


Abstract
Dopamine D2 receptor blockade has been an obligate mechanism of action present in all medications that effectively treat positive symptoms of schizophrenia (e.g., delusions and hallucinations) and have been approved by regulatory agencies since the 1950s. Blockade of 5-hydroxytrypatmine2A receptors plays a contributory role in the actions of the second generation of antipsychotic drugs, the so-called atypical antipsychotics. Nevertheless, substantial unmet medical needs remain for the treatment of negative symptoms and cognitive dysfunction. Recognition that dissociative anesthetics block the N-methyl-d-aspartate (NMDA) receptor channel has inspired a search for glutamatergic therapeutic mechanisms because ketamine and phencyclidine are known to induce psychotic-like symptoms in healthy volunteers and exacerbate the symptoms of patients with schizophrenia. Current pathophysiological theories of schizophrenia emphasize that hypofunction of NMDA receptors at critical sites in local circuits modulate the function of a given brain region or control projections from one region to another (e.g., hippocampal-cortical or thalamocortical projections). The demonstration that a metabotropic glutamate 2/3 (mGlu2/3) receptor agonist prodrug decreased both positive and negative symptoms of schizophrenia raised hopes that glutamatergic mechanisms may provide therapeutic advantages. In addition to discussing the activation of mGlu2 receptors with mGlu2/3 receptor agonists or mGlu2 receptor positive allosteric modulators (PAMs), we discuss other methods that may potentially modulate circuits with hypofunctional NMDA receptors such as glycine transporter inhibitors and mGlu5 receptor PAMs. The hope is that by modulating glutamatergic neurotransmission, the dysfunctional circuitry of the schizophrenic brain (both local circuits and long-loop pathways) will be improved.
Dopamine and serotonin, as opposed to emerging interest in glutamate, played a dominant role during the formative years of modern psychopharmacology when thinking about the pathophysiology and treatment of schizophrenia. Models derived from studying the behavioral effects of amphetamine and serotonergic hallucinogens were emphasized (Table 1). The seminal observations by Arvid Carlsson and his colleagues in the late early 1960s that first postulated functional dopamine receptor blockade as a necessary therapeutic action by the phenothiazine and butyrophenone structural classes set the stage for our current mechanistic understanding of all antipsychotic drugs currently used today (Carlsson and Lindqvist, 1963). The emergence of in vitro receptor binding and positron emission topography receptor occupancy studies in healthy volunteers and patients identified dopamine D2 receptor blockade as a critical feature for antipsychotic drugs in the clinic (Creese et al., 1976; Seeman et al., 1976; Farde et al., 1988). The early identification of dopamine D2 receptor binding sites by Seeman and Snyder in the 1970s and correlating antipsychotic efficacy to dopamine D2 receptor binding, before cloning of the D2-like family of dopamine receptors, further enriched the understanding for the molecular target that all antipsychotic drugs have in common (Creese et al., 1976; Seeman et al., 1976). Blockade of 5-hydroxytryptamine2A (5-HT2A) receptors was an important molecular target complimenting the blockade of dopamine D2 receptors for the development of atypical antipsychotic drugs (Meltzer et al., 1989; Kapur et al., 1999).
Effects of atypical antipsychotic drugs and drugs modulating targets modulating the glutamatergic system on positive and negative symptoms of schizophrenia and preclinical behavioral paradigms relevant to these symptom domains in addition to cognitive dysfunction
Clinical observations that channel-blocking N-methyl-d-aspartate (NMDA) receptor blockers induced psychotic symptoms in healthy volunteers or exacerbated the positive, negative, and cognitive dysfunction of patients with schizophrenia has been the most salient driving force behind the glutamatergic hypotheses regarding the pathophysiology and treatment of schizophrenia (Luby et al., 1959; Tamminga, 1998; Krystal et al., 1999). The initial hypothesis of glutamatergic hypofunction was strongly influenced by neuroimaging studies suggesting cortical hypofunction during rest or the performance of cognitive tasks. Later, the noncompetitive NMDA receptor antagonists were found to preferentially increase metabolism and extracellular glutamate levels in defined limbic circuits rather than over the entire cortical manner (Lahti et al., 1995; Moghaddam et al., 1997; Duncan et al., 1999; Holcomb et al., 2005).
Thus, these studies suggested that subanesthetic doses of noncompetitive NMDA receptor antagonists were activating glutamatergic neurons in the cerebral cortex and hippocampus; the glutamate released by these neurons could activate other ionotropic glutamate receptors (α-amino-3-hydroxy-5-methylisoxazole-4-propionate and kainate) or metabotropic glutamate (mGlu) receptors. Although these studies do not support a general notion that glutamate function or NMDA receptor function is necessarily decreased everywhere in the schizophrenic brain, the hypothesis remains that impaired NMDA function in important cellular compartments of the limbic forebrain may be a critical feature underlying the pathophysiology of schizophrenia. The variability from neuroimaging studies describing hyperactivation and hypoactivation in the brains of patients with schizophrenia compared with healthy volunteers suggests that the brain from a patient with schizophrenia is more aptly characterized by dysfunctional circuitry rather than simply increased or decreased activity (Callicott et al., 2003).
Together with the notion of a dysfunctional circuitry, the hypothesis that schizophrenia might be a neurodevelopmental disorder gained prominence because a variety of neuroimaging modalities indicated that substantial changes may be present even before initial treatment. The magnetic resonance imaging diffusion tensor imaging studies (Ellison-Wright and Bullmore, 2009) were especially important because they highlighted substantial abnormalities in long-loop tracts such as thalamocortical pathways that were consistent with histopathological studies defining a diminished neuropil in the prefrontal cortex and loss of cells in a number, although not all, of thalamic stereology studies. In addition to suspected developmental defects in these long-loop pathways connecting the prefrontal cortex, thalamus, striatum, hippocampus, amygdala, and dopamine-containing brainstem neurons, a better understanding of local circuit defects in schizophrenia has also emerged. Thus, currently, investigators work within the framework that schizophrenia involves dysfunctional local and long-loop network activity rather than there being simply decreased glutamatergic NMDA receptor tone in the brain (Tan et al., 2007).
How does this new view explain the earlier data that channel-blocking NMDA receptor antagonist induce psychotomimetic effects in animals and humans? The effects of the NMDA receptor antagonists could occur locally in cortical circuits in the prefrontal cortex or the hippocampus. GABAergic interneurons are known to spend a greater proportion of time in relatively depolarized states compared with pyramidal neurons and would experience a relaxation of Mg2+-induced block, allowing for cation flux through the channel. Several classes of GABAergic interneurons (e.g., chandelier and basket cells) provide a major inhibitory drive on cortical output cells, the pyramidal cells. Thus, systemic administration of noncompetitive NMDA receptor antagonists would preferentially block active NMDA channels in interneurons rather than pyramidal cells, resulting in a disinhibition of circuits. For schizophrenia, hypofunction of NMDA receptors on local GABAergic interneurons could play a key role in dysfunction of neuron ensembles, leading to disturbances of γ oscillations that may be the foundation for optimizing cortical function (Ford and Mathalon, 2008; Gonzalez-Burgos and Lewis, 2008). On the other hand, disinhibition of local circuits in areas projecting to crucial limbic circuitry such as hippocampal-prefrontal cortical pathways could account for an increase in activity in the PFC after the short-term administration of noncompetitive NMDA receptor antagonists. For schizophrenia, again, hypofunction of NMDA receptors at various nodes within these micro- and macrocircuits may contribute to disturbances of γ oscillations, leading to disturbances of cognition, affect, and bizarre delusions and hallucinations (Roopun et al., 2008). The importance of glutamate and NMDA receptors during normal central nervous system development is likely to play an important mechanistic role in the neurodevelopmental hypothesis of schizophrenia.
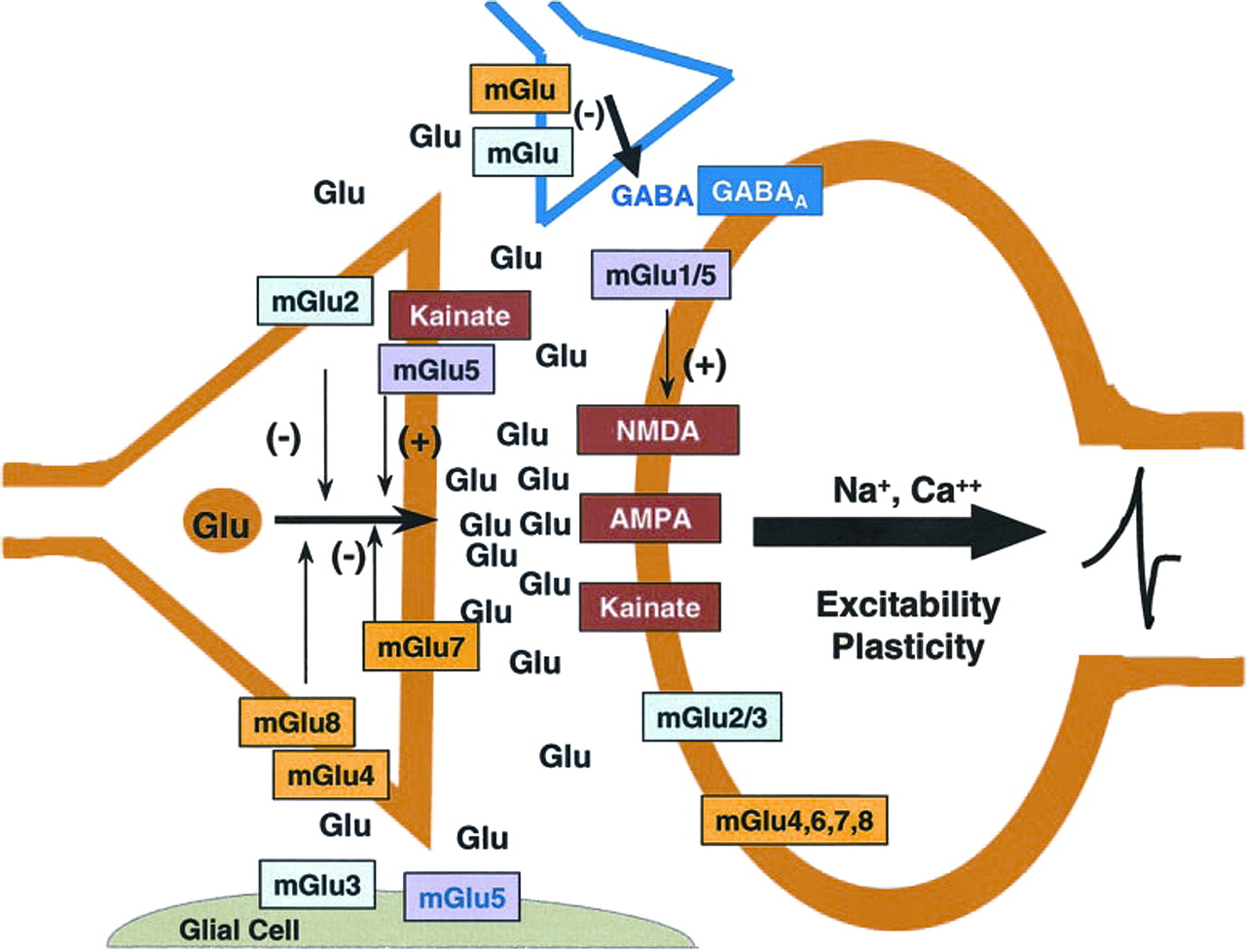
The clinical report that an mGlu2/3 receptor agonist prodrug effectively treated both positive and negative symptoms of patients with schizophrenia from a multicenter, double-blind, placebo-controlled study (Patil et al., 2007) is significant for at least several reasons. First, this is the first nondopamine D2 receptor-based treatment that has produced an apparently robust antipsychotic effect. Second, the primary effect of activating mGlu2 receptors involves the stimulation of an autoreceptor with a limbic localization in many of the regions suspected to be involved in the pathophysiology of schizophrenia (prefrontal cortex, dorsal and ventral striatum, thalamus, hippocampus, and amygdala) (Fig. 1) (Ghose and Tamminga, 2007). Thus, activation of mGlu2 receptors would be expected to decrease glutamate release from excitatory amino acid-containing axons/nerve terminals as shown in Fig. 2 (Schoepp, 2001).

Distributed dysfunctional cortical-subcortical circuits in patients with schizophrenia. Schematic of hypothetical neural systems underlying the three major symptom domains of schizophrenia: positive symptoms or psychosis, cognitive dysfunction, and negative affect. AH, anterior hippocampus; ACC, anterior cingulate cortex; mPFC, medial prefrontal cortex; DLPFC, dorsolateral prefrontal cortex; V. Pall, ventral pallidum; V Str., ventral striatum; SN/VTA, substantia nigra/ventral tegmental area. [Adapted from Ghose S and Tamminga CA (2007) Phenomenology and clinical science of schizophrenia, in Handbook of Contemporary Neuropharmacology (Sibley DR, Hanin I, Kuhar M, and Skolnick P eds) pp 251–283, John Wiley & Sons, New York. Copyright © 2007 John Wiley & Sons. Used with permission.]

Glutamatergic neurotransmission with and primary localization of mGlu and ionotropic glutamate receptors with respect to presynaptic glutamatergic nerve terminals, postsynaptic cells, presynaptic GABAergic nerve terminals, and glia. A primary function of mGlu2 receptors is as an autoreceptor for glutamatergic nerve terminals. The mGlu3 receptor is present as a heteroceptor on a number of GABAergic interneurons throughout the brain. This receptor is one of two primary mGlu receptors, along with mGlu5 receptors, that modulate the function of glia. The mGlu3 receptor is also present as a postsynaptic receptor as well. The mGlu5 receptor has been intensely studied with respect to functional positive coupling with NMDA receptors. A portion of these receptors also appear to be located on glutamatergic axons/nerve endings in addition to the glial localization noted above. [Reprinted from Schoepp DD (2001) Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther 299:12–20. Copyright © 2001 American Society for Pharmacology and Experimental Therapeutics. Used with permission.]
In this minireview, we review evidence that modulating glutamate in dysfunctional limbic circuits may be an important strategy opening up new molecular targets for the treatment of schizophrenia. The discussion first centers around orthosteric mGlu2/3 receptor agonists or mGlu2 receptor PAMs. The last two targets that are exemplified are those that might address hypofunctional NMDA receptor in the miswired dysfunctional limbic circuits, inhibitors of the glycine transporter 1 subtype, and mGlu5 receptor PAMs.
mGlu2/3 Receptor Agonists
It was more than 10 years ago that the first studies using selective mGlu2/3 receptor agonists convincingly demonstrated the antipsychotic-like potential of such agents (Moghaddam and Adams, 1998; Cartmell et al., 1999). One of the most publicized pharmacological effects of mGlu2/3 receptor stimulation is the reversal of various effects induced by psychotomimetic drugs such as PCP. For instance, in full accordance with the predominantly presynaptic function of mGlu2 receptors and their role in the negative feedback regulation of excess release of glutamate, LY354740 was shown to attenuate PCP-induced increase in glutamate release in the prefrontal cortex in rats (Moghaddam and Adams, 1998).
Cortical glutamate release is also under the control of 5-HT2A receptors. 5-HT2A receptor activation induces excitatory postsynaptic currents at apical dendrites of pyramidal neurons in the prefrontal cortex. 5-HT2A receptor-mediated increases in glutamate release have been linked to cognitive, perceptual, and affective distortions produced by hallucinogenic 5-HT2A agonists. It is noteworthy that these effects can be blocked not only by 5-HT2A receptor antagonists but by mGlu2/3 receptor agonists as well (Marek et al., 2000). These data implicate mGlu2/3 receptors in the common mechanisms underlying the actions of different types of psychotomimetic drugs such as PCP-like NMDA receptor channel blockers and lysergic acid diethylamide-like 5-HT2A receptor agonists. Physiological significance of such interactions is supported by emerging evidence on colocalization of 5HT2A and mGlu2/3 receptors in the cortex (González-Maeso et al., 2008).
In full agreement with the results of neurochemical and electrophysiological studies, one of the best documented behavioral effects of mGlu2/3 receptor stimulation is the reversal of behaviors induced by PCP-like agents and 5-HT2 receptor agonists. Reversal of PCP-induced motor hyperactivity was demonstrated for one of the first selective mGlu2/3 receptor agonists, LY354740 (Moghaddam and Adams, 1998), and was repeatedly confirmed for this and other mGlu2/3 receptor agonists in several laboratories (Cartmell et al., 1999). Various motor stereotypes induced by PCP-like drugs and 5-HT2A agonists [(±)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane] are also readily inhibited by mGlu2/3 receptor stimulation (Gewirtz and Marek, 2000; Homayoun and Moghaddam, 2008).
Increased cortical glutamate release is likely to account for the ability of systemic administration of PCP-like agents (such as MK-801) to increase the spontaneous firing rate of prefrontal cortical neurons, to decrease the variability of spike trains, and to disrupt patterns of spontaneous bursting. Stimulation of mGlu2/3 receptors was shown to reverse these effects (Homayoun et al., 2005), a finding that is well in line with the results of neurochemical studies.
At first glance, enhanced glutamatergic activity and increased firing rate of cortical neurons after short-term NMDA receptor channel blockade do not fit well with the views on hypofrontality in schizophrenia. Indeed, there are several reports on enhanced activity of brain areas involved in the pathophysiology of schizophrenia under conditions of limited cognitive load. For example, one study found greater activation in the hippocampus, thalamus, and dorsolateral prefrontal cortex in patients with schizophrenia during the sensory gating task and in response to urban noise (Tregellas et al., 2007, 2009), whereas another group reported on the elevated hippocampal and amygdala activity during the passive viewing of human faces (Holt et al., 2006). However, when the behavioral performance sets additional demands on these networks, activity will increase further, and such an above-physiological increase will now be accompanied by abnormally high or lost synchronization, phenomena also observed after short-term application of sufficiently high doses of PCP-like drugs (Sebban et al., 2002; Pinault, 2008). Such synchronization is seen as a potential facilitator of neural communication and synaptic plasticity. For the tasks, commonly used to study hypofrontality (e.g., working memory tasks), synchronized network activity is very critical because it favors information retention via increased activity of certain ensembles of neurons. Decorrelated or desynchronized activity may functionally be expressed as hypofrontality.
Another hypothetical approach to examine the relationships between neuronal activity and functional performance is best represented by the analysis of dopaminergic modulation of working memory and prefrontal cortical activity. It is currently believed that there is a certain level of dopamine receptor stimulation (more specifically, D1 receptors) that is required for optimum performance (Arnsten, 1998). Both reduced and increased dopamine receptor stimulation result in impaired cognitive performance. In other words, there is an inverted U-shaped relationship between cortical activity and cognitive performance. These studies led further to explain the existence of both hyper- and hypofrontality states in patients with schizophrenia. Callicott and colleagues (2003) suggested that schizophrenia is associated with the leftward shift in the inverted U function compared with healthy control subjects. At low working memory load, patients are thus expected to appear hyperfrontal. However, their working memory capacity is eventually breached, which results in a hypofrontal pattern.
Interestingly enough, cognitive effects of mGlu2/3 receptor agonists may also follow a biphasic dose-effect function. There were reports that short-term systemic application of selective mGlu2/3 receptor agonists such as LY354740 impairs performance in cognitive tasks in laboratory animals (Higgins et al., 2004). By following the inverted U-shaped activity-performance function hypothesis outlined above by Callicott and colleagues (2003), one could speculate that the impairing effects of mGlu2/3 receptor agonists are more likely to be observed under somewhat challenging conditions that require a significant increase in cortical activity but still result in proper cognitive performance. In contrast, one may expect then that lower-demand cognitive tasks may fail to detect the impairing effects of mGlu2/3 receptor agonists. Perhaps this explains why no cognitive-impairing effects of LY354740 were observed in humans in whom tasks may have been optimized to study cognitive impairments produced by ketamine (Krystal et al., 2005).
Within the inverted U hypothesis, it presents no difficulty to understand how a compound that may impair cognition when given alone is able to reverse cognitive deficits induced by another agent. Moghaddam and Adams (1998) demonstrated that LY354740 counteracted the detrimental effects of PCP on working memory performance in the T-maze task. It should be noted that there are negative results as well. More specifically, mGlu2/3 receptor agonists were shown not to be able to antagonize cognition-impairing effects of PCP in other studies in which experimental protocols may have been optimized to detect cognition-impairing effects of mGlu2/3 agents (Schlumberger et al., 2009). Furthermore, failures to reverse certain cognitive effects of PCP may be related to the fact that PCP-like agents are able to impair performance in behavioral tasks via a number of different mechanisms, including motor activation, behavioral disinhibition, and induction of state-dependence. Meanwhile, relationships between excessive glutamate release, synchronization loss, and hypofrontality induced by psychotomimetic drugs and/or observed in patients with schizophrenia were studied using mainly working memory tasks, and it is unclear to what extent these phenomena contribute to the effects of PCP-like agents observed in various other behavioral cognitive tasks.
A study by Moghaddam and Adams (1998) explicitly linked the reversal of PCP-induced cognitive impairment with the ability of LY354740 to attenuate PCP-induced cortical glutamate release. Taken together with work from this and other groups, these data suggest that mGlu2/3 receptor stimulation may reverse this excessive release of glutamate, restore normal neuronal activity, and thereby improve cognitive performance.
The only known clinical laboratory study on the cognitive effects of mGlu2/3 receptor stimulation in humans directly supports these expectations. Krystal and coworkers (2005) reported that LY354740 induced a dose-dependent reversal of ketamine-induced impairment of working memory. These data support the view that mGlu2/3 receptor stimulation should have beneficial effects on cognition in patients with schizophrenia, and these effects are localized specifically in the domain of working memory, one of the key domains implicated in the pathophysiology of schizophrenia. Given that treatment with LY354740 did not modulate other effects induced by ketamine infusion (e.g., reduced vigilance, increased distractibility, impaired performance on a verbal learning test, increased PANSS scores), it remains to be established whether the reversal of working memory impairments (and more generally the reversal of schizophrenia-associated hypofrontality) is predictive of clinical efficacy. Further regarding the effects of mGlu2/3 receptor agonists on cognition in humans, LY354740 and the prodrug LY544344 did not induce adverse events, suggesting cognitive impairment in more than 450 patients with generalized anxiety disorder in whom this mGlu2/3 receptor agonist and prodrug where found to produce therapeutic effects (Michelson et al., 2005; Dunayevich et al., 2008).
First, support for these predictions relative to patients with schizophrenia is provided by the results of a recent clinical study in which the mGlu2/3 receptor agonist LY404039 (administered as a prodrug known as LY2140023) reduced PANSS total, positive and negative scores in patients with schizophrenia (Patil et al., 2007). Effects of LY2140023 on cognition were not assessed in this study. However, it is noteworthy that in the Patil study, there were two cognition-related PANSS items (difficulty in abstract thinking and poor attention) that were reportedly positively affected by mGluR2/3 agonist treatment (A. Breier, personal communication). Although the effects of mGlu2/3 receptor agonists on cognition in patients with schizophrenia await more formal evaluation, these preliminary data may be taken as the first evidence confirming the expectations generated by the results of preclinical studies (Table 1). Further confirmatory studies are required to understand the therapeutic efficacy effect size for LY2140023, especially compared with aripiprazole, quetiapine, and ziprasidone. These atypical antipsychotic drugs, unlike olanzapine and risperidone, have not been found to provide a clinically significant benefit with respect to treating the positive and negative symptoms of schizophrenia over the first generation of antipsychotic drugs compared in the CATIE studies or meta-analyses.
One important question of relevance for ongoing and future drug discovery efforts is the mGlu receptor subtype mediating the therapeutic effects of LY2140023 in patients. Genetic association analyses have been consistently pointing at single-nucleotide polymorphisms in the mGlu3 receptor gene (GRM3) in several populations of patients with schizophrenia. This GRM3 polymorphism is associated with a noncoding region that does not seem to affect mGlu3 receptor expression outside of a single report suggesting altered levels of mGlu3 receptor homodimers (Corti et al., 2007). However, detailed studies conducted by Weinberger and colleagues suggested a specific molecular pathway by which the GRM3 genotype alters glutamate neurotransmission, prefrontal and hippocampal physiology, and cognition, and thereby increased the risk for schizophrenia (Egan et al., 2004). More specifically, these studies revealed a link between a high-risk GRM3 allele and reduced expression of glial glutamate transporter. Although there is not much further evidence in support of this hypothesis, a genetic silencing of glial transporter expression in mice resulted in the behavioral abnormalities that are typically observed after the treatment with psychotomimetic drugs (Karlsson et al., 2008). The most intriguing is perhaps that these behaviors were effectively antagonized by the stimulation of mGlu2/3 receptors.
However, a series of studies conducted in mGlu2 and mGlu3 receptor knockout mice clearly indicated the role of mGlu2 receptors in the antipsychotic-like effects of mGlu2/3 receptor agonists. Indeed, mGlu2/3 receptor agonists, including the mGlu2/3 agonist with demonstrated antipsychotic activity in the clinic, lose the ability to attenuate behaviors induced by psychotomimetic drugs when tested in mGlu2, but not mGlu3, knockout mice (Spooren et al., 2000; Fell et al., 2008; Woolley et al., 2008).
mGlu2 Receptor Potentiators (e.g., PAMs)
Another growing body of evidence supporting the significance of mGlu2 receptors in the antipsychotic effects of orthosteric mGlu2/3 receptor agonists comes from a number of studies that demonstrated comparable efficacy of mGlu2 receptor PAMs (Galici et al., 2005; Johnson et al., 2005). The latter class of drugs presents considerable interest for a number of reasons. For instance, orthosteric receptor agonists act at the glutamate binding site and are themselves structurally constrained glutamate analogs that set several serious limitations (e.g., restricted chemical space, lower receptor selectivity). Given the predominantly extrasynaptic location of mGlu2 receptors that is likely to control the release of glutamate by detecting excess amounts of glutamate that escape the reuptake mechanisms, the ability to selectively target these receptors offers significant advantages. Most importantly, mGlu2 receptor PAMs may not induce any appreciable effects under normal conditions but will enhance negative feedback control under the conditions of abnormally enhanced glutamate release as reflected in frequency-dependent modulation of synaptic transmission in the striatum (Johnson et al., 2005). Such ability to “recognize” pathological state(s) and/or networks could result in improved therapeutic efficacy and safety.
Available evidence seems to support such a profile of mGlu2 receptor PAMs. On the one hand, there are no signs that these drugs produce any cognitive impairment in normal subjects. This is an important phenomenon to consider because of the knockout mouse data that specifically implicated mGlu2 receptors in cognitive-impairing effects of mGlu2/3 receptor agonists (Higgins et al., 2004). On the other hand, mGlu2 receptor potentiators reproduce both antipsychotic-like (Table 1) and anxiolytic-like effects of mGlu2/3 receptor agonists (Galici et al., 2005; Johnson et al., 2005). There is much less information available to claim that positive allosteric modulation of mGlu2 receptors would be sufficient to overcome cognitive impairments associated with acute NMDA receptor channel blockade by PCP-like drugs or with glutamatergic dysbalance associated with schizophrenia. With several research groups actively working in this field and a number of fairly potent compounds already known, characterization of the cognitive effects of selective mGlu2 receptor potentiators will certainly be completed soon.
Glycine Transporter 1 Inhibitors
Among the approaches aiming at the reversal of a suspected NMDA receptor hypofunction in schizophrenia activation of the glycine-site represents the most direct one. Several small clinical trials usually restricted to one or two centers have been performed with either glycine and with d-serine, the natural (full) agonists; with d-cycloserine, a partial agonist of the glycine site; and with sarcosine, a naturally occurring low-affinity glycine transport inhibitor. Across studies, the full agonists were more effective than the partial agonist d-cycloserine, and the tested compounds were more efficacious in conjunction with typical and newer atypical antipsychotics than with clozapine (Javitt, 2008). The latter outcome has been attributed to a putative intrinsic component of clozapine enhancing glutamatergic neurotransmission (Coyle, 2006). The most prominent effect across all trials was an improvement of negative symptoms by 15% (Javitt, 2008). Positive and cognitive symptoms were significantly improved in some but not all trials. Because of the small number of patients recruited in these trials and the negative outcome of one large multicenter trial, the CONSIST study (Buchanan et al., 2007), the trials do not allow a final conclusion about the clinical effectiveness of NMDA glycine-site activation. A key problem in the interpretation of the results is the uncertainty of whether appropriate brain concentrations, and, more importantly, synaptic levels of the glycine site agonists, have been achieved. To obtain a more efficient increase of synaptic glycine and hence NMDA receptor activation, high-affinity inhibitors of the glycine transporter 1 (GlyT1) are currently being developed by several pharmaceutical companies.
GlyT1 shows a widespread distribution throughout the brain. In the cerebellum and caudal parts of the central nervous system, GlyT1 seems to be predominantly localized on glial cells and to be involved in the regulation of inhibitory glycinergic neurotransmission (Zafra and Giménez, 2008), whereas in the forebrain, it has been localized pre- and postsynaptically at glutamatergic synapses. At the molecular level, physical association with synthaxin 1A and with postsynaptic density protein 95, an NMDA receptor-associated protein, indicates a synaptic localization close to NMDA receptors. The vicinity to NMDA receptors makes GlyT1 ideally suited to modulate their activity. A number of companies have GlyT1 inhibitor programs at different stages (preclinical to clinical phase 2) and have published preclinical profiles of some of the compounds. To demonstrate expected target-related effects, the compounds were shown to increase extracellular glycine levels in the cortex (Depoortère et al., 2005; Boulay et al., 2008) and hippocampus (Martina et al., 2004) of freely moving rats and to enhance NMDA receptor activity (Martina et al., 2004; Depoortère et al., 2005; Boulay et al., 2008). The results correspond to increases of NMDA receptor-mediated currents in heterozygous GlyT1 knockout mice and forebrain-selective homozygous GlyT1 knockout mice (Tsai et al., 2004; Yee et al., 2006). Apart from enhancing synaptic activity, GlyT1 inhibition also increased long-term potentiation in line with the role of NMDA receptors in synaptic plasticity (Martina et al., 2004). Together, the data support the validity of the concept to enhance NMDA receptor activity by GlyT1 inhibition.
Preclinical antipsychotic-like effects of GlyT1 inhibitors are described in an increasing number of publications, which can only in part be summarized in this review (Table 1). Several compounds have been reported to antagonize the hyperlocomotion induced by PCP or MK-801 (Harsing et al., 2003; Depoortère et al., 2005; Boulay et al., 2008; Singer et al., 2009). In adult rats treated with PCP at neonatal age, the enhanced locomotor response to amphetamine could be reversed (Depoortère et al., 2005). The attenuation of locomotor responses may, at least in part, be mediated via an effect on dopaminergic neurotransmission. GlyT1 inhibitors have been reported to inhibit PCP and methamphetamine-induced (Drescher et al., 2006; K.U. Drescher, personal communication) and ethanol-induced (Molander et al., 2007) dopamine release in the nucleus accumbens, whereas basal dopamine in the PFC was increased (Depoortère et al., 2005). In rats continuously treated with PCP, the increased sensitivity of the DA release and the locomotor response to amphetamine could be reversed by GlyT1 inhibition (Javitt et al., 2004). These results indicate the capability to reverse evoked or sensitized mesolimbic hyperdopaminergic states and to increase basal dopamine in the PFC, suggesting a beneficial effect on positive symptoms associated with imbalanced dopaminergic activity. Support for efficacy against positive symptoms comes also from the reported reversal of amphetamine-disrupted latent inhibition (Black et al., 2009), a model thought to reflect increased salience and distractibility associated with psychotic symptoms.
By increasing dopamine in the PFC in conjunction with an expected enhancement of NMDA receptor activity, GlyT1 inhibitors are believed to reverse dysfunctional PFC-subcortical circuitry associated with negative symptoms. In support of potential efficacy against negative symptoms, we have observed the attenuation of PCP-induced social interaction deficits by different GlyT1 inhibitors (data not shown). Furthermore the two GlyT1 inhibitors SSR103800 and SSR504734 were found to reverse abnormally persistent latent inhibition induced by MK-801 (Black et al., 2009), which has been linked to impaired set-shifting associated with negative symptoms (Weiner, 2003).
Because the latter model is also believed to reflect cognitive inflexibility, the results point to the ability of GlyT1 inhibition to reinstate cognitive flexibility. Several other effects of GlyT1 inhibitors in the cognitive domain have been reported. SSR103800 (Boulay et al., 2008) and N[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine (Karasawa et al., 2008) reversed object recognition impaired by short- or long-term NMDA receptor antagonists; SSR504734 reversed impaired social discrimination in rats treated with PCP at neonatal age (Harich et al., 2007), and SSR504734 enhanced working memory performance in a continuous delayed alternation task in C57BL/6 mice (Singer et al., 2009). These effects are of relevance for improving cognition in schizophrenia because these models reflect particular schizophrenia-related cognitive deficits, such as impaired visual object memory, face recognition memory (Calkins et al., 2005), and working memory. Together, the available data describe a broad antipsychotic-like profile of GlyT1 inhibitors promising efficacy across different symptoms in schizophrenia. Little is known about the cells or circuits mediating the reported effects. Numerous examples of GlyT1 inhibitors antagonizing the effects of NMDA receptor antagonists suggest that GlyT1 inhibitors may affect cells that exhibit a particular sensitivity for NMDA receptor antagonists. An intriguingly selective PCP antagonistic effect was recently reported for SSR504734, which completely and selectively reversed the pattern of brain activation by PCP in a pharmacological magnetic resonance imaging study (Gozzi et al., 2008). The most plausible explanation for this observation is the reversal of reduced NMDA receptor activity at GABAergic interneurons suspected to have a particular sensitivity for a blockade by noncompetitive NMDA receptor antagonists (Coyle, 2004).
The reinstatement of impaired NMDA receptor activity at interneurons (but not necessarily selectively at interneurons) may be fundamental for the antipsychotic activity of GlyT1 inhibitors. It was shown in a recent study that the expression of the GluN2A (NMDA NR2A subunit) in glutamic acid decarboxylase-67 isoform-positive cells in layers 2 and 5 of the cingulate cortex of subjects with schizophrenia is dramatically reduced by more than 50% (Woo et al., 2008). The reduced GluN2A expression is probably due to a reduced NMDA receptor activity at the affected interneurons because it has been shown in other studies that GluN2A expression seems to be down-regulated by reduced glutamatergic input and vice versa. In line with this assumption, treatment of intermediate duration with MK-801 has been shown to almost completely down-regulate the expression of NMDA receptor subunits and parvalbumin in microdissected parvalbumin-positive interneuron in the rat frontal cortex (Xi et al., 2009). Whether GlyT1 inhibition is able to enhance NMDA receptor expression in GABAergic interneurons needs to be demonstrated.
There have been concerns that GlyT1 inhibition may be associated with unwanted side effects, particularly with motor impairments and respiratory effects mediated via the activation of glycine A receptors in the cerebellum and in the brainstem. It is important to note that these concerns are based on the effects of a subclass of GlyT1 inhibitors, which contain a sarcosine moiety in their structure. It has been shown that sarcosine-derived inhibitors exhibit an allosteric and irreversible inhibition, whereas nonsarcosine-derived structures were found to be reversible competitive inhibitors (Mezler et al., 2008). Consistent with an irreversible inhibition, sarcosine-derived inhibitors produce slowly increasing and sustained locomotor impairments (Perry et al., 2008), pointing to cumulating binding associated with a poorly controllable degree of inhibition. Nonsarcosine-derived structures may exhibit a more appropriate safety margin, not only because of their reversible mode of action, but also because of the competitive and surmountable transport inhibition. Different glycine concentrations may lead to subtle although critical differences in the degree of inhibition in different target regions. The competitive compounds currently in development will hopefully provide the proof of whether GlyT1 inhibition represents an efficacious antipsychotic mechanism at safe doses.
mGlu5 Receptor Potentiators
The mGlu5 receptor mRNA and protein is distributed widely throughout the limbic forebrain (e.g., cortex, hippocampus, dorsal and ventral striatum) with a relatively sparse distribution in much of the brainstem (Abe et al., 1992; Romano et al., 1995). One of the interesting relationships of the postsynaptic mGlu5 receptor is coupling to NMDA receptors through the Homer family of scaffolding proteins. Most of the immunoreactivity with antibodies recognizing mGlu5 is found on membranes of dendritic spines and shafts. One function of certain Homer proteins may be to bring the mGlu5 receptor into close physical proximity with the NMDA receptor at synaptic sites. A second cellular compartment implicated in the pathophysiology of schizophrenia that mGlu5 receptors may share with NMDA receptors are glial cells. The understanding that much of the signal in 2-deoxyglucose experiments is derived from the functional coupling of astrocytes and nerve terminals highlights the potential importance of these glial glutamate receptors in modulating local circuit activity. A third shared cellular compartment between mGlu5 receptors and NMDA receptors may be interneurons in a variety of cortical and subcortical sites. However, although NMDA receptors with various heteromeric combinations of subunits are present in almost all neurons in the brain, mGlu5 receptors may be restricted to certain subclasses of GABAergic interneurons, as seems is the case in the primate prefrontal cortex (Muly et al., 2003) and hippocampal formation (Kerner et al., 1997). A numerically minor cellular compartment in which mGlu5 receptors are also found are in axons of presumed glutamatergic neurons in which they may enhance glutamate release in the prefrontal cortex and neocortex (Romano et al., 1995; Thomas et al., 2000; Muly et al., 2003; Marek and Zhang, 2008; Musante et al., 2008). Homer1 proteins may play a regulatory role in targeting mGlu5 receptors to axonal or dendritic compartments (Ango et al., 2000).
Among the effects seen in mice with a deletion of the mGlu5 receptor, sensorimotor gating as reflected by prepulse inhibition was disrupted in several mice knockout strains with different backgrounds (Brody et al., 2004b). Neither clozapine nor haloperidol reversed the prepulse inhibition deficit in the mGlu5 receptor−/− strain at doses that were active in reversing either amphetamine- or ketamine-induced deficits in wild-type mice (Brody et al., 2004a). The effects of mGlu5 receptor PAMs in animals and mGlu5 receptor negative allosteric modulators in humans described below, however, suggest caution in overinterpreting data with transgenic mice lines.
For both the group I (mGlu1 and mGlu5) and group II (mGlu2 and mGlu3) mGlu receptors, the development of selective agonists within a group with appropriate drug-like properties has been very difficult. As a result, the focus that shifted to developing selective compounds modulating a single mGlu receptor has focused on developing positive or negative allosteric agonists/modulators. A number of series of mGlu5 receptor PAMs have been developed that enhance Ca2+ mobilization in response to glutamate in cells transfected with mGlu5 receptors. Several different allosteric binding sites have been identified for these mGlu5 receptor PAMs; one site seems to bind both PAMs and NAMs. A further complication is that different mGlu5 PAMs seem to differentially recruit different signaling pathways in cortical astrocytes (Zhang et al., 2005); this effect may relate to the two different allosteric binding sites identified. The mGlu5 receptor PAM N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide has been shown to enhance NMDA receptor activity via potentiation of mGlu5 receptor function in hippocampal slices (O'Brien et al., 2004). Both in vitro and in vivo studies supporting utility in treating cognitive dysfunction may be found in a recent report that the mGlu5 receptor PAMs facilitated both hippocampal long-term potentiation and long-term depression in addition to enhancing performance in the Morris water maze (Ayala et al., 2009). Additional evidence for a procognitive effect of mGlu5 receptor PAMs was found in the attenuation of performance impairments induced by MK-801 on an operant set-shifting task when an mGlu5 receptor PAM was administered after the channel-blocking NMDA receptor antagonist (Darrah et al., 2008). The Addex mGlu5 receptor PAM ADX47273 was also reported to attenuate natural or scopolamine-induced forgetting in the novel object recognition task (Epping-Jordan et al., 2005).
Further evidence consistent with antipsychotic action for mGlu5 receptor PAMs has been collected for different chemical platforms originating from Merck and Addex scientists, respectively, using classic antipsychotic screening tests (Table 1). For example, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5yl) benzamide and/or ADX47273 has been found to suppress amphetamine-induced hyperactivity or amphetamine-induced deficits in prepulse inhibition in rats (Epping-Jordan et al., 2005; Kinney et al., 2005; Liu et al., 2008). Several different mGlu5 receptor PAMs have also been reported to attenuate locomotor hyperactivity induced by channel-blocking NMDA receptor antagonists (Chan et al., 2008; Homayoun and Moghaddam, 2008; Liu et al., 2008), although mGlu5 receptor NAMs have been reported to enhance locomotor hyperactivity induced by channel-blocking NMDA receptor antagonists. The mGlu5 receptor PAM ADX47273 also exerted an antipsychotic-like effect in a classic screen by reducing conditioned avoidance responding in the rat (Liu et al., 2008).
Although mGlu5 receptor PAMs have not yet reached phase 2 proof-of-concept testing in patients, a putative anxiolytic drug, fenobam, was investigated in phase 2 trials that was subsequently determined to be a selective mGlu5 receptor NAM (Porter et al., 2005). Although there was modest clinical evidence for an anxiolytic effect of fenobam in patients, is was discontinued after this medication seemed to induce the derealization and depersonalization in patients with anxiety disorders. Although these observations may portend positive effects for mGlu5 receptor PAMs in the clinic for schizophrenia, it still remains unknown whether these adverse events were due to the pharmacology of the parent molecule.
Many questions remain for this target. What symptom domains of schizophrenia will mGlu5 PAMs effectively treat? Which cellular locus of action(s) will be responsible for these effects? If mGlu5 receptor PAMs are effective in treating schizophrenia, will these effects be related to coupling of mGlu5 receptors with NMDA receptors through scaffolding proteins, or will they be independent of this physiological coupling?
Conclusions
Over the last 25 years, hypotheses reflecting glutamate hypofunction and then later NMDA receptor hypofunction in schizophrenia have been reinterpreted by the growing recognition that this pathophysiological principle acts within the context of dysfunctional local circuits and miswired long-loop pathways between the prefrontal cortex, hippocampus, striatum, thalamus, amygdala, and the ventral tegmental area. A number of the leading genetic susceptibility candidates are intimately involved in glutamatergic neurotransmission. Furthermore, there has been a growing understanding in the neurodevelopmental biology for NMDA and mGlu receptors. In this context, it is not especially surprising that targets which profoundly alter the synaptic availability of glutamate and/or directly modulate NMDA receptor function demonstrate strong localization within cortical and subcortical circuits implicated in schizophrenia. It is expected that better understanding the nature by which ensembles of neurons lead to oscillatory behavior both within a given region and between cortical and subcortical regions will help us to optimize glutamatergic treatments for schizophrenia. It is also hoped that such translational work between rodents, nonhuman primates, and humans will also lead to additional breakthroughs to treat this devastating illness.
Footnotes
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.059865
-
ABBREVIATIONS:
- 5-HT2A
- 5-hydroxytryptamine2A
- NMDA
- N-methyl-d-aspartate
- mGlu2/3
- metabotropic glutamate 2/3
- ADX47273
- [S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl}-methanone]
- GluN2A
- N-methyl-d-aspartate NR2A subunits
- GlyT1
- glycine transporter subtype 1
- GRM3
- mGlu3 receptor gene
- LY354740
- (1S,2S,5R,6S)-2-aminobicylo[3.1.0]hexane-2,6-dicarboxylic acid
- LY404039
- (−)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]-hexane-4,6-dicarboxylic acid
- LY544344
- (1S,2S,5R,6S)-2[(2′S)-(2′-amino)-propionyl]aminobicyclo [3.1.0]hexane-2,6-dicarboxylic acid
- LY2140023
- methionine prodrug of (−)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]-hexane-4,6-dicarboxylic acid
- mGlu
- metabotropic glutamate
- MK-801
- dizocilpine
- MRI
- magnetic resonance imaging
- NAM
- negative allosteric modulator
- PAM
- positive allosteric modulator
- PANSS
- positive and negative symptom score
- PCP
- phencyclidine
- PFC
- prefrontal cortex
- SSR504734
- (2-chloro-N-[(S)-phenyl[(2S)]-piperidin-2-yl]methyl)-3-trifluoromethyl benzamide
- ORG 25935
- cis-N-methyl-N-(6-methoxy-1-phenyl-1,2,3,4-tetrahydronaphthalen-2-ylmethyl)aminomethylcarboxylic acid hydrochloride.
- Received July 28, 2009.
- Accepted November 23, 2009.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics
References
- Abe et al., 1992.↵
- Ango et al., 2000.↵
- Arnsten, 1998.↵
- Ayala et al., 2009.↵
- Benneyworth et al., 2007.
- Black et al., 2009.↵
- Boulay et al., 2008.↵
- Brody et al., 2004a.↵
- Brody et al., 2004b.↵
- Buchanan et al., 2007.↵
- Calkins et al., 2005.↵
- Callicott et al., 2003.↵
- Carlsson and Lindqvist, 1963.↵
- Cartmell et al., 1999.↵
- Chan et al., 2008.↵
- Corti et al., 2007.↵
- Coyle, 2004.↵
- Coyle, 2006.↵
- Creese et al., 1976.↵
- Darrah et al., 2008.↵
- Depoortère et al., 2005.↵
- Drescher et al., 2006.↵
- Dunayevich et al., 2008.↵
- Duncan et al., 1999.↵
- Egan et al., 2004.↵
- Ellison-Wright and Bullmore, 2009.↵
- Epping-Jordan et al., 2005.↵
- Farde et al., 1988.↵
- Fell et al., 2008.↵
- Ford and Mathalon, 2008.↵
- Galici et al., 2005.↵
- Gewirtz and Marek, 2000.↵
- Geyer and Ellenbroek, 2003.
- Ghose and Tamminga, 2007.↵
- Gonzalez-Burgos and Lewis, 2008.↵
- González-Maeso et al., 2008.↵
- Gozzi et al., 2008.↵
- Harich et al., 2007.↵
- Harsing et al., 2003.↵
- Higgins et al., 2004.↵
- Holcomb et al., 2005.↵
- Holt et al., 2006.↵
- Homayoun et al., 2005.↵
- Homayoun and Moghaddam, 2008.↵
- Javitt, 2008.↵
- Javitt et al., 2004.↵
- Johnson et al., 2005.↵
- Kapur et al., 1999.↵
- Karasawa et al., 2008.↵
- Karlsson et al., 2008.↵
- Kerner et al., 1997.↵
- Kinney et al., 2005.↵
- Kłodzinska et al., 2002.
- Krystal et al., 2005.↵
- Krystal et al., 1999.↵
- Lahti et al., 1995.↵
- Liu et al., 2008.↵
- Luby et al., 1959.↵
- Marek et al., 2000.↵
- Marek and Zhang, 2008.↵
- Martina et al., 2004.↵
- Meltzer et al., 1989.↵
- Mezler et al., 2008.↵
- Michelson et al., 2005.↵
- Moghaddam et al., 1997.↵
- Moghaddam and Adams, 1998.↵
- Molander et al., 2007.↵
- Muly et al., 2003.↵
- Musante et al., 2008.↵
- O'Brien et al., 2004.↵
- Patil et al., 2007.↵
- Perry et al., 2008.↵
- Pinault, 2008.↵
- Porter et al., 2005.↵
- Romano et al., 1995.↵
- Roopun et al., 2008.↵
- Rorick-Kehn et al., 2007.
- Sánchez and Arnt, 2000.
- Schlumberger et al., 2009.↵
- Schoepp, 2001.↵
- Sebban et al., 2002.↵
- Seeman et al., 1976.↵
- Singer et al., 2009.↵
- Spooren et al., 2000.↵
- Takamori et al., 2003.
- Tamminga, 1998.↵
- Tan et al., 2007.↵
- Thomas et al., 2000.↵
- Tregellas et al., 2007.↵
- Tregellas et al., 2009.↵
- Tsai et al., 2004.↵
- Weiner, 2003.↵
- Woo et al., 2008.↵
- Woolley et al., 2008.↵
- Xi et al., 2009.↵
- Yee et al., 2006.↵
- Zafra and Giménez, 2008.↵
- Zhang et al., 2005.↵




{kind=link}
{kind=link}