-mediated epidermal growth factor receptor transactivation, which conferred an antiapoptotic effect in vitro and in vivo that was dependent on both β-arrestin 1 and 2 (Noma et al., 2007). Similar to our observations, the authors in this study reasoned that this dual requirement for each isoform might not only reflect the need for heterodimerization but also distinct roles for β-arrestin 1 and 2 in protection against programmed cell death.
HSP27 is a member of the small HSP family characterized by low monomeric molecular mass and a conserved α-crystallin domain near the C terminus (Garrido, 2002). Under normal conditions, HSP27 is found in the cell as a large oligomeric structure with a molecular mass of approximately 800 kDa. HSP27 oligomerization is a dynamic process that is dependent upon numerous physical and chemical parameters, which include temperature, pH, ionic strength, exposure to stress, and phosphorylation status of the monomer. Likewise, the unique cytoprotective functions of HSP27 can be associated with its quaternary structure as well as the degree of monomer phosphorylation (Rogalla et al., 1999). Phosphorylation by mitogen-activated protein kinase-activated protein kinase-2 (MK2) resulting in the dissociation of large HSP27 oligomers has been described previously (Thériault et al., 2004). Cytoprotective mechanisms that involve phosphorylation-induced HSP27 monomers include inhibiting Daxx enhancement of Fas-induced apoptosis or regulating activation of the antiapoptotic kinase Akt (Charette et al., 2000; Rane et al., 2003). Specific biophysical measurements for the latter demonstrate that phosphorylation of HSP27 is essential for dissociating Akt from the MK2-p38-Akt signaling complex (Zheng et al., 2006). However, no information in the literature suggests a direct interaction of β-arrestin with these phosphorylated HSP27 monomers. Conversely, as with other small HSPs, HSP27 also exhibits ATP-independent chaperone activity in an effort to reduce protein aggregate generation, by sequestering or refolding proteins during times of cellular stress (Garrido, 2002). Specifically for HSP27, only large nonphosphorylated oligomeric structures have been shown to exhibit chaperone activity in vitro (Lelj-Garolla and Mauk, 2006). Moreover, a specific mechanism for sequestering cytosolic cytochrome c, effectively inhibiting the intrinsic apoptosis pathway by disrupting formation of the apoptosome, has been demonstrated for HSP27 both in vitro and in vivo (Bruey et al., 2000). Whether or not βAR activation is regulating the stability of large oligomeric HSP27 structures through β-arrestin is currently being investigated.
It is noteworthy that another cytoprotective function for HSP27 has been described involving the ubiquitin-proteasome degradation pathway (Parcellier et al., 2003). In this investigation, the enhanced cytoprotective activity of NF-κB was facilitated by degradation of phosphorylated and polyubiquitinated I-κBα through an HSP27/I-κBα/26S proteasome complex. The authenticity of this mechanism to adjust cellular levels for the cell division protein kinase inhibitor p27Kip1, which regulates cell viability in response to stressful stimuli, has also been documented (Parcellier et al., 2006). Of further importance, β-arrestin isoforms are also polyubiquitinated, which is a requirement for their endocytotic function, serving as adaptors for selective ubiquitination by bringing substrate specific E3 ligases into close proximity with receptor proteins (Shenoy et al., 2001). In this example, ubiquitination of the β2AR subtype leads to its down-regulation through proteasomal destruction. This β-arrestin-mediated recruitment of an as-yet-unidentified E3 ligase leading to ubiquitination of the β2AR subtype, demonstrates an agonist-dependent specificity of β-arrestins to selectively target proteins for degradation. Moreover, association of the RING finger E3 ligase Mdm2 with β-arrestin oligomers has been shown to shuttle this ubiquitin ligase out of the nucleus into the cytoplasm thereby regulating apoptotic signaling pathways (Boularan et al., 2007). Consequently, the β-arrestin/HSP27 complex described in our study, through either distinct isoform-dependent pathways or by mediating subcellular localization of proteasomal complexes, could facilitate agonist-selective degradation of proteins important for the progression of programmed cell death. However, the precise molecular mechanism by which this β-arrestin/HSP27 signalosome regulates apoptosis is currently being explored. Nevertheless, agonist-dependent interaction of the potent apoptotic regulator HSP27 with β-arrestin represents a critical new complex to be investigated and eventually exploited, as a modulator of apoptosis in diseases where programmed cell death plays an essential role.
Acknowledgments
We thank ShreeKantra S. Poudel for technical assistance. The polyclonal β-arrestin antibody (A1CT) was generously provided by Robert J. Lefkowitz (Howard Hughes Medical Institute and the Departments of Medicine and Biochemistry, Duke University Medical Center, Durham, NC).
Footnotes
-
This work was supported in part by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK062865]; the National Institutes of Health National Institute of General Medical Sciences [Grant GM66726]; National Institutes of Health Biomedical Research Infrastructure Networks (BRIN) program [Grant RR016471]; the National Institutes of Health Centers of Biomedical Research Excellence (CO-BRE) program [Grant RR017699]; and by a Thesis Grant, Faculty of Graduate Studies, Mahidol University.
-
L.R and E.B.H. contributed equally to this work
-
ABBREVIATIONS: HSP, heat shock protein; AR, adrenergic receptor; 7TMR, seven-transmembrane receptor; HSP27, 27-kDa heat shock protein; RNAi, RNA interference; DMEM, Dulbecco's modified Eagle's medium; ISO, isoproterenol; [125I]CYP, (-)3-[125I]iodocyanopindolol; CGP 20712A, 1-[2-((3-carbamoyl-4-hydroxy)phenoxy)ethylamino]-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]-2-propanol methanesulfonate; ICI 118,551, erythro-dl-1-(7-methylindan-4-yloxy)-3-isopropylaminobutan-2-ol; SR 59230A, 3-(2-ethylphenoxy)-1-[(1S)-1,2,3,4-tetrahydronaphth-1-ylaminol]-(2S)-2-propanol oxalate; PAGE, polyacrylamide gel electrophoresis; IP, immunoprecipitated; LC/MS/MS, liquid chromatography/tandem mass spectrometry; HA, hemagglutinin; PBS, phosphate-buffered saline; siRNA, small interfering RNA; STS, staurosporine; TUNEL, terminal deoxynucleotidyl transferase biotin-dUTP nick-end labeling; PI, propidium iodide; RFU, relative fluorescence units; SR 59230A; PI3K, phosphoinositide 3-kinase; MAPK, mitogen-activated protein kinase; MK2, mitogen-activated protein kinase-activated protein kinase-2; Akt, protein kinase B.
-
↵1 Current affiliation: Sanford Research/University of South Dakota, Cardiovascular Research Institute, Sioux Falls, South Dakota.
-
↵2 Current affiliation: Case Center for Proteomics, School of Medicine, Case Western Reserve University, Cleveland, Ohio.
- Received November 12, 2008.
- Accepted January 21, 2009.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}