


Abstract
In the forebrain, synaptic glycine concentrations are regulated through the glycine transporter GlyT1. Because glycine is a coagonist of the N-methyl-d-aspartate (NMDA) receptor (NMDAR), which has been implicated in schizophrenia, inhibition of GlyT1 is thought to provide an option for the treatment of schizophrenia. In support of this hypothesis, GlyT1 inhibitors facilitate in vivo NMDAR function and demonstrate antipsychotic-like effects in animal models. Among the specific GlyT1 inhibitors, substituted N-methyl-glycine (sarcosine) derivatives (e.g., (R)-N[3-(4′fluorophenyl)-3-(4′phenyl-phenoxy)propyl]-sarcosine [NFPS], (R)-N[3-phenyl-3-(4′-(4-toluoyl)phenoxy)-propyl]sarcosine [(R)-NPTS], and (R,S)-(±)N-methyl-N-[(4-trifluoromethyl)phenoxy]-3-phenyl-propylglycine [Org24589]), and non-sarcosine-containing inhibitors, such as 2-chloro-N-[(S)-phenyl[(2S)-piperidin-2-yl] methyl]-3-trifluoromethyl benzamide, monohydrochloride (SSR504734), have been described. In the present study, we analyzed the mode of interaction of these compounds with GlyT1 by using electrophysiological measurements in Xenopus laevis oocytes, and with two binding assays, using [3H](R)-NPTS or 2-chloro-N-[(S)-phenyl[(2S)-N-methylpiperidin-2-yl]-methyl]-3-trifluoromethyl benzamide monohydrochloride ([3H]N-methyl-SSR504734) as radioligands. Inhibition of electrogenic glycine transport by sarcosine-based compounds was apparently irreversible and independent of glycine concentration. The latter indicates a noncompetitive mode of action. In contrast, both SSR504734 and N-methyl-SSR504734 exhibited reversible and competitive inhibition of glycine transport. In GlyT1-expressing membranes, the binding of the novel radioligand [3H]N-methyl-SSR504734 to a single site on GlyT1 was competitively displaced by glycine and SSR504734 but noncompetitively by sarcosine-based compounds. Inversely, [3H](R)-NPTS binding was competitively inhibited by sarcosine-based compounds, whereas glycine, SSR504734, and N-methyl-SSR504734 noncompetitively decreased maximal binding. Our data indicate that besides exerting an apparently irreversible or reversible inhibition, GlyT1 inhibitors differ by exhibiting either a noncompetitive or competitive mode of inhibition. The divergent modes of inhibition may significantly affect the efficacy and tolerability of these drugs.
Glycine acts as a neurotransmitter at inhibitory glycine receptors (glycine A receptors) as well as an essential coagonist of the N-methyl-d-aspartate (NMDA)-type glutamate receptor. The extracellular glycine concentration in the brain is regulated by the high-affinity glycine transporters GlyT1 and GlyT2 (Betz et al., 2006).
GlyT1 and GlyT2 possess different expression patterns, pharmacology, and transport stoichiometries. Because of its stoichiometry of 2 Na+/1 Cl-/1 glycine, GlyT1 is ideally suited to transport glycine into and out of the cell throughout a large concentration range and at different membrane potentials, thereby controlling synaptic glycine concentrations (Roux and Supplisson, 2000). Besides its expression in caudal regions of the central nervous system, GlyT1 is also present in the forebrain. There, it is localized in astrocytes as well as in pre- and postsynaptic terminals of glutamatergic synapses, where GlyT1 was shown to be physically associated with PSD95, an NMDAR-associated protein (Cubelos et al., 2005). These findings support a crucial role of GlyT1 in controlling NMDAR activity by regulating synaptic glycine concentrations.
Hypofunction of NMDAR is thought to be implicated in schizophrenia (Millan, 2005; Coyle, 2006). The most compelling evidence for a pathophysiological role of impaired NMDAR originates from the discovery of several schizophrenia susceptibility genes linked to NMDAR (Harrison and Weinberger, 2005; Morrison and Pilowsky, 2007). Another line of evidence is provided by the behavioral and neurophysiological effects of NMDAR antagonists, which resemble key aspects of schizophrenia (Javitt and Zukin, 1991; Lahti et al., 1995; Morris et al., 2005). Enhancing NMDAR activity by increasing glycine levels via GlyT1 inhibition has been demonstrated (Bergeron et al., 1998) and is being pursued as an approach for the treatment of schizophrenia (for reviews, see Lechner, 2006; Lindsley et al., 2006).
GlyT1 inhibitors belong to diverse structural classes (Lechner, 2006; Lindsley et al., 2006). Like sarcosine, substituted sarcosine derivatives, such as (R)-N-[3-phenyl-3-(4′-(4-toluoyl)-phenoxy)-propyl]-sarcosine [(R)-NPTS], (R)-N[3-(4′fluorophenyl)-3-(4′phenyl-phenoxy)propyl]-sarcosine [NFPS (ALX5407)], and (R,S)-(±)N-methyl-N-[(4-trifluoromethyl)phenoxy]-3-phenyl-propylglycine (Org24598), inhibit GlyT1, but not GlyT2. Of these, NFPS and Org24598 have been used to evaluate the concept of facilitating NMDAR signaling by GlyT1 inhibition. The compounds have been shown to enhance extracellular glycine levels (Atkinson et al., 2001), to facilitate NMDAR activity (Chen et al., 2003), and to enhance long term potentiation (LTP) in the rat hippocampus in vivo (Kinney et al., 2003). Further in vivo studies revealed an antipsychotic-like profile in animal models, including inhibition of PCP-induced hyperlocomotion (Harsing et al., 2003), reversal of impaired prepulse inhibition, and a pattern of c-fos expression resembling that of clozapine (Kinney et al., 2003). While showing promising properties in disease-related models, sarcosine-based compounds have also been reported to exert toxic effects (Harsing et al., 2006) by an unknown mechanism. This may be due to apparent irreversible GlyT1 inhibition, which has been reported for NFPS (Aubrey and Vandenberg, 2001). A nonsarcosine, reversible GlyT1 inhibitor [2-chloro-N-[(S)-phenyl[(2S)-piperidin-2-yl]methyl]-3-trifluoromethyl benzamide, monohydrochloride (SSR504734)] has been described (Depoortère et al., 2005). The compound was shown to elevate brain glycine levels, enhance glutamatergic neurotransmission, and to be effective in several models predictive of antipsychotic activities (Depoortère et al., 2005). Despite this extensive characterization, a detailed analysis of the molecular mode of action at GlyT1 was not released.
Here we compared the mechanism of interaction of two non-sarcosine-based GlyT1 inhibitors (SSR504734 and N-methyl-SSR504734) with those of three sarcosine-based compounds [NFPS, (R)-NPTS, and Org24589]. In particular, we investigated whether the two classes of compounds affect the same or different binding sites at the transporter, and whether the interaction site is orthosteric or allosteric to the functional glycine site.
To this end, we studied the inhibitory mechanisms of the compounds by measuring the electrogenic glycine transport in GlyT1-expressing Xenopus laevis oocytes. Furthermore, we developed a new binding assay based on [3H]N-methyl-SSR504734 and employed the recently described binding assay using [3H](R)-NPTS (Lowe et al., 2003). We outline that these structural classes of GlyT1 inhibitors act at different binding sites on GlyT1. The implications of these findings for the pharmacological properties of GlyT1 inhibitors will be discussed.
Materials and Methods
Materials. (R)-NPTS was synthesized at Abbott as described by Lowe (2002). NFPS (ALX 5407) and Org24598 were commercially available, and SSR504734 was synthesized at Abbott according to procedures described by described by Dargazanli et al. (2003).
For 2-chloro-N-[(S)-phenyl[(2S)-N-methylpiperidin-2-yl]-methyl]-3-trifluoromethyl benzamide monohydrochloride (N-methyl-SSR504734), which is a methyl derivative of SSR504734, the tritium label was introduced as follows: a solution of SSR504734 (4 mg, 0.01 mmol) in tetrahydrofuran (2 ml) was charged to a 5-ml round-bottomed flask equipped with a magnetic stirrer bar. To this solution, aqueous formaldehyde (50 μl) and triethylamine (0.1 ml) were added followed by Pd/C (10%, 8 mg), and this mixture was tritiated (tritium, 1.9 Ci) in a Trisorber (IN/US Systems, Tampa, FL) for 15 h. The excess tritium was pushed back in a recovery bed, and the reaction flask was purged with helium. The reaction mixture was diluted with methanol and the catalyst was filtered. The labile tritium was removed by treating with methanol (3 × 5 ml) and concentrated. The residue was taken in methanol (10 ml), and radioactivity was measured as 89 mCi. The solvent was concentrated, and the residue was purified by preparative HPLC. The crude product was evaporated to dryness, and the residue was dissolved in 1.2 ml of acetonitrile with 0.1% TFA. Purification was accomplished by HPLC on an Agilent 1100 series HPLC system with ChemStation software (Agilent, Palo Alto, CA). Approximately 450 μl of this sample was injected onto a Luna C 18 column (5 μm, 250 × 10 mm i.d.; Phenomenex, Torrance, CA) with a gradient mobile phase of 2% to 95% B for 35 min (mobile phase A = 0.1% TFA/water; mobile Phase B = 0.1% TFA/acetonitrile) at a flow rate of 4 ml/min with an UV detection at 254 nm. The fractions containing [3H]N-methyl-SSR504734 were collected at approximately 18 min using an Agilent fraction collector. The product-containing fractions were pooled and solvents were removed by evaporation in vacuo. The residue (9 mCi, 99% pure) was dissolved in 4 ml of 200-proof ethanol. The specific activity was determined to be 11.4 Ci/mmol from the ratio of the mass spectroscopic measurement of the isotopic molecular ions. Tritium labeling of (R)-NPTS ([3H](R)-NPTS) was carried out by Biotrend Chemikalien GmbH (Köln, Germany).
Expression of hGlyT1 in Mammalian Cells. HEK293 and CHO cells were stably transfected with a pcDNA3 expression plasmid encoding the full-length human GlyT1c transporter. Cell clones hGlyT1c_5_CHO and hGlyT1c_48_HEK293 resistant to G418 were selected for binding and functional uptake studies. Human splice variants GlyT1a, GlyT1b, and GlyT1c (Kim et al., 1994) in pcDNA3 expression plasmid were transiently expressed in FreeStyle HEK293 (HEK293-F) cells (Invitrogen).
Functional Expression in X. laevis Oocytes. Female X. laevis (Nasco, Fort Atkinson, WI), were anesthetized in solution with 0.2% Tricaine (Sigma, St. Louis, MO) and 2 g/l sodium hydrogen carbonate (Sigma), ovary lobes were removed, and oocytes were released from the follicle tissue with collagenase (type I, 2 mg/ml for 2 h; Roche Applied Science, Mannheim, Germany). Stage V and VI oocytes were selected by hand and each oocyte was injected with 20 nl of cDNA solution (10 ng/ml in water) into the nucleus. Alternatively, cRNA was transcribed with T7 RNA polymerase from pGemHeJuel plasmids (Liman et al., 1992) containing either human GlyT1c or human GlyT2a and capped with 5′-7-methyl guanosine using the mMessage mMachine kit (Ambion Inc., Austin, TX). Oocytes were injected with 50 nl (300 ng/ml) RNA solution. The cells were then incubated for 2 to 5 days at 18°C in Barth medium [88 mM NaCl, 1 mM KCl, 0.82 mM MgSO4, 0.33 mM Ca(NO3)2, 0.41 mM CaCl2, 2.4 mM NaHCO3, and 5 mM Tris-HCl, pH 7.4] supplemented with gentamicin (50 mg/ml; Sigma). Oocytes were used for membrane preparations and electrophysiological uptake measurements.
Membrane Preparation from GlyT1c-Expressing X. laevis Oocytes and Recombinant CHO and HEK293 Cells. GlyT1c-expressing cells were pelleted and washed twice in ice-cold PBS, containing 2 mM EDTA and snap-frozen in liquid nitrogen or ethanol/dry ice. The thawed cell pellets were resuspended in ice-cold sucrose buffer (0.25 M sucrose, 10 mM HEPES, 1 mM PMSF, 5 μg/ml pepstatin A, 3 mM EDTA, and 0.025% bacitracin, pH 7.4) and homogenized in 5-ml aliquots by sonication in a tissue homogenizer. Cell breakage was monitored by light microscopy. The homogenate was centrifuged for 10 min at 1000g at 4°C to pellet remaining unbroken cells. The sucrose buffer supernatant was centrifuged at 60,000g at 4°C for 1 h. The resulting pellet was resuspended in 20 ml of ice-cold 20 mM Tris-HCl, pH 7.4, containing 5 μg/ml pepstatin A, 0.1 mM phenylmethylsulfonyl fluoride, and 3 mM EDTA and centrifuged again at 60,000g at 4°C for 1 h. After a final resuspension of the pellet by help of sonication, the protein concentration was determined and the suspension was snap-frozen in aliquots and stored at -80°C.
[3H](R)-NPTS or [3H]N-methyl-SSR504734 Radioligand Binding Assays. Radioligand binding to human GlyT1c transporter-expressing membranes was measured in duplicate in a total volume of 200 μl in 96-well plates. To 100 μl of membrane suspension in assay buffer (120 mM NaCl, 2 mM KCl, 10 mM HEPES, 1 mM MgCl2, and 1 mM CaCl2, pH 7.5), 80 μl of [3H](R)-NPTS (0.5 nM final) or [3H]N-methyl-SSR504734 was added in assay buffer, yielding a final membrane protein concentration of 50 μg/ml. In competition experiments, 10 μl of buffer or unlabeled compound solution were added. The final DMSO concentration was 1% in all cases. Nonspecific binding was determined in the presence of 10 μM Org24598 (or its racemate Org24461) for [3H](R)-NPTS or 10 μM SSR504734 for [3H]N-methyl-SSR504734. After incubation at room temperature for 1 h, the incubation mixture was harvested (Tomtec Mach III U Harvester) through 96-well GF/B filter plates (Perkin-Elmer Life and Analytical Sciences, Waltham, MA), presoaked for 1 h with 40 μl per well of 0.1% polyethylenimine. After washing twice with ice-cold buffer (50 mM Tris-HCI, pH 7.4), plates were dried and 35 μl of scintillator (BetaplateScint; PerkinElmer Life and Analytical Sciences) were added per well. The radioactivity was determined by liquid scintillation spectrometry in a MicroBeta (PerkinElmer Life and Analytical Sciences) plate counter.
Data Analysis. For binding of [3H](R)-NPTS or [3H]N-methyl-SSR504734 to cell membranes, the calculation of Kd and Bmax values from the saturation binding assays and of the IC50 values from the displacement binding was performed by iterative nonlinear regression analysis adapted from the Ligand program (Munson and Rodbard, 1980). Radioligand saturation binding and displacement curves in the presence or the absence of tested compounds were fitted using a one- or a two-site fit, and the more suitable model was identified by a partial F-test (De Lean et al., 1982). Apparent Ki values were calculated from the IC50 values using the Cheng-Prusoff equation (Cheng and Prusoff, 1973).
Glycine Uptake in Recombinant hGlyT1 Expressing Cells. Human GlyT1c expressing recombinant hGlyT1c_5_CHO or hGlyT1c_ 48-HEK293 cells or FreeStyle HEK293-F cells transiently transfected with human GlyT1a, GlyT1b, or GlyT1c were plated at 20,000 cells per well in 96-well Cytostar-T scintillation microplates (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) and cultured to subconfluence for 24 h. For glycine uptake assays, the culture medium was aspirated and the cells were washed once with 100 μl of HBSS (Invitrogen, Carlsbad, CA) containing 5 mM l-alanine (Merck, Darmstadt, Germany). Eighty microliters of HBSS buffer was added, followed by 10 μl of inhibitor or vehicle (10% DMSO) and 10 μl of [3H]glycine (TRK71; GE Healthcare). The final glycine concentration was 250 nM (if not stated otherwise). The plates were placed in a Wallac Microbeta (PerkinElmer Life and Analytical Sciences) and continuously counted by solid-phase scintillation spectrometry up to 3 h. Nonspecific uptake was determined in the presence of 10 mM unlabeled glycine or 10 μM Org24598 (IC50 calculations were made by four-parametric logistic nonlinear regression analysis (Prism; GraphPad Software, San Diego, CA), using determinations within the range of linear increase of [3H]glycine incorporation.
Glycine Uptake in Mouse Astrocytes. Experimental procedures were approved by Abbott's Animal Welfare Office and were performed in accordance with the German national guidelines as well as recommendations and policies of the U.S. National Institutes of Health “Principles of Laboratory Animal Care” (1996 edition). Animal housing and experiments were conducted in the facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Primary mouse astrocytes were prepared from C57BL6 mice (Janvier, Le-Genest-St-Isle, France) and cultured in Dulbecco's modified Eagle's medium with 10% fetal calf serum and 1% PenStrep under 10% CO2. Twenty-four hours before the experiment, the medium was substituted by serum-free Neurobasal/B27 medium, and cells were plated at 15,000 cells per well in 96-well Cytostar-T scintillation microplates (GE Healthcare). After washing the cells once with 100 μl of HBSS (Invitrogen) containing 5 mM l-alanine (Merck), the glycine uptake assay was performed as described above.
Electrophysiological Measurements in X. laevis Oocytes. The measurement of the membrane current on whole oocytes was carried out as described previously (Methfessel et al., 1986; Mezler et al., 2001). The cells were penetrated with two microelectrodes with a resistance <1.5 MΩ, filled with 1.5 M potassium acetate plus 0.1 M KCl) in an acrylic chamber. The membrane potential was held at -60 mV with a voltage-clamp amplifier (TEC 03X, npi Electronic, Tamm, Germany), and the membrane current was recorded at 20 Hz. The measurement chamber was continuously perfused with frog Ringer solution (115 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, and 10 mM HEPES, pH 7.2). All compounds were diluted in frog Ringer solution, and measurements were at room temperature. To administer compounds, the software-driven perfusion was switched between reservoirs with the appropriate solutions. The flow rate of 5 ml/min resulted in an exchange of solutions at the oocyte within 1 to 2 s. Data acquisition and analysis were performed with CellWorks 5.5 software (npi Electronic) and Prism software.
Results
To study differences in their interaction kinetics, GlyT1 inhibitors from two different structural classes were investigated: the first class comprised SSR504734 and N-methyl-SSR504734 and the second class comprised the N-methylglycine (sarcosine) derivatives (sarcosine-based compounds) NFPS, (R)-NPTS, and Org24598 (Fig. 1).
All Tested Compounds Are Potent and Selective GlyT1 Inhibitors without Splice-Variant Selectivity. Steady-state inhibition experiments of [3H]glycine uptake in mouse astrocytes and CHO cells recombinantly expressing human GlyT1c demonstrated that all tested GlyT1 inhibitors completely blocked [3H]glycine uptake with nanomolar potency. In mouse astrocytes, the rank order was NFPS > (R)-NPTS > N-methyl-SSR504734 > Org24598 ≫ SSR504734 (Table 1, first column). The rank order of potency was similar in the recombinant system, expressing the human GlyT1c transporter (Table 1, middle column); in general, however, the potency of inhibition was reduced. All compounds tested, as well as the substrates glycine and sarcosine, displaced binding of the new radioligand [3H]N-methyl-SSR504734 (Table 1, right column). To rule out substantial differences in the inhibitory potency of the compounds for the GlyT1 subtypes, we tested the inhibition of glycine uptake for cells expressing either hGlyT1a, hGlyT1b, or hGlyT1c. Org24598 and SSR504734 were used as representatives for sarcosine-based and non-sarcosine-based inhibitors and were tested for inhibition of [3H]glycine uptake in GlyT1-expressing HEK293-F cells, transiently transfected with the three human GlyT1 splice variants. The IC50 values for the different subtypes were not significantly different (p > 0.05 by Kruskal-Wallis test; data not shown). Based on these and previously published results, the subsequent experiments were carried out with hGlyT1c only. None of the compounds demonstrated inhibition of GlyT2-mediated glycine transport in radioactive uptake experiments in recombinant GlyT2-expressing cells or electrophysiological measurements in X. laevis oocytes (data not shown).
All compounds tested are potent inhibitors of GlyT1 and displace [3H]N-methyl-SSR504734 binding
Inhibition of [3H]glycine uptake by native mouse astrocytes (left column) and recombinant mammalian cells with stable expression of human GlyT1c (hGlyT1c_5_CHO cells; middle column). Cells were incubated with 200 nM of [3H]glycine and the uptake was measured after 90 or 120 min within the linear course of incorporation. Data represent geometric means and their 95% confidence limits (n = 2 to 6). All inhibitors tested, as well as the substrates glycine and sarcosine, compete for the binding of the new radioligand [3H]N-methyl-SSR504734 (right column).

Two classes of GlyT1 inhibitors. Chemical structure of sarcosine-based and non-sarcosine-based GlyT1 inhibitors used in these studies. (R)-NPTS, NFPS, and Org24598 contain a sarcosine-moiety (marked light gray). This moiety is absent in SSR504734 and its N-methyl-derivative N-methyl-SSR504734.
SSR504734 and N-methyl-SSR504734 Are Potent, Reversible and Orthosteric Inhibitors of Electrogenic GlyT1 Transport in X. laevis Oocytes.X. laevis oocytes have been described as a useful system to functionally express and characterize GlyT1 (Roux and Suplisson, 2000; Aubrey and Vandenberg, 2001). To evaluate the inhibitory properties of SSR504734, we first stimulated the glycine transport through human GlyT1c by adding glycine (20 μM) at a concentration corresponding approximately to its KM, followed by a coapplication of 20 μM glycine and 3 μM SSR504734, and a washout period, in which 20 μM glycine was applied in the absence of SSR504734 (Fig. 2A). SSR504734 proved to be an effective and reversible inhibitor of electrogenic glycine transport also in the X. laevis oocyte system. The compound blocked the transport of 20 μM glycine with an on-rate (t½on) of 11 ± 1 s (n = 3) and the inhibition of glycine transport was readily reversible with an off-rate (t½off) of 40.2 ± 8.3 s (n = 3).
If the inhibitor binding site is orthosteric to the site bound by glycine, it should be possible to displace the inhibitor with increasing glycine concentrations, and the high substrate concentration should protect the transporter from inhibition (surmountability). Nonsurmountable compounds would pre-clude the agonist to attain the maximal signal. Therefore, we evaluated the inhibition of the compound by coapplication of a defined concentration of the inhibitor together with a low (10 μM; approximately 1/2 KM) and a high (3 mM, approximately 150× KM) concentration of glycine. In our surmountability protocol, after a control stimulus with 20 μM glycine, hGlyT1c was activated by glycine (10 μM or 3 mM), followed by a coapplication of 3 μM SSR504734 together with 10 μM and 3 mM glycine, respectively (Fig. 2B-D). Although at 10 μM glycine, the current is blocked by 82 ± 2% (n = 5), the inhibition is significantly (p < 0.0001; t test) reduced with 3 mM glycine. Only a residual inhibition of 36 ± 3% (n = 6) is left under these conditions (Fig. 2B).
To further validate a potential competitive mode-of-action for SSR504734, we tested the glycine dependence of the inhibition in more detail. Because SSR504734 inhibition is quickly reversible, we were able to test the compound at concentrations of 0, 1, 3, and 10 μM together with a glycine concentration response curve. The results for 0 and 10 μM are shown (Fig. 3A and B, top). Without inhibitor, the EC50 of glycine is 19.4 ± 5.7 μM (Fig. 3A; n = 7). At higher inhibitor concentrations, the glycine EC50 is shifted to larger values. For example, at 10 μM SSR504734, the EC50 of glycine is 592 ± 40 μM (Fig. 3B; n = 4). According to a modified Gaddum/Schild model (Lazareno and Birdsall, 1993), a nonlinear regression analysis of glycine concentration-response curves with increasing concentrations of SSR504734 resulted in a calculated Schild slope of 1 when subjected to global fitting (Fig. 3C, top). These data suggest that inhibition of SSR504734 is competitive to glycine. Consequently, transformation into a linear Schild Plot resulted in a slope of 0.91 (Fig. 3C, bottom). The Kb of SSR504734 determined from global nonlinear fitting was 214 nM (Fig. 3C, top). This value corresponds well to the IC50 value of this compound in the radioactive uptake assay in hGlyT1c-expressing CHO cells (see Table 1, middle column).

SSR504734 is a reversible and surmountable inhibitor of GlyT1 transport. A, inhibition of 3 μM SSR504734 was evaluated in the presence of 20 μM glycine (bold line) by addition of the inhibitor in the open state of the transporter (thin line). The compound blocked the glycine transport current with t½on = 11 ± 1 s (n = 3) and inhibition could be reversed with 20 μM glycine at t½off = 40.2 ± 8.3 s (n = 3). B, quantitative evaluation of glycine transport inhibition by SSR504734, calculated from C and D. Although at 10 μM glycine the inhibitor blocked the glycine transport current by 82 ± 2% (n = 5), the glycine current of 3 mM glycine was only blocked by 36 ± 3% (n = 6). C, GlyT1-expressing oocytes were stimulated with 20 μM glycine (1), 10 μM glycine (2), and 10 μM glycine + 3 μM SSR504734 (3; dashed line). D, GlyT1-expressing oocytes were stimulated with 20 μM glycine (1), 3 mM glycine (2), and 3 mM glycine + 3 μM SSR504734 (3; dashed line). Values are means ± S.D.; significance was tested by unpaired t test; holding potential was -60 mV.
To facilitate the generation of a radioligand for GlyT1, we synthesized N-methyl-SSR504734. In X. laevis oocytes, similar to SSR504734, 3 μM N-methyl-SSR504734 blocked the glycine-induced current through GlyT1c (Fig. 4) with a t½ on of 12 ± 0.3 s (n = 4). Although the t½ off with 868.4 ± 36 s (n = 4) was relatively slow, the inhibition clearly proved to be reversible. A representative trace of such an experiment is shown in Fig. 4A. Because of the slow off-rate of N-methyl-SSR504734, a direct Schild evaluation was not feasible. Therefore, the rate of inhibition was tested in separate experiments with two glycine concentrations as described above (surmountability). Whereas 1 μM N-methyl-SSR504734 almost completely blocked the glycine current at 10 μM glycine (inhibition 91 ± 5% (n = 4); Fig. 4B,C), the inhibition was largely alleviated at 3 mM glycine [residual inhibition, 26 ± 4% (n = 5); Fig. 4, B and D]. These findings are equivalent to the surmountability of SSR504734, for which we have validated the competitive inhibition with the Schild analysis.
Sarcosine-Based Compounds Are Apparently Irreversible and Noncompetitive Inhibitors of Electrogenic GlyT1 Transport in X. laevis Oocytes. NFPS has previously been reported to be a nonreversible and nonsurmountable inhibitor of GlyT1 transport (Aubrey and Vandenberg, 2001). Because of the structural similarity, we evaluated whether these properties were also shared by other compounds of this class.
In a first set of experiments, we verified that all three sarcosine-based inhibitors are apparently irreversible inhibitors of GlyT1 in the X. laevis oocyte system (Fig. 5). At a concentration of 1 μM, all three compounds blocked the transport of 20 μM glycine quickly [t½on values of 23 ± 4 s (NFPS, n = 4), 59 ± 4 s (NPTS, n = 4), and 70 ± 2 s (Org24598, n = 4)] and almost completely. The blockade was apparently irreversible, because an application of 20 μM glycine for 10 min did not reduce the inhibition of the compounds; also, after washout, 20 μM glycine did not induce an increased signal but the current still is either completely absent (Fig. 5A for NFPS) or corresponds to the residual current initially left by the inhibitor [Fig. 5, B (for NPTS) and C for (Org24598)].
Furthermore, we evaluated whether the sarcosine-based inhibitors are competitive or noncompetitive blockers of glycine transport. To avoid an influence of apparent irreversibility on the following glycine current, the sarcosine-based inhibitors were tested in the surmountability protocol. NFPS (500 nM) blocked the glycine current induced by 10 μM and 3 mM glycine to the same extent (91 ± 4%, n = 5; and 90 ± 4%, n = 5, respectively; Fig. 6A-C). This indicates that inhibition by NFPS is not surmountable by high glycine concentrations and suggests a noncompetitive inhibition of glycine transport. Likewise, 2 μM (R)-NPTS (Fig. 6D) and 1.5 μM Org24598 (Fig. 6E) inhibition was not surmountable. For (R)-NPTS, inhibition was 96 ± 3% at both glycine concentrations [10 μM (n = 7) and 3 mM (n = 4); Fig. 6D]; for Org24598, the inhibition amounted to 93 ± 8% (n = 8) and 96 ± 1% (n = 6) for 10 μM and 3 mM glycine, respectively (Fig. 6E).
Characterization of the New Radioligand [3H]N-methyl-SSR504734—Saturation Isotherms and Binding Kinetics. Our functional studies suggest that SSR504734 and N-methyl-SSR504734 are competitive inhibitors of glycine transport. Therefore, we assumed that SSR504734 and N-Methyl-SSR504734 bind to the glycine binding site of the transporter. For studying the interaction of inhibitors and glycine through binding studies, only tritiated (R)-NPTS and NFPS had been described as radioligands. To provide a radioligand for the proposed glycine binding site, we radiolabeled N-methyl-SSR504734. Saturation binding experiments with [3H]N-methyl-SSR504734 revealed a mean Kd of 8.1 nM for different preparations of X. laevis oocyte membranes or 3.3 nM for hGlyT1c from CHO cell membranes. All inhibitors tested, as well as the substrates glycine and sarcosine, concentration-dependently displaced [3H]N-methyl-SSR504734 binding to human GlyT1c expressing CHO cell membranes (Table 1, right column).

SSR504734 inhibition of GlyT1 is competitive for glycine transport in X. laevis oocytes. A, top, concentration-response curve of glycine transport in a X. laevis oocyte expressing GlyT1c. Bottom, the EC50 of glycine on GlyT1 in oocytes was 19.4 ± 5.7 μM (n = 7). B, top, concentration-response curve of glycine transport in an oocyte expressing GlyT1c. Besides a first and last 20 μM glycine stimulus (bold lines), at each concentration of the concentration-response determination, glycine was supplemented with 10 μM SSR504734 (thin lines). The EC50 value of glycine in the presence of 10 μM SSR504734 was right-shifted to 592 ± 40 μM (n = 4). C, top, Schild analysis of curve shifts with increasing concentrations of 1, 3, and 10 μM SSR504734. Global fitting resulted in a calculated Schild slope of 1. Bottom, accordingly, transformation into a linear Schild Plot resulted in a slope of 0.9). The Kb determined from global nonlinear fitting was 214 nM. n = 4-7; holding potential was -60 mV.
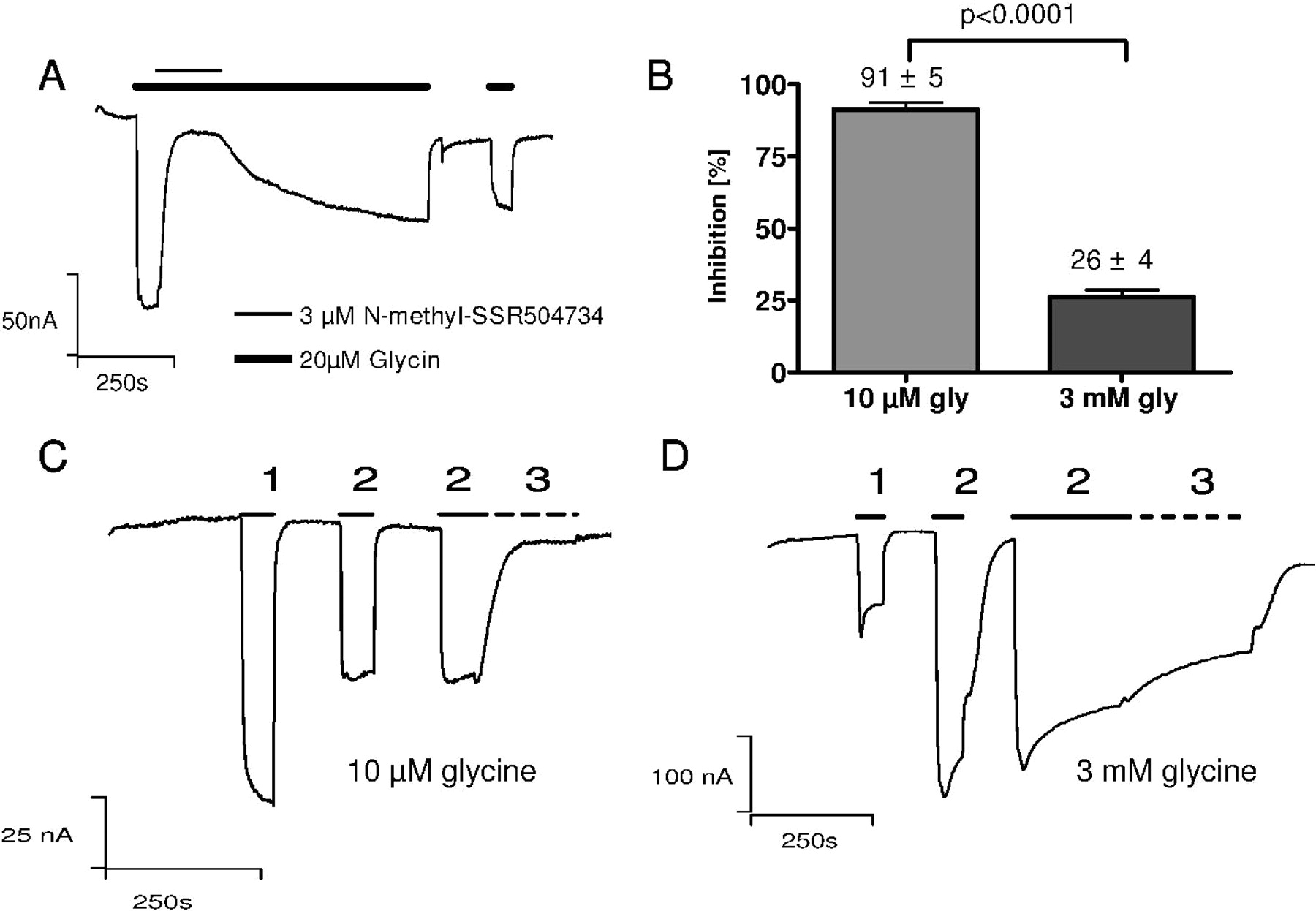
N-methyl-SSR504734 inhibition is reversible and surmountable with high glycine concentrations. A, N-methyl-SSR504734 inhibition was evaluated in the presence of 20 μM glycine (bold bars) by addition of the inhibitor at 3 μM in the open state of the transporter (thin bar). The compound monophasically blocked the glycine transport current with t½on = 12.3 ± 0.3 s (n = 4) and inhibition could be reversed with 20 μM glycine at t½off = 868.4 ± 36 s (n = 4). (B) Quantitative evaluation of glycine transport inhibition by N-methyl-SSR504734, calculated from C and D. Although at 10 μM glycine the inhibitor blocked the glycine transport current by 91 ± 5% (n = 5), the glycine current of 3 mM glycine was only blocked by 26 ± 4% (n = 4). C, GlyT1-expressing oocytes were stimulated with 20 μM glycine (1), 10 μM glycine (2) and 10 μM glycine + 1 μM N-methyl-SSR504734 (3; dashed line). D, GlyT1-expressing oocytes were stimulated with 20 μM glycine (1), 3 mM glycine (2), and 3 mM glycine + 1 μM N-methyl-SSR504734 (3; dashed line). Because of the long transport times, for calculation of inhibition of D, the reduction of the current with inhibitor was corrected for the reduction of the current with 3 mM glycine alone at the same time of wash-in. Values are means ± S.D.; significance, unpaired t test; holding potential was -60 mV.

Sarcosine-containing compounds are apparently irreversible inhibitors of GlyT1. Inhibition of NFPS, NPTS, and Org24598 was evaluated in the presence of 20 μM glycine (bold bars) by addition of the inhibitor at 1 μM in the open state of the transporter (thin bar). A, NFPS (1 μM) blocked the glycine transport current with t½ on of 23 ± 4 s (n = 4). B, NPTS (1 μM) blocked the glycine transport current with t½ on of 59 ± 4 s (n = 4). C, Org24598 (1 μM) blocked the glycine transport current with t½ on 70 ± 2 s (n = 4). For all three compounds, the t½ off could not be determined. Also a 2-min washout with normal frog Ringer solution did not reconstitute the transport activity, probed by application of 20 μM glycine.
Displacement of [3H]N-methyl-SSR504734 Binding by Sarcosine-Containing and Non-Sarcosine-Containing Compounds. More specifically, we measured [3H]N-methyl-SSR504734 saturation-binding isotherms in the absence and presence of increasing concentrations of the sarcosine-based inhibitors SSR504734 and glycine in recombinant CHO membranes expressing human GlyT1c, thereby testing for a competitive or noncompetitive mode of displacement.
SSR504734, like glycine, significantly increased the Kd of the [3H]N-methyl-SSR504734 binding while leaving the Bmax unaffected (Fig. 7, A and B, Table 2). These data support a competitive interaction of these compounds at the [3H]N-methyl-SSR504734 binding site, in support of the functional data.
Allosteric or competitive characteristics of GlyT1 inhibitors in saturation binding experiments of [3H]N-methyl-SSR504734 and [3H](R)-NPTS to recombinant human GlyT1c-expressing CHO membranes
SSR504734 and glycine demonstrate competitive displacement of [3H]N-methyl-SSR504734 and increase the apparent Kd without relevant effect on the Bmax. In contrast, (R)-NPTS behaves as a noncompetitive inhibitor. Noncompetitive displacement of [3H](R)-NPTS binding was demonstrated for SSR504734 and N-methyl-SSR504734 by decrease of Bmax, which was also observed as a trend for glycine. Values represent geomeans and 95% confidence limits (n = 2-4). ANOVA was used to assess significance of changes in Kd and Bmax obtained from nonlinear regression analysis of 1-site saturation binding isotherms of the respective radioligand in the absence or presence of increasing concentrations of test compound; values without confidence limits were not included in the ANOVA analysis.
In contrast, as a representative of the sarcosine-based compounds, (R)-NPTS decreased Bmax and increased the Kd, indicating a mixed-model inhibition mechanism of [3H]N-methyl-SSR504734 displacement (Fig. 7C, Table 2). Similar results were obtained for NFPS (data not shown).
Noncompetitive Displacement of [3H](R)-NPTS Binding by the Glycine-Competitive Non-Sarcosine-Containing N-Methyl-SSR504734. For sarcosine-based GlyT1 inhibitors such as (R)-NPTS, the functional data in X. laevis oocytes, together with the displacement of [3H]N-methyl-SSR504734, suggest an allosteric binding site distinct from the glycine binding site. [3H](R)-NPTS was used as a radioligand to cross-check the binding of N-methyl-SSR504734, SSR504734, and glycine. Binding of [3H](R)-NPTS to recombinant CHO membranes expressing human GlyT1c revealed a Kd value of 2.2 nM when the data were fitted to a one-site model (data not shown).
As demonstrated in Table 2 and Fig. 7, D and F, increasing concentrations of SSR504734 and N-methyl-SSR504734 resulted in a significant decrease of the Bmax of [3H](R)-NPTS binding, although the Kd remained unchanged. With glycine (Fig. 7E), a similar trend was observed, although the Bmax decrease did not reach statistical significance. Thus, SSR504734 and N-methyl-SSR504734 interact noncompetitively with [3H](R)-NPTS. Conversely, these findings are consistent with the competitive displacement of [3H]N-methyl-SSR504734 by SSR504734 and glycine. Similar data were obtained using X. laevis oocyte membranes (data not shown), indicating that the binding properties of the inhibitors to hGlyT1c are independent of the expression system. Further evidence for different binding sites of [3H](R)-NPTS and [3H]N-methyl-SSR504734 comes from a comparison of the potency to block radioligand binding. When generating the Ki ratio for [3H]N-methyl-SSR504734 over [3H](R)-NPTS binding displacement, the sarcosine-containing compounds display ratios greater than 1 [17 for ORG24598, 8 for (R)-NPTS, and 3 for NFPS], whereas N-methyl-SSR504734 (0.15), SSR504734 (0.4), and glycine (0.75) display ratios less than 1.
Discussion
In our studies, we investigated the interaction mode of different high affinity GlyT1 inhibitors with the transporter. We show that the compounds can be differentiated according to their competitive or noncompetitive interaction with the glycine binding site. Using SSR504734 and its N-methyl derivative, we describe for the first time a competitive interaction of a high-affinity GlyT1 inhibitor with the glycine binding site.
All tested compounds were potent inhibitors of glycine transport in native (mouse astrocytes) and recombinant systems expressing human GlyT1. Furthermore, like glycine, all inhibitors blocked binding of both radioligands, [3H](R)-NPTS and [3H]N-methyl-SSR504734 to recombinant GlyT1-expressing mammalian cell membranes, as well as to X. laevis oocyte membranes. For studying whether the compounds exert a competitive or noncompetitive mode of inhibition, we employed two types of experiments. We first investigated the surmountability of inhibition of the electrogenic GlyT1 transport by high glycine levels. In these experiments, inhibition of SSR504734 and its N-methyl derivative were surmountable by high glycine, indicating that the inhibitor binding site is orthosteric to the site bound by glycine. The SSR504734-induced shift of the glycine concentration response curve by linear and nonlinear Schild analysis confirmed that this compound was competitive for glycine. We also identified the sarcosine-based compounds as noncompetitive inhibitors, as they exert inhibition independent of glycine concentration and are therefore nonsurmountable. Because the inhibitor was applied under conditions of active transport, glycine could directly compete with the sarcosine-based inhibitor. Under these conditions, the apparently irreversible properties of these compounds should not play a role.

Sarcosine-containing compounds are nonsurmountable inhibitors of GlyT1 in X. laevis oocytes. Oocytes expressing GlyT1c were stimulated with 20 μM glycine (1), 10 μM glycine (2), and 10 μM glycine + 500 nM NFPS (3, dashed line) (A); 20 μM glycine (1), 3 mM glycine (2), and 3 mM glycine + 500 nM NFPS (3, dashed line) (B). C, under both conditions—at 10 μM glycine and 3 mM glycine—NFPS strongly blocked the glycine-induced current by 91 ± 4% (at 10 μM glycine) and 90 ± 4% (at 3 mM glycine) respectively. D, under identical conditions, inhibition of 2 μM NPTS at 10 μM and 3 mM glycine was equal, at 96 ± 3%. E, 1.5 μM ORG24598 inhibited the glycine transport current at 10 μM by 93 ± 8% and at 3 mM glycine by 96 ± 1%. Values in C, D, and E are means ± S.D., n = 5 in all cases. The inhibition at 10 μM and 3 mM glycine current in C, D, and E were not statistically different (Student's t test). Holding potential was -60 mV.
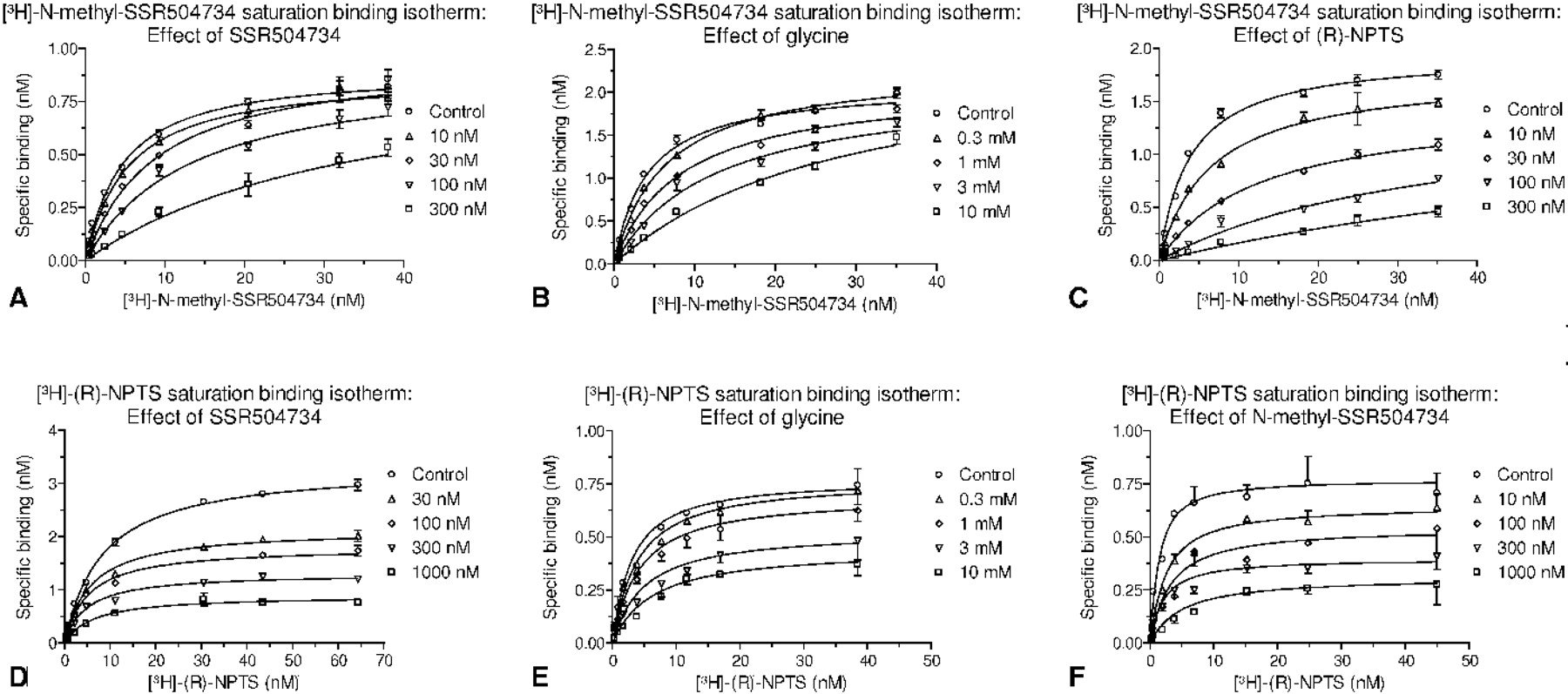
Allosteric or competitive characteristics of GlyT1 inhibitors. In saturation binding experiments of [3H]N-methyl-SSR504734 to recombinant human GlyT1c-expressing CHO membranes, saturation binding isotherms were examined. Changes in saturation binding isotherms of [3H]N-methyl-SSR504734 with increasing compound concentrations demonstrate competitive displacement by SSR504734 (A) and glycine (B) but noncompetitive displacement by (R)-NPTS (C). Changes in saturation binding isotherms of [3H](R)-NPTS with increasing compound concentrations demonstrate noncompetitive displacement by SSR504734 (D), glycine (E), and N-methyl-SSR504734 (F). Graphs depict values (mean and S.E.M.) and nonlinear regression lines (one-site model) from representative saturation binding experiments used for the calculations of the values given in table 2.
These findings were corroborated with a second set of experiments where we studied the effects of the transport inhibitors on the binding parameters of two GlyT1 radioligands, [3H](R)-NPTS and [3H]N-methyl-SSR504734. Glycine and SSR504734 shifted the Kd of [3H]N-methyl-SSR504734 to higher values without decreasing the Bmax. This confirms the competitive interaction observed in the functional experiments and indicates that [3H]N-methyl-SSR504734 occupies a binding site orthosteric to the site bound by glycine. Inversely, as indicated by a decrease in Bmax, the effects of these ligands revealed a noncompetitive interaction on [3H](R)-NPTS binding, indicating that this ligand binds to an allosteric site at the transporter. Contrary to the glycine-competing ligands, the sarcosine-based compounds increased the Kd for [3H](R)-NPTS, consistent with a competitive inhibition of [3H](R)-NPTS binding by these compounds. Like-wise, sarcosine-based inhibitors decreased the Bmax of the [3H]N-methyl-SSR504734, again indicative for a noncompetitive interaction of this inhibitor class with the glycine binding site.
The finding that sarcosine-based compounds bind to a site distinct from the substrate (glycine) site corresponds to findings in earlier reports (Aubrey and Vandenberg, 2001; Mallorga et al., 2003), describing a noncompetitive interaction between the sarcosine-based GlyT1 inhibitor NFPS with glycine. In a more recent study, mutations of amino acids of the putative substrate (glycine) binding site in the transmembrane domain 6 (TM6) of the transporter affected substrate affinity and selectivity (glycine versus sarcosine affinity). However, these mutations only marginally decreased the affinity of NFPS (Vandenberg, 2006). In another study, employing GlyT1-GlyT2 chimeric transporters, the transmembrane domains TM1 and TM3 were found to be involved in NFPS binding (Núñez et al., 2005). Together, these data are consistent with a binding site for sarcosine-based structures distinct from the substrate binding site. Conversely, it has been reported that sarcosine increased the Kd of [3H](R)-NFPS binding and decreased the Bmax. These data were interpreted as potentially overlapping binding sites of sarcosine and NFPS (Mallorga et al., 2003). Similar to the data for sarcosine, our data with (R)-NPTS provide some hint for a potential dual binding of the sarcosine-based inhibitors both to the allosteric binding site and the orthosteric site. The reason for this interpretation is the increase of the Kd for the [3H]N-methyl-SSR504734 binding by (R)-NPTS. However, this effect could also be due to a noncompetitive interaction affecting the ligand affinity and therefore the binding of sarcosine-based compounds to the substrate binding site remains hypothetical. Furthermore, if there were any direct glycine-competitive interaction of the sarcosine-based compounds, its contribution to the overall compound affinity should be small, because mutations within sequences involved in glycine binding had only a small effect on NFPS binding (Vandenberg, 2006).
The major new finding in our studies is that SSR504734, as well as its N-methyl-derivative, are glycine-competitive inhibitors. This demonstrates that binding to the orthosteric and the allosteric binding sites is separable. Because the binding of all compounds to either site affects the Bmax of ligands at the other site, the two sites seem to be closely coupled. This may be either a conformational coupling or an interaction based on spatial proximity. Compounds may bind to one site and prevent the binding of ligands to the other site by spatial hindrance.
The different modes of interaction described here may significantly affect the pharmacological profile of GlyT1 inhibitors. One important differentiating characteristic is the dependence of the degree of inhibition on the glycine concentration. While inhibition of GlyT1 by noncompetitive inhibitors is not sensitive for the glycine concentration, the degree of inhibition of glycine-competitive inhibitors greatly depends on the concentration of glycine. The glycine concentration is extremely different between different compartments within the brain, as well as between different physiological situations. Competitive inhibitors may therefore exhibit a stronger blockade in the intrasynaptic compartment, where the glycine concentration is low, and a weaker inhibition in extrasynaptic compartments, where a higher concentration of glycine partly displaces the inhibitor. An argument in favor of a selective, synaptic blockade with glycine-competitive inhibitors is the neuron-specific GlyT1 knockout mouse (Yee et al., 2006; Sanderson and Bannermann, 2007; Singer et al., 2007). Because neuronal GlyT1 is predominantly localized in pre- and postsynaptic aspects of glutamatergic terminals (Cubelos et al., 2005), neuron-specific GlyT1 knockout affects mainly synaptic glycine transport. Such animals exhibit a more favorable procognitive phenotype than mice with a global heterozygous GlyT1 knockout, which affects the neuronal and the astrocytic transporter. Therefore, compared with noncompetitive inhibitors, glycine-competitive inhibitors, having increased potential in the synaptic compartment, may be advantageous to improve cognitive performance in schizophrenia.
Competitive and noncompetitive GlyT1 inhibitors may also exert different effects in situations with strongly increased glycine concentrations, such as ischemia or epileptic activities. Indeed, SSR504734 has been shown to be neuroprotective in a model of focal cerebral ischemia, whereas NPTS exacerbated ischemic damage in the same experiment (Szabo, 2005). So far, the role of glycine in situations such as ischemia is poorly understood, especially because it may have very complex effects involving activation of intrasynaptic and extrasynaptic NMDA receptors, as well as glycine A receptors. There is increasing evidence that intrasynaptic in contrast to extrasynaptic NMDA receptors provide a pro-survival signal for neurons (Soriano et al., 2006; Zhang et al., 2007), and it is tempting to speculate that a differential effect on synaptic glycine may be responsible for the different effects of SSR504734 and NPTS in ischemia.
All considerations on the potential impact of the mode of interaction are hypothetical, however, and need to be substantiated by experiments comparing this specific compound property. Sarcosine-based compounds are not suitable as representatives of noncompetitive inhibitors, because apart from binding to an allosteric site, they are functionally apparently irreversible (Atkinson et al., 2001; Aubrey and Vandenberg, 2001). To investigate the impact of the mode of inhibition on the action of the drug in vivo, it is therefore necessary to identify noncompetitive reversible GlyT1 inhibitors and to compare them with glycine-competitive and reversible GlyT1 inhibitors, such as N-methyl-SSR504734 or SSR504734.
Acknowledgments
We thank Kerstin Bieser, Barbara Brychcy, Gisela Kreutzenberer, Tamara Nicklis, Gertraud Obradovic, Sandra Rogall, Marion Schanzenbächer, Marion Metz-Garecht, and Srirajan Vaidyanathan for expert technical assistance. Thanks to Bruce W. Suber for labeling the radioligand [3H]N-Methyl-SSR504734. We thank Alfred Hahn and Peer Jacobson for critically reading the manuscript.
Footnotes
-
M.M. and W.H. contributed equally to this work.
-
This work was presented in part in Mezler M, Hornberger W, van Gaalen MM, Wicke KM, Mueller R, Schmidt M, Braje W, and Behl B (2005) Reversibility of glycine transporter GlyT1 inhibition comparing two chemical classes. Soc Neurosci Abstr31:1022.9.
-
ABBREVIATIONS: NMDA, N-methyl-d-aspartate; NMDAR, N-methyl-d-aspartate receptor; GlyT1, glycine transporter subtype 1; (R)-NPTS, (R)-N[3-phenyl-3-(4′-(4-toluoyl)phenoxy)-propyl]sarcosine; NFPS, (R)-N[3-(4′fluorophenyl)-3-(4′phenyl-phenoxy)propyl]-sarcosine (ALX 5407); SSR504734, 2-chloro-N-[(S)-phenyl[(2S)-piperidin-2-yl]methyl]-3-trifluoromethyl benzamide, monohydrochloride; N-methyl-SSR504734, 2-chloro-N-[(S)-phenyl[(2S)-N-methylpiperidin-2-yl]-methyl]-3-trifluoromethyl benzamide monohydrochloride; Org24598, (R,S)-(±)N-methyl-N-[(4-trifluoromethyl)phenoxy]-3-phenyl-propylglycine; HPLC, high-performance liquid chromatography; TFA, trifluoroacetic acid; HEK, human embryonic kidney; HEK293-F, FreeStyle HEK293 cells; HBSS, Hanks' balanced salt solution; CHO, Chinese hamster ovary; ANOVA, analysis of variance.
- Received June 4, 2008.
- Accepted September 23, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}