


Abstract
Previous studies suggest that selective antagonists of specific subtypes of muscarinic acetylcholine receptors (mAChRs) may provide a novel approach for the treatment of certain central nervous system (CNS) disorders, including epileptic disorders, Parkinson's disease, and dystonia. Unfortunately, previously reported antagonists are not highly selective for specific mAChR subtypes, making it difficult to definitively establish the functional roles and therapeutic potential for individual subtypes of this receptor subfamily. The M1 mAChR is of particular interest as a potential target for treatment of CNS disorders. We now report the discovery of a novel selective antagonist of M1 mAChRs, termed VU0255035 [N-(3-oxo-3-(4-(pyridine-4-yl)piperazin-1-yl)propyl)-benzo[c][1,2,5]thiadiazole-4 sulfonamide]. Equilibrium radioligand binding and functional studies demonstrate a greater than 75-fold selectivity of VU0255035 for M1 mAChRs relative to M2-M5. Molecular pharmacology and mutagenesis studies indicate that VU0255035 is a competitive orthosteric antagonist of M1 mAChRs, a surprising finding given the high level of M1 mAChR selectivity relative to other orthosteric antagonists. Whole-cell patch-clamp recordings demonstrate that VU0255035 inhibits potentiation of N-methyl-d-aspartate receptor currents by the muscarinic agonist carbachol in hippocampal pyramidal cells. VU0255035 has excellent brain penetration in vivo and is efficacious in reducing pilocarpine-induced seizures in mice. We were surprised to find that doses of VU0255035 that reduce pilocarpine-induced seizures do not induce deficits in contextual freezing, a measure of hippocampus-dependent learning that is disrupted by nonselective mAChR antagonists. Taken together, these data suggest that selective antagonists of M1 mAChRs do not induce the severe cognitive deficits seen with nonselective mAChR antagonists and could provide a novel approach for the treatment certain of CNS disorders.
Muscarinic acetylcholine receptors (mAChRs) are G protein-coupled receptors (GPCRs) that are widely expressed in the central nervous system (CNS) and are critical for the modulation of activity in multiple brain circuits (Langmead et al., 2008). Previous studies suggest that mAChRs play important roles in a broad range of CNS functions, including attention and cognitive function, nociception, regulation of sleep/wake cycles, motor control, and arousal. Furthermore, mAChR ligands have been proposed to have potential efficacy in a wide variety of CNS disorders, including chronic and neuropathic pain, sleep disorders, epilepsy, schizophrenia, Alzheimer's disease, Parkinson's disease (PD), and dystonia (Bymaster et al., 2003b; Langmead et al., 2008).
Based on the heterogeneous distribution and diverse physiological roles of mAChRs, the opportunity exists for developing therapeutic agents that selectively interact with individual mAChR subtypes involved in specific CNS functions. The mAChRs are members of the family A GPCRs and include five subtypes, termed M1 to M5. Although each of the mAChR subtypes can couple to multiple signaling pathways in different systems, the M1, M3, and M5 mAChR subtypes often couple to Gq and activate phospholipase C, whereas M2 and M4 couple to Gi/o and associated effector systems, such as ion channels and adenylyl cyclase (Felder et al., 2000; Langmead et al., 2008). Unfortunately, the orthosteric (ACh) binding site of the mAChR is highly conserved, which has led to difficulty in the development of highly selective ligands for individual mAChR subtypes (Felder et al., 2000). This lack of subtype-selective pharmacological reagents for mAChRs has prevented development of a clear understanding of the physiological roles of individual mAChR subtypes.
Of the mAChRs, M1 is among the most heavily expressed in forebrain and midbrain regions and has been proposed to play important roles in memory and attention mechanisms, motor control, and regulation of sleep wake cycles (Felder et al., 2000). Based on the potential role of M1 in seizure activity and motor control, it has been postulated that highly selective M1 antagonists may have potential utility in the treatment of some epileptic disorders as well as certain movement disorders, including PD and dystonia (Hamilton et al., 1997; Pisani et al., 2007). Previous studies suggest that M1 is not involved in most of the peripheral actions of mAChR ligands, suggesting that M1-selective antagonists are not likely to induce the peripherally mediated adverse effects of nonselective mAChR antagonists (Bymaster et al., 2003a; Langmead et al., 2008). However, mAChR antagonists have also been long known to induce severe impairments in cognitive function (Drachman and Leavitt, 1974). Based on the postulated roles of M1 in different forms of learning and memory, it is possible that M1-selective antagonists could induce impairments in cognitive function that are similar to those of nonselective mAChR antagonists. However, the role of M1 mAChRs in the processes of memory acquisition and consolidation are not entirely clear. It is noteworthy that M1 mAChR knockout mice display relatively subtle changes in cognitive function, and hippocampus-dependent learning remains largely intact these animals (Miyakawa et al., 2001; Anagnostaras et al., 2003). Furthermore, studies with mice in which each of the other mAChR subtypes has been genetically deleted suggest that multiple mAChR subtypes participate in cholinergic regulation of learning and memory (for review, see Wess, 2004). Based on these observations, it is possible that M1-selective antagonists may provide a viable approach to treatment of certain CNS disorders and may not induce the adverse effects observed with nonselective mAChR antagonists. Unfortunately, the lack of highly selective antagonists of M1 and other mAChR subtypes has made it impossible to test this hypothesis. We now report discovery and detailed characterization of VU0255035 as a highly selective M1 antagonist. The pharmacokinetic profile and CNS penetration of VU0255035 makes this compound well suited for in vivo studies in animal models. It is noteworthy that VU0255035 inhibits induction of generalized seizures by the mAChR agonist pilocarpine but does not mimic the cognitive-impairing effects of the mAChR antagonist scopolamine in a behavioral measure of hippocampus-dependent learning. Together, these data raise the exciting possibility that the use of selective M1 mAChR antagonists as therapeutics may not induce severe cognitive deficits.
Materials and Methods
Cell Culture and Transfections. Chinese hamster ovary (CHO) cells stably expressing rat M1 (rM1) were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and cultured according to ATCC recommendations. CHO cells stably expressing human M2 (hM2), hM3, and hM5 were generously provided by A. Levey (Emory University School of Medicine, Atlanta, GA). rM4 cDNA, provided by T. I. Bonner (National Institutes of Health, Bethesda, MD), was used to stably transfect CHO-K1 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). To make stable hM2 and rM4 cell lines for use in calcium mobilization assays, these cells were also stably transfected with a chimeric G protein (Gqi5) using Lipofectamine 2000. rM1, hM3, and hM5 cells were grown in Ham's F-12 medium containing 10% heat-inactivated fetal bovine serum (FBS), 20 mM HEPES, and 50 μg/ml G418 sulfate. hM2-Gqi5 and rM4-Gqi5 cells were grown in the same medium also containing 500 μg/ml Hygromycin B. The rM1-Y381A CHO cell line was generated as described previously (Jones et al., 2008).
Primary High-Throughput Screening. The primary HTS was performed as described previously (Lewis et al., 2008).
Calcium Fluorescence Measurement. CHO cells expressing rM1, hM2-Gqi5, hM3, rM4-Gqi5, or hM5 were plated at 4 × 104 cells per well in standard growth media (Ham's F12 medium supplemented with 10% fetal bovine serum and 20 mM HEPES) in 96-well plates 24 h before assay and were incubated overnight at 37°C in 5% CO2. On the day of the assay, media was removed and calcium assay buffer [Hanks' balanced salt solution (HBSS; Invitrogen, Carlsbad, CA), 20 mM HEPES, 2.5 mM probenecid (Sigma, St. Louis, MO), pH 7.4] containing 1.8 μM Fluo4-AM dye (Invitrogen) was added. Cells were incubated for 45 min (37°C, 5% CO2) for dye loading. Fluo4-AM dye was removed and replaced with 60 μl of calcium assay buffer. Cells were maintained at room temperature for the assay. For calcium fluorescence measurement of antagonist potency, antagonist concentration-response curves (20 μl, 5×) were added 20 s after the beginning of data collection and an EC80 concentration of agonist (20 μl, 5×) was added 90 s later via a Flexstation II (Molecular Devices, Sunnyvale, CA). Fluorescence imaging continued for a total of 160 s acquisition time using an excitation wavelength of 488 nm, an emission wavelength of 525 nm, and a cutoff wavelength of 515 nm. Agonist and antagonist were added at a speed of 52 μl/s, and calcium flux was measured using a Flexstation II at 25°C. For Schild analyses, fixed concentrations of antagonist were added manually (20 μl, 5×) so that the agonist concentration-response curve was added via a Flexstation II after a 15-min incubation with antagonist. All of the peaks of the calcium response were normalized to the response to a maximally effective concentration of ACh (ECmax). The concentration of ACh that elicited a response that is 80% of max (EC80 value) was determined for every separate experiment, allowing for a response varying from 70 to 90% of the maximum peak. These maximum values were fit using GraphPad Prism version 4.0 to a four-parameter logistic equation to determine IC50 values and Schild Dose-Ratios.
Hippocampal Phosphoinositide Hydrolysis. Phosphoinositide (PI) hydrolysis in hippocampal slices was measured as described previously (Conn and Sanders-Bush, 1986) with a few modifications. In brief, cross-chopped (350 × 350 μM) slices of male (6-9-week-old) Sprague-Dawley rat hippocampus were incubated with 95% O2/5% CO2 bubbled Krebs buffer (108 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3 and 10 mM glucose). The tissue was allowed to recover for 30 min with shaking at 37°C. After tissue recovery, the tissue was combined, washed with warm Krebs buffer, and 25 μl of gravity-packed slices were incubated with 175 μl of Krebs buffer containing 0.5 μCi of [myo-3H]inositol (PerkinElmer Life and Analytical Sciences, Waltham, MA) for 45 min. VU0255035 or vehicle controls were added and incubated for 15 min, followed by the addition of 10 mM LiCl and incubation for an additional 15 min. Finally, an EC80 concentration of carbachol (CCh) was added, followed by an additional 45-min incubation. The reaction was terminated by the addition of 900 μl of chloroform/methanol (1:2). The aqueous and organic phases were separated by addition of 300 μl of chloroform and 300 μl of water, vortexing, and allowing the phases to separate by gravity. The aqueous phase was added to anion exchange columns (AG 1-X8 Resin, 100-200 mesh, formate form; Bio-Rad Laboratories, Hercules, CA) and [3H]inositol phosphates were eluted and measured by liquid scintillation counting. Data were fit with Prism (ver. 4.0; GraphPad Software, San Diego, CA) to a four-parameter logistic equation to determine IC50 values.
Membrane Preparation and Equilibrium Radioligand Binding. Membranes were prepared and equilibrium radioligand binding assays were performed as described previously (Shirey et al., 2008).
Electrophysiology. Transverse hippocampal slices were prepared from Sprague-Dawley rats (postnatal day 17-25). Rats were anesthetized with isoflurane and decapitated. The brain was rapidly removed from the skull and submerged in ice-cold modified artificial cerebrospinal fluid (ACSF), which was oxygenated with 95% O2/5% CO2 and composed of 230 mM sucrose, 2.5 mM KCl, 0.5 mM CaCl2, 6 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM d-glucose. The brain was blocked in the horizontal plane, glued to the stage of a Vibratome (Vibratome, St. Louis, MO) that was filled with ice-cold modified ACSF, and cut at 290 μm. Slices were then incubated in oxygenated normal ACSF (126 mM NaCl, 2.5 mM KCl, 3 mM CaCl2, 1 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM d-glucose) at 31-32°C for 30 min and maintained at room temperature afterward until transferred individually to a fully submerged recording chamber, which was continuously perfused with oxygenated ACSF at ∼30°C.
Whole-cell recordings were made from visually identified hippocampal CA1 pyramidal neuron soma under an upright microscope (BX50WI; Olympus, Lake Success, NY). A low-power objective (4×) was used to identify CA1 region of the hippocampus, and a 40× water immersion objective coupled with Hoffman optics and video system was used to visualize individual pyramidal cells. A Multi-Clamp 700B amplifier (Molecular Devices, Sunnyvale, CA) was used for voltage-clamp recordings. Patch pipettes (4-6 MΩ) were prepared from borosilicate glass (World Precision Instruments, Sarasota, FL) using a Narishige vertical patch pipette puller (Narishige, Tokyo, Japan) and filled with the pipette solution containing 61.5 mM potassium gluconate, 65 mM CsCl, 3.5 mM KCl, 1 mM MgCl2, 0.5 mM CaCl2, 10 mM HEPES, 5 mM EGTA, 2 mM Mg-ATP, and 0.2 mM Na-GTP. The pH of the pipette solution was adjusted to 7.3 with 1 M KOH, and osmolality was adjusted to ∼295 mOsm/kg. Cells were voltage-clamped at -60 mV, and NMDA receptor-mediated currents were induced by pressure ejection of 1 mM NMDA to the soma of the recorded cell through a patch pipette using a Picospritzer II (General Valve, Fairfield, NJ). The experiment was carried out in the presence of tetrodotoxin (1 μM) to block voltage-gated sodium channels. All drugs were bath-applied. Data acquisition and analysis were performed using a PC computer equipped with pCLAMP software (Molecular Devices). Data are presented as percentage of the control value or percentage potentiation. The percentage potentiation was defined as [Imax/Icontrol - 1] × 100, where Icontrol was the average amplitude of NMDA receptor currents of four trials immediately before application of CCh or VU0255035 and Imax is the maximum current amplitude during drug application. Statistical analysis of electrophysiology data were performed using two-tailed paired or unpaired Student's t test as appropriate, and p values less than 0.05 (p < 0.05) were considered to be significant. Data are presented as mean ± S.E.M.
Animals. Experiments were conducted in accordance with the National Institutes of Health regulations on animal care and approved by the Institutional Animal Care and Use Committee, Vanderbilt University Medical Center. Subjects were housed in groups of two to four per cage in a large colony room under a 12-h light/dark cycle (lights on at 6:00 AM) with food and water provided ad libitum.
Pilocarpine-Induced Seizures. Hybrid mice (C57Bk:129Sv; 2-6 months old) were used for these studies. The susceptibility of these mice to pilocarpine-induced seizures was assessed with a single injection of pilocarpine (280 mg/kg i.p.). VU0255035 was prepared in 5% lactic acid, diluted to 10 mM with H2O, and the pH was adjusted to 6.5 to 7.0 using 1 N NaOH. The 10 mM VU0255035 stock was then filtered using a 0.2-μm filter and diluted to a 2 mM stock with 9% saline. Each mouse was first injected with methylscopolamine nitrate (1 mg/kg i.p.) to block the peripheral effects of pilocarpine. Immediately thereafter, each mouse was injected with either vehicle or VU0255035 (10 mg/kg i.p.), followed 30 min later with an injection of pilocarpine (280 mg/kg i.p.). Seizures induced by pilocarpine were recorded by a digital camcorder and scored later for each 5-min period. The scoring was based on the modified Racine scale, as described at the following stages: 0, no abnormality; 1, exploring, sniffing, and grooming ceased, becoming motionless; 2, forelimb and/or tail extension, appearance of rigid posture; 3, myoclonic jerks of the head and neck, with brief twitching movement, or repetitive movements with head bobbing or “wet-dog shakes”; 4, forelimb clonus and partial rearing, or occasional rearing and falling; 5, forelimb clonus, continuous rearing and falling; 6, tonic-clonic movements with loss of posture tone, often resulting in death. The seizure scores for the first 45 min after pilocarpine injection for the control group and VU0255035 treated group were analyzed using two-way ANOVA.
Pharmacokinetic Analysis. Male Sprague-Dawley rats (Harlan, Indianapolis, IN) weighing approximately 250 g were used for the pharmacokinetics (PK) studies. VU0255035 was administered at a dose of 10 mg/kg i.p. The dosing solution was prepared in 5% lactic acid [8.5% (v/v)] in water and the pH was adjusted to 6.5 using 1 N NaOH. Blood and brain tissue samples were collected at 0.5, 1, 2, 4, and 8 h after dosing. The blood samples were collected by cardiac puncture and processed to separate plasma. The animals were decapitated to collect the whole-brain tissue samples. Both plasma and brain tissues were immediately frozen in dry ice and stored in -80°C until analysis. Before analysis, the brain samples were washed to remove residual blood, weighed, and homogenized in 5 ml of phosphate-buffered saline using ultrasonic homogenizer. The plasma and brain homogenate samples were processed by acetonitrile precipitation method and analyzed using liquid chromatography/tandem mass spectrometry (LC/MS-MS). The liquid chromatograph separation was carried out on a Luna ODS column (5 μm; 2.1 mm × 5 cm; Phenomenex, Torrance, CA) at a flow rate of 0.3 ml/min. The gradient program was used with the mobile phase, combining solvent A (95:5 0.1% formic acid in water/acetonitrile) and solvent B (95:5 acetonitrile/0.1% formic acid in water) as follows: 0% B for 0 to 2 min, 0 to 100% B for 2 to 2.5 min, 100% B for 2.5 to 3.5 min, 100 to 0% B for 3.5 to 4.0 min, and finally equilibration for 2 min before injection of next sample. Mass spectrometry was carried out using a mass spectrometer (ThermoFinnigan TSQ Quantum Ultra; Thermo Fisher Scientific, Waltham, MA) in positive ion mode. The software Xcalibur (ver. 2.0; Thermo Fisher Scientific) was used to control the instrument and collect data. The electrospray ionization source was fitted with a stainless steel capillary (100 μm i.d.). Nitrogen was used as both the sheath gas and the auxiliary gas. The ion transfer tube temperature was 300°C. The spray voltage, tube lens voltage, and pressure of sheath gas and auxiliary gas were optimized to achieve maximal response using the test compounds mixing with the mobile phase A (50%) and B (50%) at a flow rate of 0.3 ml/min. Collision-induced dissociation was performed on the VU0255035 and internal standard (VU178) under 1.0 mTorr of argon. Selected reaction monitoring was carried out using the transitions m/z 433 to 164 for VU0255035, and m/z 310 to 223 for VU178. The calibration curves were constructed and linear response was obtained in the range of 1 to 500 ng/ml by spiking known amounts of analytes in blank brain homogenates and plasma. The concentrations were expressed as nanograms per milliliter or nanograms per gram of tissue. The concentration-time data were analyzed by noncompartmental analysis using WinNonlin 5.2 (Pharsight Inc., Mountain View, CA)
Contextual Fear Conditioning. Contextual fear conditioning studies were conducted in conditioning chambers housed in a sound-attenuating cubicle (Med Associates, St. Albans, VT) with a fluorescent light mounted on the back wall of the cubicle to provide illumination for the chamber. Stainless steel grid floors connected to shock scramblers and generators delivered the 0.5-mA unconditioned stimulus. A digital video camera mounted on the front wall of the cubicle was interfaced with a personal computer equipped with Video Freeze software (MED-VFC-RS; Med Associates), which incorporated a pixel-based method to measure and quantify freezing behavior. Based on previous studies, the motion threshold was set at 120 arbitrary units, 5-s freeze duration. All trials were recorded at 30 frames per second. In addition, 1 ml of a 10% vanilla extract solution was applied to the conditioning chamber grid floor to provide an olfactory cue.
Male Sprague-Dawley rats weighing 240 to 270 g were handled and injected with vehicle for 2 days before training. On training day, rats were habituated for 30 min in the training room. For the scopolamine dose-response studies, rats were pretreated for 15 min with either vehicle (0.9% saline s.c.) or a dose of scopolamine (0.03-0.3 mg/kg s.c.) before the training session. Based on the PK studies with VU0255035, rats were pretreated for 30 min with either vehicle (5% lactic acid [8.5% (v/v)] in water i.p., pH adjusted to 6.5 using 1 N NaOH) or a dose of VU0255035 (3-30 mg/kg i.p.) before the training session. During the training session, rats were then placed in the conditioning chamber. Rats were then given a 2-min habituation, followed by four tone/shock pairing trials. The tone (30 s, 5 kHz, 70 dB) presented through a speaker initiated each trial and coterminated with an electric foot-shock (1 s, 0.5 mA). A 45-s intertrial interval separated each tone/shock pairing trials. After the fourth and final tone/shock pairing, the rat remained in the chamber for an additional 45 s without tone or shock stimuli. All rats received the same conditioned freezing training protocol.
Approximately 24 h after training, the magnitude of contextual fear conditioning response was tested by placing the rats back into the same conditioning chambers with identical visual and odor cues and measuring freezing behavior in the absence of any auditory or shock stimuli for a 7-min period equivalent to the duration of the training session. Memory of the fear response 24 h after training was assessed by recording the amount of freezing response in the testing chamber environment. Freezing was defined as a motionless posture except for respiratory movements and was calculated as the percentage of freezing behavior for the entire testing session. Data were analyzed using a one-way analysis of variance and, if significant (p < 0.05), all dose groups were compared with the vehicle group using a Dunnett's test.
Medicinal Chemistry Methods
General. All NMR spectra were recorded on a Bruker 400 MHz instrument. 1H chemical shifts are reported in δ values in parts per million downfield from tetramethylsilane as the internal standard in DMSO. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, coupling constant (Hz). 13C chemical shifts are reported in δ values in parts per million with the DMSO carbon peak set to 39.5 ppm. Low-resolution mass spectra were obtained on a mass spectrometer with electrospray ionization (6130; Agilent, Palo Alto, CA). High-resolution mass spectra were recorded on a hybrid quadrupole time-of-flight mass spectrometer fitted with electrospray sources (Q-ToF API-US; Waters, Milford, MA). Analytical thin-layer chromatography was performed on silica gel GF 250-μm plates (Analtech, Newark, DE). Analytical HPLC was performed on an Agilent 1200 series with UV detection at wavelengths of 214 and 254 nm along with evaporative light scattering detection (ELSD). Preparative purification was performed on a CombiFlash Companion (Teledyne ISCO, Lincoln, NE). Solvents for extraction, washing, and chromatography were HPLC grade. All reagents were purchased from Aldrich Chemical Co. and were used without purification. All polymer-supported reagents were purchased from Biotage, Inc. (Charlottesville, VA)
Standard Experimental Procedures for Key Compounds
Methyl 3-(benzo[c][1,2,5]thiadiazole-4-sulfonamido)propanoate. To a solution of β-alanine methyl ester (2.36 g, 17.0 mmol) in CH2Cl2 (70 ml) was added DIPEA (6.56 ml, 37.4 mmol) and 2,1,3-benzothiadiazole-4-sulfonyl chloride (2) (4.01 g, 17.0 mmol). The reaction was stirred at room temperature for 20 h. The reaction was partially concentrated under vacuum and purified by column chromatography (silica gel) using 0 to 55% EtOAc in hexanes to afford a crude orange solid. Triturated with CH2Cl2/hexanes (1:2, 70 ml) to afford the title compound as a pale yellow solid (3.81 g, 74%): m.p., 119.6 to 119.9°C; 1H NMR (400 MHz, DMSO-d6) δ 8.38 (dd, J = 9.0, 1.0 Hz, 1H), 8.19 (dd, J = 7.0, 1.0 Hz, 1H), 7.94 (t, J = 6.0 Hz, 1H), 7.85 (dd, J = 9.0, 7.0 Hz, 1H), 3.44 (s, 3H), 3.19 (q, J = 6.5 Hz, 2H), 2.46 (t, J = 6.5 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 171.5, 155.4, 149.1, 132.6, 130.9, 129.3, 126.4, 51.6, 39.03, 34.5; analytical LC-MS (J-Sphere80-C18, 3.0 × 50.0 mm, 4.1-min gradient, 5% [0.05% TFA/CH3CN]:95% [0.05% TFA/H2O]:2.31 min, >99% (214 nM, 254 nm, ELSD), m/z [M + Na] = 324.0; HRMS calculated for C10H12N3O4S2 [M+ H], 302.0305; found, 302.0269.
3-(Benzo[c][1,2,5]thiadiazole-4-sulfonamido)propanoic acid (3). To a solution of methyl 3-(benzo[c][1,2,5]thiadiazole-4-sulfonamido)propanoate (3.70 g, 12.2 mmol) in tetrahydrofuran (45 ml) was added MeOH (10 ml) and 2 N NaOH (10 ml). The reaction was stirred at room temperature for 4 h, quenched upon addition of 2 N HCl (45 ml), and extracted with EtOAc (2 × 100 ml). The combined organic extracts were dried over MgSO4, concentrated under vacuum, and triturated with CH2Cl2/hexanes (1:1, 70 ml) to afford acid 3 as an off-white solid (2.54 g, 72%); m.p. 176.1 to 176.3°C; 1H NMR (400 MHz, DMSO-d6) δ 8.37 (dd, J = 9.0, 1.0 Hz, 1H), 8.19 (dd, J = 7.0, 1.0 Hz, 1H), 7.92-7.79 (m, 2H), 3.33 (br s, 1H), 3.15 (q, J = 7.0 Hz, 2H), 2.32 (t, J = 7.0 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 172.6, 155.4, 149.2, 132.6, 130.9, 129.3, 126.4, 39.1, 34.6; analytical LC-MS (J-Sphere80-C18, 3.0 × 50.0 mm, 4.1 min gradient, 5% [0.05% TFA/CH3CN]:95% [0.05% TFA/H2O]:1.99 min, >99% (214 nM, 254 nm, ELSD), m/z [M + Na] = 310.0; HRMS calculated for C9H10N3O4S2 [M+H], 288.0113; found, 288.0110.
General Library Synthesis Protocol. To a solution of acid 3 (50 mg, 0.17 mmol) and DIPEA (46 μl, 0.34 mmol) in dichloromethane (2 ml) in a 4-ml vial was added polymer-supported dicyclohexylcarbodiimide (283 mg, 0.34 mmol, 1.2 mmol/g), HOBt (27 mg, 0.2 mmol) and one of 24 amines (HNR1R2) and rotated at room temperature for 20 h. Then, MP-carbonate (151 mg, 0.5 mmol, 3.3 mmol/g) was added to scavenge excess 3 and the HOBt. The reactions were filtered into 13 × 100-mm test tubes, and the resin was washed (3 × 5 ml) with dichloromethane. The combined organic layers were dried down on a nitrogen evaporator, and the compounds (4) were purified to >98% by preparative LC-MS on an Agilent 1200 Prep mass-directed LC-MS purification system.
N-(3-Oxo-3-(4-(pyridine-4-yl)piperazin-1-yl)propyl)benzo[c]-[1,2,5]thiadiazole-4 sulfonamide (5b, VU0255035). To a solution of acid 3 (2.00 g, 6.94 mmol) and DIPEA (2.43 ml, 13.9 mmol) in dimethylformamide (10 ml) was added 1,2-dichloroethane (1.55 g, 8.33 mmol), HOBt (937 mg, 6.94 mmol), and 1-(4-pyridyl)piperazine (1.36 g, 8.33 mmol) and stirred at room temperature for 20 h. The reaction was diluted with water (80 ml) and extracted with EtOAc (2 × 100 ml). The combined organic extracts were washed with water (2 × 80 ml) and brine (100 ml), dried over MgSO4, and concentrated under vacuum. The residue was triturated with CH2Cl2/hexanes (1:1, 40 ml) to afford amide VU0255035 (5b) as a light tan solid (1.45 g, 48%): m.p. 159.7 to 160.2°C; 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, J = 9.0 Hz, 1H), 8.20 (d, J = 7.0 Hz, 1H), 8.16 (d, J = 6.0 Hz, 2H), 7.85 (dd, J = 9.0, 7.0 Hz, 1H), 7.71 (br s, 1H), 6.80 (d, J = 6.0 Hz, 2H), 3.51 to 3.40 (m, 4H), 3.35 to 3.28 (m, 4H), 3.21 to 3.12 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 169.1, 155.4, 154.7, 149.5, 149.2, 132.5, 130.9, 129.3, 126.3, 108.6, 45.6, 45.3, 44.2, 41.7, 41.4, 33.0; analytical LC-MS (J-Sphere80-C18, 3.0 × 50.0 mm, 4.1-min gradient, 5% [0.05% TFA/CH3CN]:95% [0.05% TFA/H2O]:1.95 min, >99% (214 nM, 254 nm, ELSD), m/z [M + H] = 433.1; HRMS calculated for C18H21N6O3S2 [M + H], 433.1117; found, 433.1117.
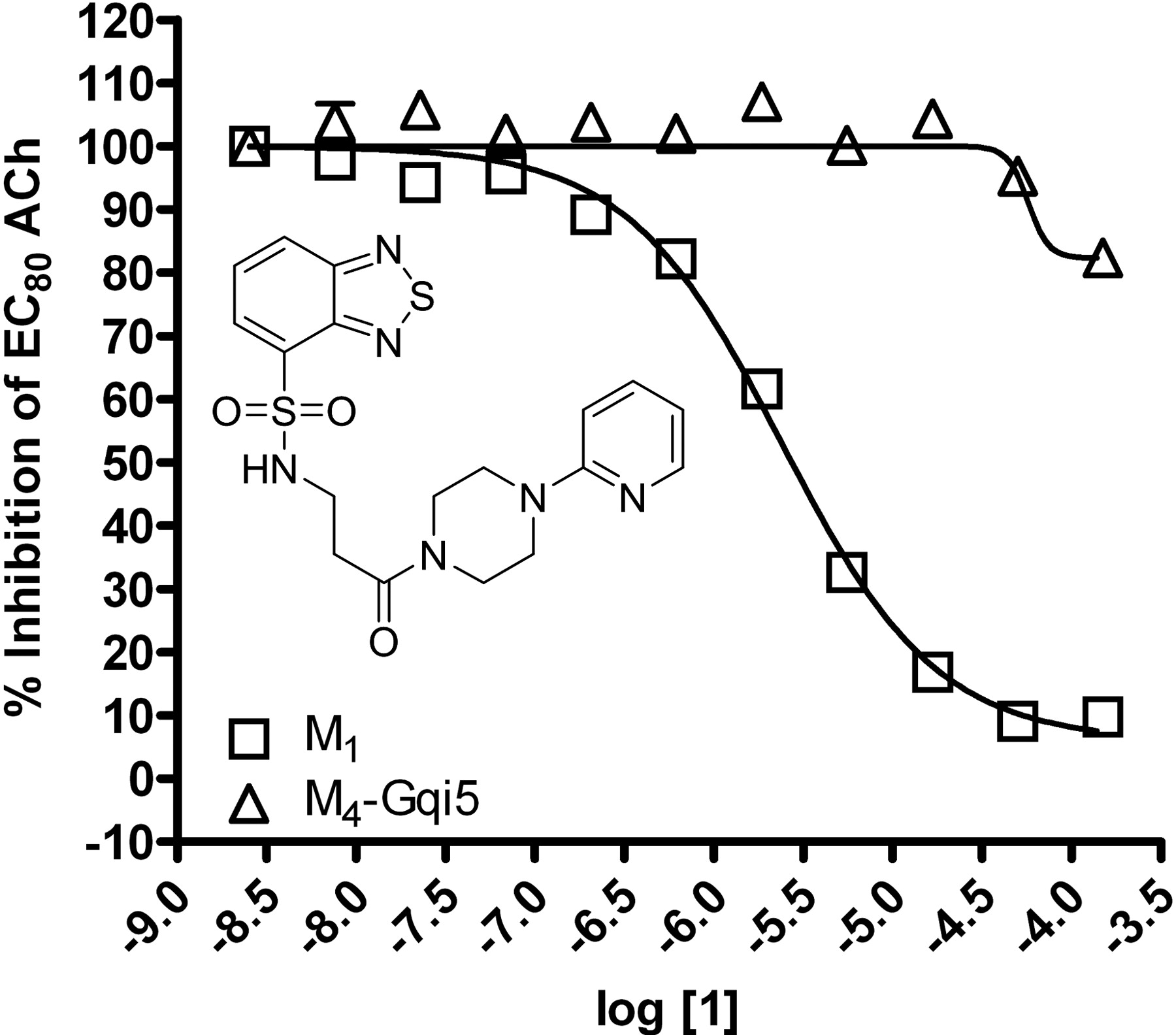
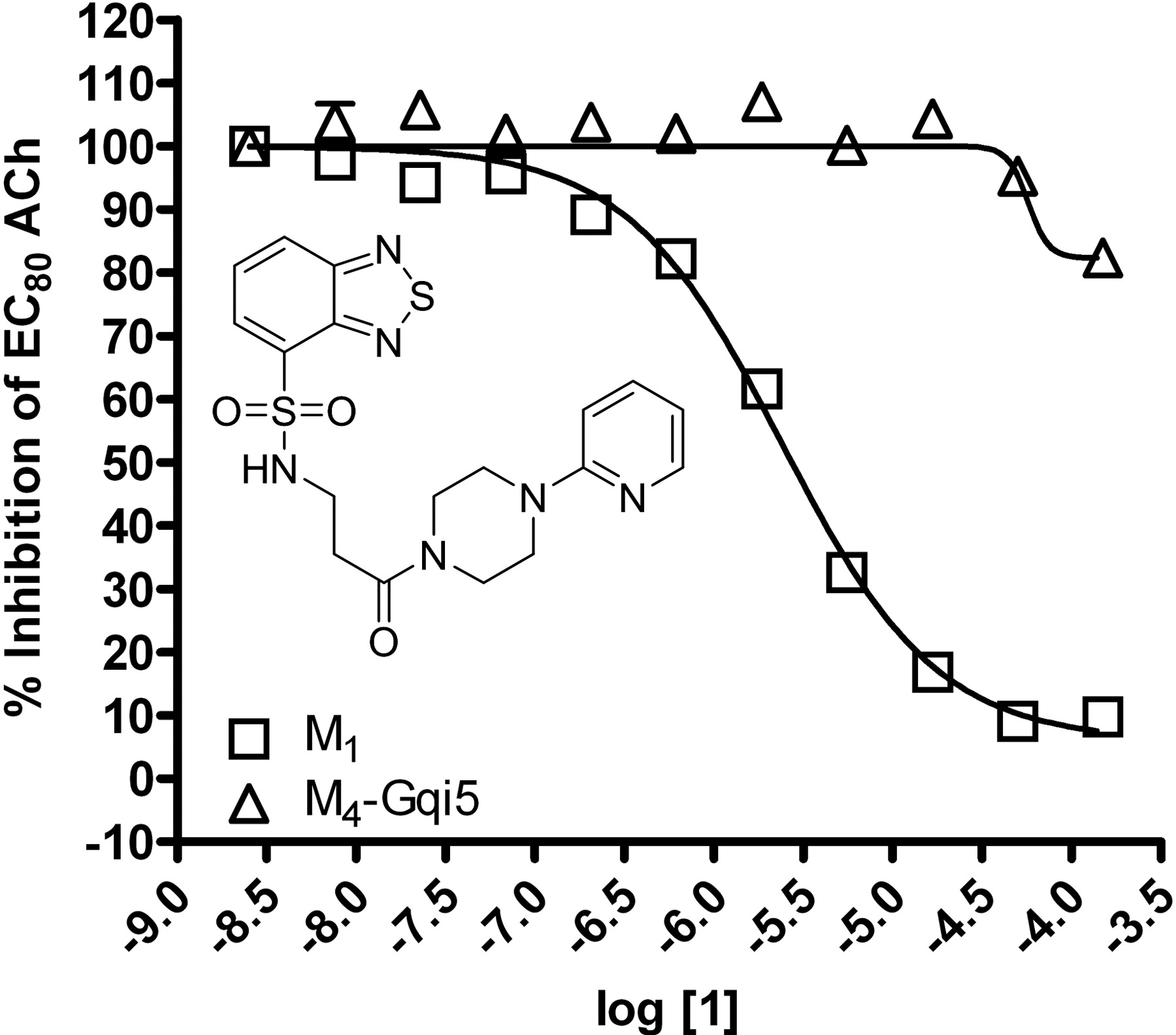
HTS lead 1 displays M1 versus M4 mAChR selective antagonism. Inset, structure of compound 1. CRCs of compound 1 were performed in the presence of an EC80 concentration of ACh for each receptor in a calcium mobilization assay. Data were normalized to the maximum response to 10 μM ACh and are presented as the percentage of the EC80 ACh response. The IC50 for compound 1 inhibition of an EC80 ACh response is 2.4 ± 1.2 μM for M1. The M4 mAChR calculated IC50 is greater than 150 μM. All data points represent the mean of three independent experiments performed in triplicate, and error bars represent S.E.M.
Results
Identification of Novel M1 Antagonists Using an HTS Approach. To search for novel antagonists of M1, we initiated a high-throughput screen of a small-molecule library within the Vanderbilt screening and chemistry center of the NIH Molecular Libraries Screening Center Network (MLSCN) (http://www.vanderbilt.edu/mlscn/Templates/index.htm) (Lewis et al., 2008). A small molecule library of 63,656 compounds from the MLSCN collection was screened against CHO cells expressing the rM1 mAChR by using a real-time cell-based calcium-mobilization assay (Z′ averaged 0.7). This effort identified 2179 primary M1 antagonist hits, of which 1665 compounds were available from BioFocus-DPI (South San Francisco, CA) for retest. After retests, 723 hits were confirmed, demonstrating a 43% re-test rate. To eliminate compounds that may inhibit calcium mobilization responses by a mechanism that is not specific to mAChR activation, the 723 verified hits were counter-screened against a CHO cell line expressing the metabotropic glutamate receptor 4 (mGluR4) and the chimeric G protein Gqi5. This secondary screen eliminated nine of the original hits. The remaining hits were tested in triplicate as 10-point concentration response curves (CRCs) against both rM1 CHO cells and a CHO cell line expressing rM4 and the chimeric G protein Gqi5 to identify compounds selective for M1 versus M4 mAChRs. The majority of compounds displayed no subtype selectivity. However, compound 1 (Fig. 1, inset), based on a N-(3-piperazin-1-yl)-3-oxopropyl)benzo[c][1,2,5]thiadiazole-4-sulfonamide scaffold, was selective for M1 versus M4 mAChRs. Compound 1 inhibited a submaximal (EC80) concentration of ACh at M1 with an IC50 of 2.4 ± 1.2 μM (Fig. 1). In contrast, compound 1 did not induce robust inhibition of an EC80 of ACh at M4 mAChRs (IC50 of >150 μM), demonstrating a greater than 60-fold selectivity for M1 versus M4 mAChRs. Although this was promising, compound 1 had relatively low potency at inhibiting M1. Thus, we initiated a chemical lead optimization effort to improve M1 potency while maintaining selectivity.
Chemical Optimization of HTS Lead 1. For chemical optimization of HTS lead 1, analogs of 1 were synthesized in a library format according to Supplemental Fig. 1. For our initial efforts, we explored alternative amides in place of the 2-pyridylpiperizine moiety. The synthesis began by treating commercially available benzo[c]thiadiazole-4-sulfonyl chloride 2 with β-alanine methyl ester under standard amide coupling conditions to afford, after saponification with LiOH/tetrahydrofuran/MeOH/H2O, the acid 3 in 55% yield for the two steps. Subsequent coupling with a diverse collection of amines under standard solution phase parallel synthesis conditions employing polymer-supported reagents and scavengers provided 24 analogs 4 (Kennedy et al., 2008). All analogs were purified by mass-directed HPLC to analytical purity (Leister et al., 2003).
Chemically Optimized Compounds Are Potent and Selective M1 mAChR Antagonists. This library of compounds was initially evaluated for activity at inhibiting the response of M1 to an EC80 concentration of ACh. The structure-activity relationship for this series was relatively flat, with functionalized piperazine amides represented by structure 5 being the only active congeners of structure 4 (Fig. 2A). As shown in Fig. 2B, a simple phenyl piperazine analog 5c (IC50 > 10 μM), was nearly inactive at M1, relative to compound 1. Functionalized phenyl piperazine analogs, such as the 3-OMePh congener 5e (IC50 > 10 μM) and the 2-CNPh derivative 5f (IC50 > 10 μM), also demonstrated limited M1 mAChR activity. In addition, aliphatic piperazine analogs, such as i-Pr 5h (IC50 > 10 μM), were devoid of any mAChR antagonist activity. Piperazinyl pyridine isomers proved to be the most interesting analogs. Relative to the 2-pyridyl analog compound 1, the 3-pyridyl congener, 5a (IC50 = 3.2 ± 1.6 μM), possessed roughly equivalent activity at M1, but also maintained selectivity versus M2-M5 (data not shown). The 4-pyridyl variant, 5b (IC50 = 309.1 ± 100.5 nM), provided an ∼8-fold increase in M1 potency relative to the HTS hit 1. Compound 5b was resynthesized on a gram scale, renamed VU0255035, and further evaluated.
The next critical step was to confirm that VU0255035 maintains mAChR subtype selectivity. The potency of VU0255035 for inhibition of an EC80 ACh response was determined for M1-M5 mAChRs (Fig. 3). VU0255035 induced only weak inhibition of M2, M3, M4, and M5 responses to ACh with IC50 values >10 μM at each subtype. Thus, VU0255035 had an IC50 of 132.6 ± 28.5 nM at M1 upon retest of the resynthesized compound and exhibited a 75-fold or greater functional selectivity for the M1 mAChR relative to other mAChR subtypes. VU0255035 has greater functional selectivity than any previous small molecule M1 antagonist and rivals the mAChR selectivity profile of the MT7 snake venom toxin (Karlsson et al., 2000).

Chemical optimization generates compounds that antagonize M1 mAChRs with greater potency than HTS lead 1. A, functionalized piperazine amides represented by structure 5. B, CRCs of compounds were performed in the presence of an EC80 concentration of ACh for M1 mAChRs in a calcium mobilization assay. Data were normalized to the maximum response to 10 μM ACh and are presented as a percentage of the EC80 ACh response. The IC50 for 5a inhibition of an EC80 ACh response is 3.2 ± 1.6 μM. The IC50 for 5b inhibition of an EC80 ACh response is 309.1 ± 100.5 nM. All other calculated IC50 values are greater than 10 μM. All data points represent the mean of three independent experiments performed in triplicate, and error bars represent S.E.M.

VU0255035 selectively antagonizes the M1 mAChR relative to other mAChRs. CRCs of VU0255035 were performed in the presence of an EC80 concentration of ACh for each receptor in a calcium mobilization assay. Data were normalized to the maximum response to 10 μM ACh and are presented as a percentage of the EC80 ACh response. The IC50 for VU0255035 inhibition of an EC80 ACh response is 132.6 ± 28.5 nM for M1. All other calculated IC50 values are greater than 5 μM. All data points represent the mean of five independent experiments performed in triplicate, and error bars represent S.E.M.
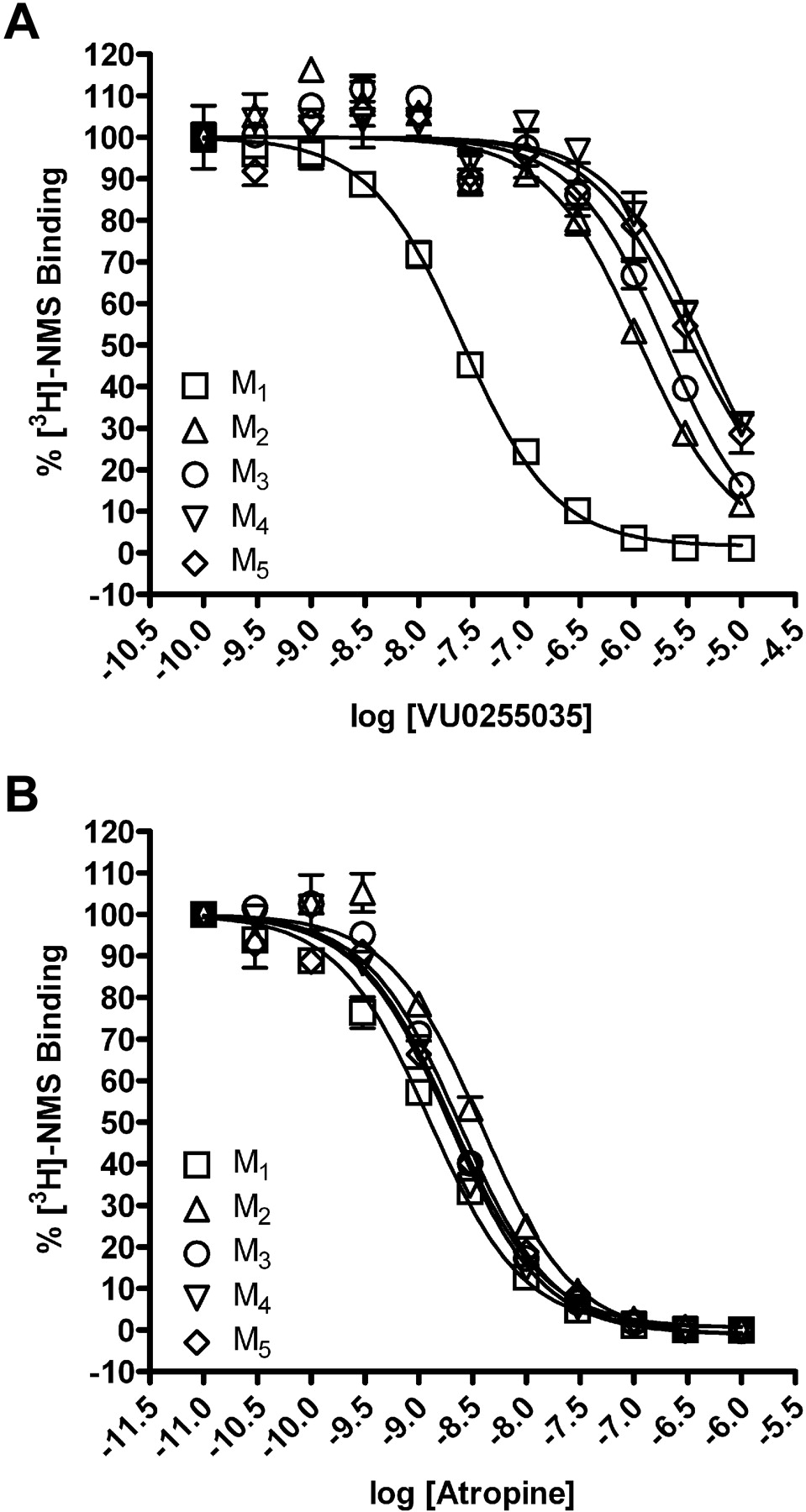
VU0255035 Is a Competitive Antagonist of M1 mAChRs. In previous efforts, we and others have had great success in achieving high subtype selectivity for mAChR activators by targeting allosteric sites rather than the orthosteric ACh binding site (Conn et al., 2009). To determine whether VU0255035 binds to the orthosteric binding site, we performed equilibrium radioligand binding studies. Membranes prepared from CHO cells expressing M1-M5 mAChRs were incubated with 0.1 nM l-[N-methyl-3H]scopolamine ([3H]NMS), an orthosteric site antagonist, in the absence or presence of increasing concentrations of either atropine (Messer, 2002) or VU0255035. This concentration of [3H]NMS is nonsaturating (Shirey et al., 2008), and all specific binding data represent less than 10% of the total binding. Atropine displaced [3H]NMS binding in membranes from cells expressing each of the mAChR subtypes with high affinity, displaying no subtype selectivity, consistent with what has been previously reported (Fig. 4B and Table 1) (Bymaster and Falcone, 2000). VU0255035 also displaced [3H]NMS binding from membranes expressing each of the mAChR subtypes (Fig. 4A). However, in contrast to atropine, VU0255035 had a much higher affinity for the orthosteric site of M1 mAChRs than for M2 to M5, showing greater than a 45-fold selectivity relative to M2 and even greater selectivity for M1 relative to the other mAChR subtypes (Table 1).
Apparent affinity values for VU0255035 and atropine for the competition of [3H]NMS binding from M1-M5 mAChRs
Ki values of VU0255035 and atropine were determined based on competition binding experiments with 0.1 nM [3H]NMS at each mAChR. The data represent the mean of three independent experiments performed in triplicate, and error bars represent S.E.M.
We then performed saturation binding experiments with increasing concentrations of [3H]NMS in the absence or presence of 10, 30, or 100 nM VU0255035. Scatchard analysis was performed (Fig. 5) to determine whether VU0255035 reduces [3H]NMS binding in a manner that is consistent with competitive interaction with the [3H]NMS binding site. Nonspecific binding was determined in the presence of 1 μM atropine. Scatchard analysis revealed data consistent with binding of [3H]NMS to a single site in the absence of antagonist (linear regression, r2 = 0.99). The Kd value of [3H]NMS in the absence of antagonist was 0.17 nM, consistent with previous studies (Shirey et al., 2008). Increasing concentrations of VU0255035 maintained a linear Scatchard regression line (linear regression, r2 = 0.99 for 10 nM VU0255035, r2 = 0.98 for 30 nM VU0255035, r2 = 0.99 for 100 nM VU0255035) and had no effect on the predicted receptor density (Bmax) but altered the slope of the regression line (Fig. 5). The apparent affinity of [3H]NMS was reduced by VU0255035, with Kd values of 0.25, 0.37, or 0.76 nM for 10, 30, or 100 nM VU0255035, respectively. The reduction in apparent Kd with no accompanying change in linearity of the regression line or the apparent Bmax is consistent with a competitive interaction of VU0255035 with the [3H]NMS binding site.
We further evaluated the question of whether VU0255035 inhibits M1 mAChRs by a competitive or noncompetitive mechanism in functional studies by performing a Schild analysis. Concentration-response relationships of ACh-induced increases in calcium mobilization in the absence or presence of increasing concentrations of VU0255035 (100 nM, 300 nM, 1 μM, 3 μM) (Fig. 6A) were evaluated. The maximum response to ACh was not significantly altered by increasing concentrations of VU0255035, but VU0255035 induced parallel rightward shifts in the ACh concentration response relationships. Analysis of the data using a Schild analysis (Arunlakshana and Schild, 1959) (Fig. 6B) revealed a Schild regression line with a slope that was not statistically different from unity (linear regression slope = 0.95 ± 0.16) and a calculated Kd of 33 nM for VU0255035. These data are consistent with a competitive interaction of VU0255035 with the orthosteric ACh site. We also performed a Schild analysis of the effect of VU0255035 on the response of M1 to TBPB (Fig. 6C), an allosteric agonist of M1 (Jones et al., 2008). In contrast with the Schild analysis using ACh, Schild analysis of effects of VU0255035 on the response to TBPB revealed a Schild regression slope statistically different from unity (linear regression slope = 0.69 ± 0.06) (Fig. 6D). These data are consistent with the hypothesis that VU0255035 does not interact with the same site as the allosteric agonist TBPB and that VU0255035 is acting as a competitive orthosteric site antagonist.

VU0255035 competes with [3H]NMS binding at M1 to M5 mAChRs and displays M1 selectivity. A, binding of 0.1 nM [3H]NMS was displaced by VU0255035 at M1 to M5 mAChRs. B, binding of 0.1 nM [3H]NMS was displaced by atropine at M1 to M5 mAChRs. For all experiments, specific binding represented less than 10% of the total radioligand binding. All data points represent the mean of three independent experiments performed in triplicate, and error bars represent S.E.M.
To further validate that VU0255035 is acting as an orthosteric antagonist, we determined the effect of VU0255035 on a mutant version of the M1 muscarinic receptor containing a point mutation that renders the receptor less sensitive to acetylcholine or orthosteric antagonists (Spalding et al., 2006; Jones et al., 2008). Mutation of tyrosine 381 to alanine (Y381A) in the M1 mAChR causes a large rightward shift in the potency of the orthosteric agonist ACh. The EC50 values for ACh were 2.5 ± 0.3 nM for wild-type M1 (WT) versus 13.7 ± 1.6 μM for the Y381A mutant (Y381A) (Fig. 7A). In contrast to ACh, the potency of an allosteric agonist of the M1 mAChR N-desmethylclozapine (NDMC) (Sur et al., 2003) is unaffected by the Y381A mutation. The EC50 values for NDMC were 140.5 ± 26.1 nM for WT versus 162.4 ± 6.1 nM for Y381A (Fig. 7A). If VU0255035 acts as a competitive orthosteric site antagonist, its potency should be decreased by the Y381A mutation. Consistent with this, VU0255035 potency was dramatically reduced in cells expressing M1-Y381A compared with cells expressing WT M1 (Fig. 7B). The IC50 values for VU0255035 versus ACh were 53.3 ± 7.7 nM for WT versus 38.9 ± 0.2 μM for Y381A, demonstrating a rightward shift in potency of more than 500-fold versus ACh. The IC50 values for VU0255035 versus NDMC were 57.3 ± 3.5 nM for WT versus 33.4 ± 8.4 μM for Y381A, also demonstrating a rightward shift in potency of more than 500-fold versus NDMC. Together, these data demonstrate that Y381A mutation equivalently affects VU0255035 potency, whether measured versus ACh or NDMC. These data are consistent with the data from radioligand binding and Schild analysis studies and indicate that VU0255035 is acting as a competitive orthosteric M1 mAChR antagonist.
VU0255035 Inhibits Carbachol-Induced Increases in PI Hydrolysis in Rat Hippocampal Slices. To verify that VU0255035 can block M1-mediated responses in native tissue preparations, we determined effects of this compound on activation of PI hydrolysis by mAChR agonists in hippocampal slices. The PI hydrolysis responses to nonselective mAChR agonists, such as CCh, are nearly abolished in M1 knockout mice, suggesting that this response is mediated by M1 mAChRs (Porter et al., 2002). We performed initial studies measuring PI hydrolysis in rat hippocampal slices with a range of concentrations of CCh to determine a submaximal concentration of CCh (EC80) to use (data not shown). VU0255035 blocked the PI hydrolysis response to an EC80 concentration of CCh in rat hippocampal slices in a concentration-dependent manner (Fig. 8) with an IC50 value of 2.4 ± 1.0 μM.

VU0255035 reduces [3H]NMS binding to M1 mAChRs in a competitive manner. Scatchard analysis demonstrates that VU0255035 dose-dependently decreases [3H]NMS binding affinity but does not alter the Bmax. Saturation binding experiments were performed on membranes from M1 expressing CHO cells in the absence or presence of 10, 30, or 100 nM VU0255035. In the absence of VU0255035, the Bmax was 562 ± 51 fmol/mg of protein. With 10, 30, and 100 nM VU0255035-treated samples, the Bmax was 554 ± 67, 583 ± 60, or 538 ± 54 fmol/mg of protein, respectively. There was no significant difference between the Bmax values of [3H]NMS binding for the vehicle treated or any of the VU0255035 treated samples (Student's t test). Linear regression lines were generated from three independent experiments performed in duplicate. Error bars represent S.E.M.
VU0255035 Inhibits CCh-Induced Potentiation of NMDA Receptor Currents in Hippocampal CA1 Pyramidal Cells. Activation of mAChRs in hippocampal pyramidal cells and in other brain regions has been demonstrated to result in the potentiation of NMDA receptor-mediated currents and the mAChR-mediated potentiation of NMDA currents in CA1 pyramidal cells in the hippocampus has been demonstrated to be mediated by activation of M1 mAChRs (Marino et al., 1998; Rouse et al., 1999). Thus, we determined the effect of VU0255035 on NMDA receptor (NMDAR)-mediated inward currents in CA1 pyramidal cells. Cells were voltage-clamped at -60 mV and NMDA receptor currents (INMDA) were evoked by pressure ejection of NMDA (1 mM) applied in the region of the cell soma. As has been shown previously (Marino et al., 1998), bath application of the cholinergic agonist CCh (10 μM) induced an increase in the peak amplitude of NMDA-evoked currents in pyramidal cells (Fig. 9A). The increased amplitude of NMDAR currents peaked at 163.6 ± 17.1% of baseline (n = 9, p = 0.0072) approximately 2 min after initial application of CCh and decayed rapidly. To determine whether VU0255035 could antagonize the CCh effect of potentiating NMDAR currents, we bath-applied VU0255035 (5 μM) before the application of CCh. As illustrated in Fig. 9B and summarized in Fig. 9C, VU0255035 (5 μM) completely blocked the CCh-induced potentiation of NMDAR currents with a peak potentiation of 10.3 ± 7.7% in the presence of VU0255035 (n = 7) compared with a peak potentiation of 63.6 ± 17.1% observed in control (n = 9; p = 0.016), consistent with VU0255035 antagonizing M1 mAChRs. VU0255035 by itself had no significant effect on NMDAR currents (107.4 ± 8.5% of baseline, n = 7, p = 0.382).
VU0255035 Inhibits Pilocarpine-Induced Seizures and Reduces Pilocarpine-Induced Mortality in Mice. The data presented above provide strong evidence that VU0255035 is a highly selective orthosteric antagonist of M1 and that this compound is effective as an M1 antagonist in the hippocampal formation. To further evaluate the selectivity of this compound, we determined the activity of VU0255035 in binding to a large panel of GPCRs, ion channels, transporters, and kinases using a MDS Pharma screen of the LeadProfilingScreen series of potential targets (MDS Pharma Services, King of Prussia, PA). VU0255035 was devoid of significant activity at all targets included in this screen (Supplemental Table 1) when assayed at a concentration of 10 μM. Thus, VU0255035 is highly selective for M1 relative to other mAChR subtypes or multiple other known targets and could provide an unprecedented opportunity to determine the effects of selective blockade of M1 in vivo. To evaluate VU0255035 as a potential reagent for in vivo studies, we performed PK studies to determine whether VU0255035 achieves reasonable brain exposure when dosed systemically. After intraperitoneal administration, VU0255035 was rapidly absorbed into systemic circulation and into brain (Supplemental Fig. 2). The maximum concentration in the brain and plasma (Tmax) was achieved within 0.5 h. The brain/plasma (area under the curve for the 8-h dosage) ratio was determined to be 0.48, indicating that VU0255035 shows excellent properties in terms of its ability to cross the blood brain barrier. The Cmax of this compound was 1307.89 ± 327.69 ng/ml for plasma and 251.32 ± 168.32 ng/g for the brain. The elimination half-life (T½) of VU0255035 was 1.29 h for the plasma and 2.58 h for the brain. Although these PK studies do not provide information about oral bioavailability or other PK parameters after oral dosing, these data suggest that VU0255035 has PK properties that make it useful as a research tool for intraperitoneal dosing.
Having established that VU0255035 achieves acceptable brain penetration after intraperitoneal administration, we evaluated the effects of this compound in blocking pilocarpine-induced seizures in vivo. Pilocarpine, a mixed M1/M4 agonist, has been demonstrated to induce seizures via the M1 mAChR subtype (Hamilton et al., 1997). To determine whether VU0255035 can inhibit pilocarpine-induced seizures in mice, C57Bk:129Sv mice, 2-6 months old, were dosed with methylscopolamine nitrate (1 mg/kg i.p.) to block the peripheral effects of pilocarpine, followed immediately by injections of either vehicle or VU0255035 (10 mg/kg i.p.). Finally, 30 min after the vehicle or VU0255035 injection, the mice were dosed with an injection of pilocarpine (280 mg/kg i.p.). Seizures were graded based on a modified Racine scale for the first 45 min after pilocarpine injection as outlined under Materials and Methods. These time points were chosen based on pharmacokinetic analysis and corresponded to the maximal brain exposure of VU0255035. VU0255035 caused a statistically significant decrease in pilocarpine-induced seizure scores as analyzed by two-way ANOVA, p < 0.0001 for the time course. In addition, post hoc analyses of each time point demonstrated significant differences at the 35-min (p < 0.05) and 40-min time points (p < 0.01) after pilocarpine injection (Fig. 10). Furthermore, VU0255035 reduced pilocarpine-induced mortality 24 h after pilocarpine treatment with 67.5% mortality in the vehicle-treated control subjects versus 25% mortality in the VU0255035-treated group. Together, these data demonstrate that VU0255035 antagonizes the M1 mAChR in vivo.
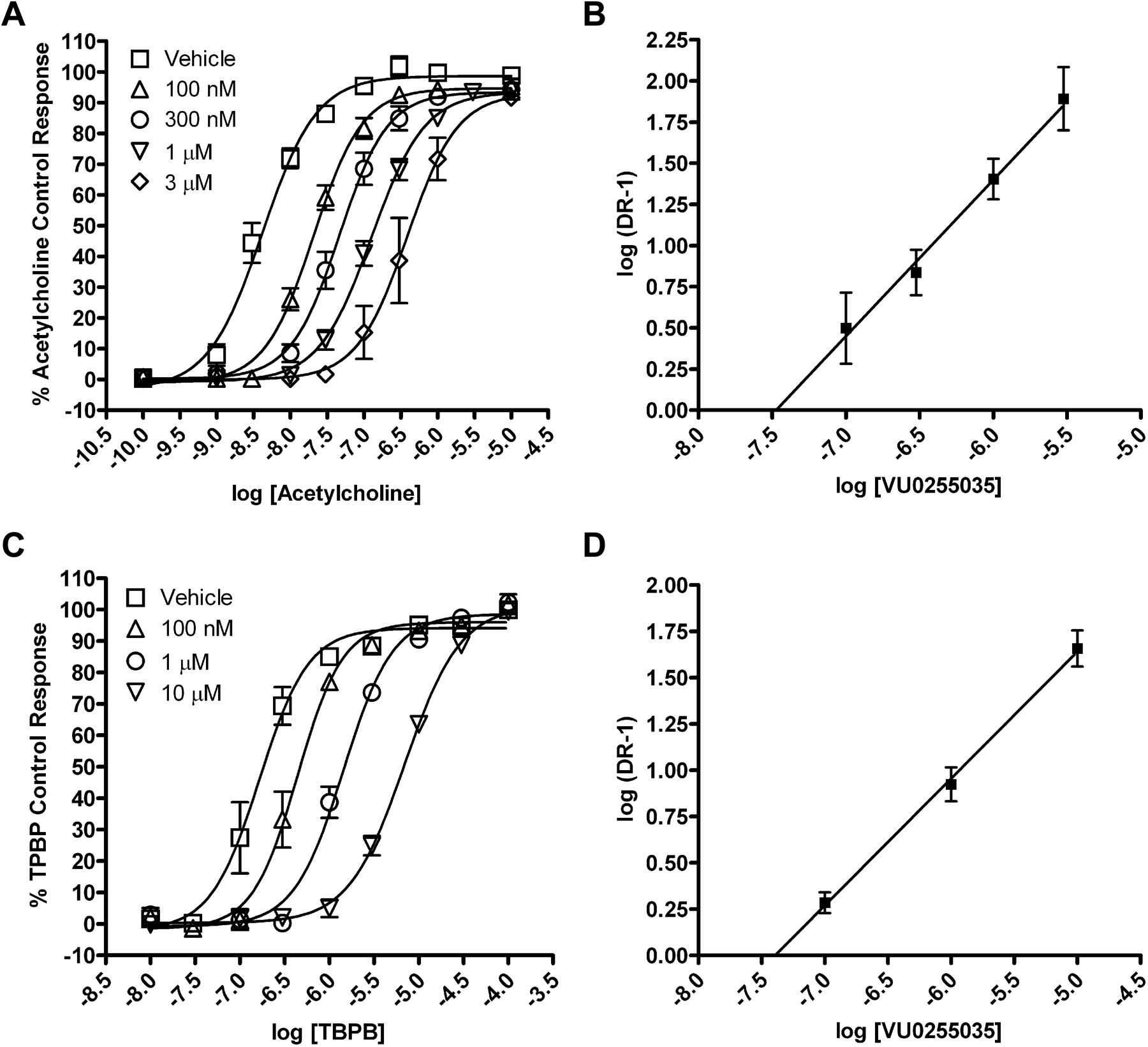
VU0255035 competitively antagonizes the M1 mAChR functional response to ACh but does not competitively antagonize the M1 mAChR response to TBPB. A, ACh CRCs in the absence or presence of 100 nM, 300 nM, 1 μM, or 3 μM VU0255035 in a calcium mobilization assay. B, Schild regression of the dose ratios derived from the VU0255035 antagonism of ACh. The slope of this regression is 0.95 ± 0.16 with a calculated Kd of 33 nM. C, TBPB CRCs in the absence or presence of 100 nM, 1 μM, or 10 μM VU0255035 in a calcium mobilization assay. D, Schild regression of the dose ratios derived from the VU0255035 antagonism of TBPB. The slope of this regression is 0.69 ± 0.06. Values represent the mean ± S.E.M. of four to six experiments conducted in triplicate.
VU0255035 Does Not Disrupt the Acquisition of Contextual Fear Conditioning, a Preclinical Model of Learning and Memory. A major potential concern with the use of M1 antagonists as potential therapeutic agents is that these compounds may impair cognitive function. This is based on the severe cognition-impairing effects of nonselective mAChR antagonists and the possibility that blockade of M1 could play a role in impairing cognitive function. Having demonstrated the efficacy of VU0255035 for reversal of pilocarpine-induced seizures, we next wanted to evaluate the dose-dependent effects of VU0255035 on the acquisition of contextual fear conditioning, a model of hippocampus-dependent learning and memory in which rats exhibit increased freezing behavior or passive fear response in the context or environment in which they previously experienced an adverse stimulus such as a mild foot-shock (Phillips and LeDoux, 1992). Previous studies have shown that the nonselective muscarinic antagonist scopolamine produces dose-dependent deficits in the acquisition of the contextual fear conditioning response (Anagnostaras et al., 1995). However, before the development of VU0255035, the role of M1 muscarinic receptors in this form of hippocampus-dependent learning and memory remained unclear as M1 mAChR knockout mice have been shown to exhibit either normal (Miyakawa et al., 2001) or enhanced (Anagnostaras et al., 2003) contextual fear acquisition, suggesting that M1 mAChRs may not be required for memory formation or for initial stability of memory in hippocampus.
Within the dose range tested (3.0-30.0 mg/kg), VU0255035 had no effect on the acquisition of the contextual fear conditioning response (Fig. 11A) consistent with the lack of deficits on contextual freezing behavior reported in the M1 knockout mice (Miyakawa et al., 2001; Anagnostaras et al., 2003). In contrast, scopolamine produced dose-dependent deficits in contextual fear response as shown by a significant decrease in freezing behavior, consistent with published findings (Miyakawa et al., 2001; Anagnostaras et al., 2003) (Fig. 11B).

The VU0255035 antagonism of M1 mAChR responses are right-shifted by Y381A mutation. A, CRCs of ACh and NDMC were performed on both wild-type (WT) and Y381A-mutated M1 mAChRs in a calcium mobilization assay. Data were normalized to either the maximum response to 1 μM ACh (WT) or to 10 mM ACh (Y381A) and are presented as the percentage of maximal ACh response. The EC50 values for ACh were 2.5 ± 0.3 nM (WT) or 13.7 ± 1.6 μM (Y381A). The EC50 values for NDMC were 140.5 ± 26.1 nM (WT) or 162.4 ± 6.1 nM (Y381A). B, CRCs of VU0255035 were performed in the presence of an EC80 concentration of ACh or NDMC for WT and Y381A M1 mAChRs in a calcium mobilization assay. Data were normalized to the percentage of either the EC80 ACh response or the EC80 NDMC response. The IC50 values for VU0255035 versus ACh were 53.3 ± 7.7 nM (WT) or 38.9 ± 0.2 μM (Y381A). The IC50 values for VU0255035 versus NDMC were 57.3 ± 3.5 nM (WT) or 33.4 ± 8.4 μM (Y381A). All data points represent the mean of three independent experiments performed in triplicate, and error bars represent S.E.M.

VU0255035 antagonizes PI hydrolysis in rat hippocampal slices induced by the mAChR agonist CCh. CRCs of VU0255035 were performed in the presence of an EC80 concentration of CCh for induction of PI hydrolysis in rat hippocampal slices. Data were normalized to the maximum response to 100 μM CCh and are presented as the percentage of the EC80 CCh response. The IC50 for VU0255035 inhibition of an EC80 CCh response is 2.4 ± 1.0 μM. All data points represent the mean of six independent experiments performed in triplicate and error bars represent S.E.M.
Discussion
Cholinergic pathways provide neuromodulatory systems involved in regulating multiple aspects of brain function. Based on this broad influence of cholinergic systems in the CNS, it is surprising that there have not been greater advances in development of therapeutic agents that target cholinergic signaling. A major challenge with regulation of cholinergic signaling is that all cholinergic agents developed thus far have dose-limiting adverse effects that prevent widespread use in the clinic (Brocks, 1999; Holden and Kelly, 2002; Wess et al., 2007; Langmead et al., 2008). In theory, agents that selectively activate or block individual subtypes of ACh receptors could provide beneficial effects of modulating specific aspects of cholinergic systems without inducing the broad range of peripheral and central adverse effects. For instance, multiple efforts have focused on discovery and development of highly selective agonists or allosteric activators of M1 or M4 for treatment of Alzheimer's disease or schizophrenia (for review, see Conn et al., 2009).
Although discovery of selective activators of mAChRs has been a major focus of previous efforts, multiple studies suggest that selective antagonists of individual mAChR subtypes may also have important utility for treatment of CNS disorders. For instance, nonselective mAChR antagonists such as trihexyphenidyl have clearly established efficacy in the treatment of certain movement disorders, such as PD and dystonia (Pisani et al., 2007). Although the precise mechanism of action of mAChR antagonists in treatment of PD and dystonia is not known, it is likely that these drugs act by modulating activity in the basal ganglia motor circuit by actions in the striatum (Pisani et al., 2007). M1 and M4 mAChRs are the most likely candidates for mediating these motor effects of mAChR antagonists (Hersch et al., 1994; Potter and Purkerson, 1995; Santiago and Potter, 2001; Potter et al., 2004). In addition, selective antagonists of mAChR subtypes have potential utility in treatment of obesity (Maresca and Supuran, 2008), drug dependence (Langmead et al., 2008), and certain epileptic disorders (Hamilton et al., 1997). Of these, selective M1 antagonists represent an especially attractive target for treatment of movement disorders and some forms of epilepsy. Discovery and characterization of a highly selective M1 antagonist represents a major break-through and provides the first selective small molecule antagonist for the M1 mAChR subtype. Furthermore, optimization of our original HTS hit to yield VU0255035 as a systemically active M1 mAChR antagonist allows use of this compound for in vivo studies of the behavioral effects of selective blockade this receptor subtype. Consistent with previous studies suggesting that M1 is the major mAChR subtype involved in induction of seizures by excessive activation of cholinergic systems, VU0255035 reduced pilocarpine-induced seizures.

VU0255035 blocks the CCh-induced potentiation of NMDAR currents in CA1 pyramidal cells. A, representative traces of NMDA-evoked currents obtained from a typical experiment (top) and time course of normalized amplitude of NMDAR currents before, during and after application of 10 μM CCh (n = 9) (bottom). B, representative traces of NMDAR currents (top) and time course of normalized amplitude of NMDAR currents in control, during application of VU0255035 (5 μM) and application of VU0255035 with 10 μM CCh, and washout (n = 7) (bottom), showing the ability of VU0255035 to block CCh-induced potentiation of NMDAR currents. C, summary of the peak potentiation of NMDAR currents by 10 μM CCh in control (n = 9) and in the presence of VU0255035 (n = 7) (p = 0.016). Data represent mean ± S.E.M.
Although M1 mAChR antagonists have been postulated to have potential utility in treatment of CNS disorders, a major concern that has reduced focus on discovery and development of mAChR antagonists is the possibility that these agents could induce severe cognitive disturbances. Anticholinergics have long been associated with induction of transient cognitive deficits in humans (Drachman and Leavitt, 1974) and in animal models (Hagan et al., 1987; Fornari et al., 2000). Despite their efficacy in some CNS disorders, impairments in cognitive function, along with peripheral adverse effects, have prevented widespread clinical use of mAChR antagonists. Although blockade of M2 and M3 mAChRs is thought be responsible for most peripheral adverse effects (Wess et al., 2007), blockade of M1 may play a major role in impaired cognitive function. M1 is highly expressed in the hippocampus and other forebrain regions (Levey et al., 1991) where mAChRs play critical roles in regulation of neuronal excitability and synaptic transmission (Wess et al., 2007; Shirey et al., 2008). Based on early findings that M1 is heavily expressed in these regions, this receptor was postulated to play a critical role in learning and memory. However, more recent studies reveal that mAChR regulation of excitability of hippocampal pyramidal cells is not altered in M1 mAChR knockout mice (Rouse et al., 2000), suggesting that M1 is not the major mAChR subtype responsible for regulating excitability of these cells. Furthermore, although M1 is the primary mAChR subtype involved in modulation of NMDA receptor currents in these cells (Marino et al., 1998; Jones et al., 2008), this receptor is not responsible for acute modulation of transmission at excitatory or inhibitory synapses in the hippocampus (Rouse et al., 1999; Jones et al., 2008; Shirey et al., 2008). Thus, it is likely that multiple mAChR subtypes are critical for regulating hippocampal and cortical function and that M1 does not play as dominant a role as was once assumed. Consistent with this, studies in M1 mAChR knockout mice revealed that performance in hippocampus-dependent learning tasks including Morris water maze and contextual fear acquisition remain intact or may be enhanced after deletion of the M1 gene (Miyakawa et al., 2001; Anagnostaras et al., 2003). Furthermore, Anagnostaras et al. (2003) found that scopolamine induced comparable impairments in the Morris water maze paradigm in M1 mAChR knockout and WT mice, indicating that other non-M1 mAChRs clearly must play a critical role in hippocampus-dependent learning and memory (Anagnostaras et al., 2003). Our present finding that VU0255035 does not induce deficits in contextual fear acquisition is consistent with the M1 mAChR knockout mouse data and provides initial evidence that selective M1 antagonists may not induce the same severity of cognitive deficits associated with the nonselective anticholinergics. In future studies, it will be important to determine the effects of VU0255035 in a range of models of other forms of cognitive function to gain a more complete understanding of the roles of M1 in different forms of learning and memory and further assess the potential adverse effect liability of M1-selective antagonists. In addition, VU0255035 provides a valuable tool to allow further studies of the physiological roles of M1 in the hippocampus, cortex, and other brain regions heavily modulated by cholinergic afferents.

VU0255035 blocks pilocarpine-induced seizures in mice. For these studies, C57Bk:129Sv mice, 2-6 months old, were dosed with methylscopolamine nitrate (1 mg/kg i.p.) to block the peripheral effects of pilocarpine, followed immediately thereafter with injections of either vehicle or VU0255035 (10 mg/kg i.p.). Finally, 30 min after the vehicle or VU0255035 injection, the mice were dosed with an injection of pilocarpine (280 mg/kg i.p.). Seizures were graded based on a modified Racine scale as outlined under Materials and Methods for the first 45 min after pilocarpine injection. The seizure scores for the first 45 min after pilocarpine injection for the control group and VU0255035-treated group were analyzed using two-way ANOVA. VU0255035 caused a statistically significant decrease in pilocarpine-induced seizure scores as analyzed by two-way ANOVA, p < 0.0001. Post hoc analyses of individual timepoints demonstrated a significant difference at the 35 min (*, p < 0.05) and 40 min (**, p < 0.01) time points. All data points represent the mean of eight mice for the control group and six mice for the VU0255035 group.
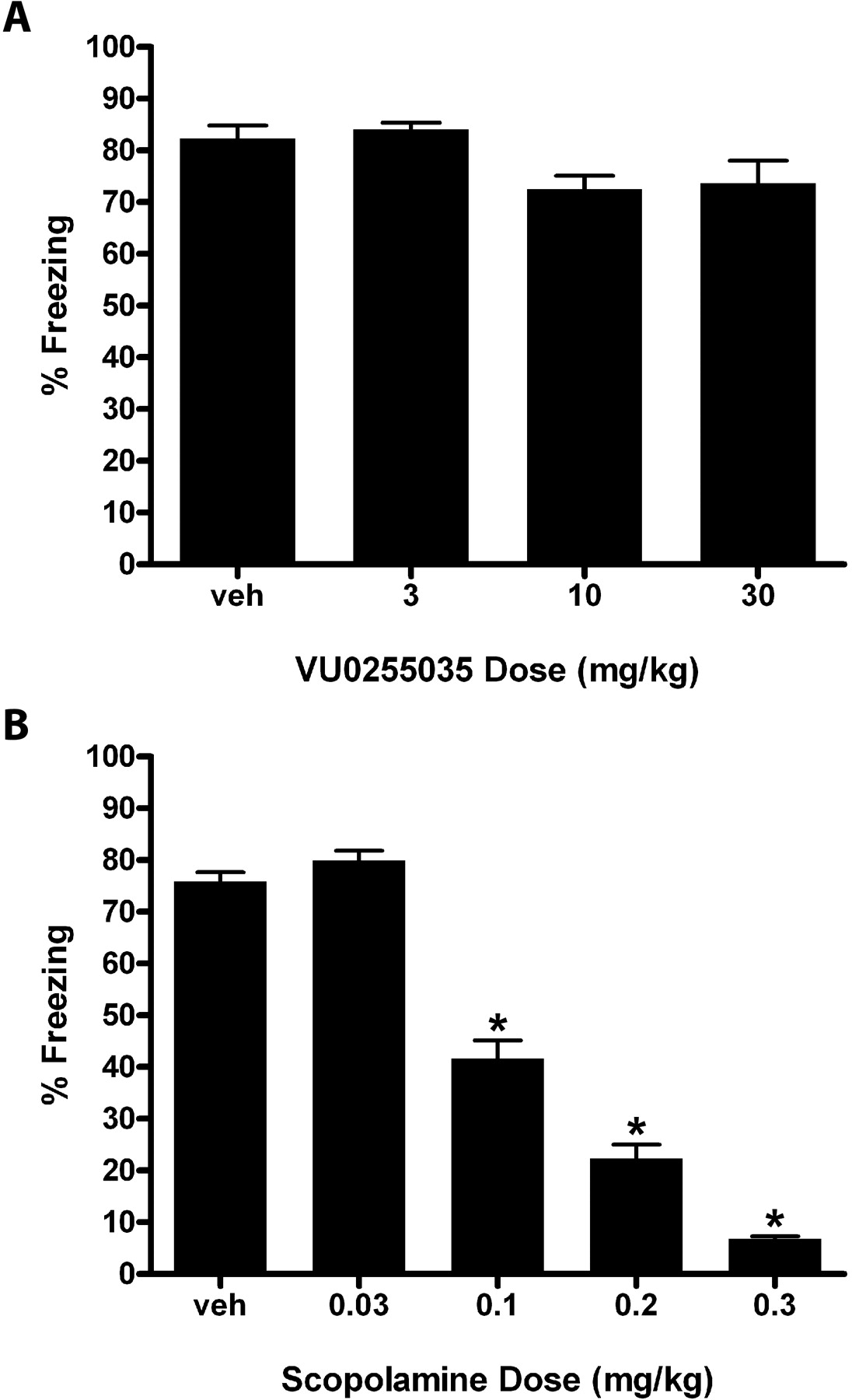
VU0255035 does not affect acquisition of a contextual fear conditioning response whereas scopolamine induces deficits. A, VU0255035 (3.0-10.0 mg/kg i.p.) had no effect on the acquisition of contextual fear. B, Scopolamine (0.03-0.3 mg/kg s.c.) produced a robust dose-dependent disruption in the acquisition of contextual fear-conditioning response as shown by a decrease in the percentage of time spent freezing in the same context environment as the training session, significant after doses of 0.1, 0.2, and 0.3 mg/kg by a Dunnett's comparison with vehicle. Results are expressed as mean percentage freezing behavior ± S.E.M. (n = 6-10 rats per treatment group). *, p < 0.05 compared with the vehicle control group.
It was somewhat surprising that we were able to achieve the high M1-subtype selectivity observed with VU0255035 with a compound that targets the orthosteric (ACh) binding site. The orthosteric site of the mAChRs is highly conserved (Felder et al., 2000), and previous efforts to develop this level of M1-subtype selectivity with orthosteric ligands have been largely unsuccessful. In contrast, recent efforts to develop ligands for less conserved allosteric sites on mAChRs have yielded highly selective positive allosteric modulators of individual mAChR subtypes (for review, see Conn et al., 2009). Given the functional selectivity of VU0255035 for M1 relative to M2-M5 mAChR subtypes, we initially expected that VU0255035 was probably acting as an allosteric antagonist of M1 mAChRs. However, multiple studies, including Scatchard analysis, Schild analysis, and mutagenesis studies, provide strong evidence that VU0255035 is a competitive orthosteric M1 antagonist. These data do not rule out the possibility that VU0255035 binds to a site that partially overlaps with the orthosteric binding site. Additional mutagenesis studies and binding studies will be necessary to fully evaluate the binding pocket for VU0255035.
In summary, VU0255035 is a highly selective orthosteric antagonist of M1 mAChRs. This compound demonstrates selective M1 mAChR antagonism both in vitro and in vivo as demonstrated through in vitro functional studies, the inhibition of PI hydrolysis induced by CCh in hippocampal slices, and the inhibition CCh-induced NMDA receptor potentiation in the hippocampus. The in vivo efficacy of VU0255035 for the inhibition of pilocarpine-induced seizures and lack of effect on hippocampus-dependent contextual fear acquisition, combined with pharmacokinetic data indicating that VU0255035 is rapidly taken up into the brain, provide valuable data demonstrating its utility as a tool to evaluate the role of M1 mAChRs antagonists as a novel approach for the treatment of PD, dystonia, and other movement disorders and that the use of selective M1 mAChR antagonists as therapeutics may not induce severe cognitive deficits. VU0255035 is a MLSCN probe available for free; use the identifier CID24768606.
Footnotes
-
This work was supported by the National Institutes of Health National Institute of Mental Health [Grants 3U54-MH074427, 3U54-MH074427-02S1, 1XO1-MH077606-01, 1U54-MH084659] (first three to C.D.W.; last to C.W.L.); the Dystonia Medical Research Foundation (to Z.X.); and a PhRMA Foundation Award (to D.J.S.).
-
ABBREVIATIONS: mAChR, muscarinic acetylcholine receptor; GPCR, G protein-coupled receptor; CNS, central nervous system; PD, Parkinson's disease; ACh, acetylcholine; r, rat; h, human; CHO, Chinese hamster ovary; HTS, high-throughput screening; NMDA, N-methyl-d-aspartate; PI, phosphoinositide; VU0255035, N-(3-oxo-3-(4-(pyridine-4-yl)piperazin-1-yl)propyl)benzo[c][1,2,5]thiadiazole-4 sulfonamide; CCh, carbachol; ACSF, artificial cerebrospinal fluid; PK, pharmacokinetics; HPLC, high-performance liquid chromatography; DIPEA, N,N-diisopropylethylamine; DMSO, dimethyl sulfoxide; LC, liquid chromatography; MS, mass spectrometry; HRMS, high resolution mass spectrometry; ELSD, evaporative light scattering detection; HOBt, N-hydroxybenzotriazole; mGluR, metabotropic glutamate receptor; CRC, concentration response curve; NMS, N-methyl-scopolamine; NDMC, N-desmethylclozapine; WT, wild type; NMDAR, NMDA receptor; ANOVA, analysis of variance; CS, conditioned stimulus; TBPB, 1-(1′-2-methylbenzyl)-1,4′-bipiperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one; VU178, (4-((3-fluorophenyl)ethynyl)phenyl)(morpholino)methanone; MLSCN, Molecular Libraries Screening Center Network.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Accepted April 30, 2009.
- Received March 25, 2009.
- U.S. Government work not protected by U.S. copyright





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}