


Abstract
σ Ligands modulate opioid actions in vivo, with agonists diminishing morphine analgesia and antagonists enhancing the response. Using human BE(2)-C neuroblastoma cells that natively express opioid receptors and human embryonic kidney (HEK) cells transfected with a cloned μ opioid receptor, we now demonstrate a similar modulation of opioid function, as assessed by guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding, by σ1 receptors. σ Ligands do not compete opioid receptor binding. Administered alone, neither σ agonists nor antagonists significantly stimulated [35S]GTPγS binding. Yet σ receptor selective antagonists, but not agonists, shifted the EC50 of opioid-induced stimulation of [35S]GTPγS binding by 3- to 10-fold to the left. This enhanced potency was seen without a change in the efficacy of the opioid, as assessed by the maximal stimulation of [35S]GTPγS binding. σ1 Receptors physically associate with μ opioid receptors, as shown by coimmunoprecipitation studies in transfected HEK cells, implying a direct interaction between the proteins. Thus, σ receptors modulate opioid transduction without influencing opioid receptor binding. RNA interference knockdown of σ1 in BE(2)-C cells also potentiated μ opioid-induced stimulation of [35S]GTPγS binding. These modulatory actions are not limited to μ and δ opioid receptors. In mouse brain membrane preparations, σ1-selective antagonists also potentiated both opioid receptor and muscarinic acetylcholine receptor-mediated stimulation of [35S]GTPγS binding, suggesting a broader role for σ receptors in modulating G-protein-coupled receptor signaling.
σ Receptors were originally proposed by Martin and colleagues (1976) based on studies with the benzomorphan (±)SKF-10047, but they are now defined as nonopioid, nonphencyclidine, haloperidol-sensitive, naloxone-inaccessible, (+)-benzomorphan-selective binding sites (Quirion et al., 1992) with two subtypes, σ1 and σ2, based on their binding-selectivity profiles (Bowen et al., 1993). σ Binding sites are widely distributed across species and are present in almost all tissues, with highest expression in the central nervous system, immune system, kidney, and liver (Gundlach et al., 1986; Walker et al., 1992; Bowen, 2000) and in a wide range of tumors (Vilner et al., 1995; Ryan-Moro et al., 1996; Aydar et al., 2004; Spruce et al., 2004).
The σ1 binding site has been cloned from guinea pig (Hanner et al., 1996), human (Kekuda et al., 1996), mouse (Seth et al., 1997; Pan et al., 1998), and rat (Seth et al., 1998; Mei and Pasternak, 2001), showing greater than 80% amino acid homology to each other but no structural homology with any traditional receptor family. σ Receptors physically associate with diverse cellular systems, including lipid microdomains (Hayashi and Su, 2003), Kv1.4 potassium channels (Aydar et al., 2002) and NMDA receptors (Monnet et al., 1996; Martina et al., 2007), and it has been suggested recently that they have endoplasmic reticulum chaperone functions (Hayashi and Su, 2007). However, many fundamental questions regarding the functional significance of σ binding sites remain unanswered.
In vivo, σ1 receptors modulate opioid and orphanin FQ analgesic sensitivity with different tonic levels of σ activity among strains of mice and rats (Chien and Pasternak, 1993, 1994, 1995; Rossi et al., 1996, 1997; Mei and Pasternak, 2007). In these studies, haloperidol potentiated μ, δ, κ1, κ3, and orphanin FQ analgesia. Although haloperidol has high affinity for both σ1 and dopamine D2 receptors, the continued ability of haloperidol to potentiate opioid analgesia in dopamine D2 receptor knockout mice further supports a direct σ action (King et al., 2001).
Opioid receptors are heterotrimeric G-protein-coupled receptors (GPCRs) whose functionality is determined by macromolecular receptor assemblies comprising receptors and G-proteins as well as associated cellular factors such as regulators of G-protein signaling, various receptor activity-modifying proteins, and G-protein-coupled receptor kinases (McLatchie et al., 1998; Pitcher et al., 1998; Hollinger and Hepler, 2002). The complex network of protein-protein interactions involved in GPCR signaling implicates a growing list of cytosolic and membrane-bound signal-modifying proteins involved in various cellular second-messenger systems. What is more, allosteric drug interactions add to the complexity of regulating ternary structures comprising GPCR and associated factors (Christopoulos and Kenakin, 2002).
Here, we demonstrate a physical and functional association of σ1 receptors with μ opioid receptors. We show that σ1 receptors directly associate with opioid receptors and that through this association, σ-selective antagonists can potentiate opioid-induced cell signaling.
Materials and Methods
Chemical Compounds and Peptide Ligands.
BD1047 was obtained from Tocris Bioscience (Ellisville, MO). Morphine, [d-Pen2, d-Pen5]enkephalin (DPDPE), [d-Ala2, MePhe4,Gly(ol)5]enkephalin (DAMGO), fentanyl, (+)SKF-10047 and (+)pentazocine were gifts from the Research Technology Branch of the National Institute on Drug Abuse (National Institutes of Health, Bethesda, MD). Methacholine and haloperidol were purchased from Sigma-Aldrich (St. Louis, MO), whereas [3H]DAMGO and [3H]diprenorphine were purchased from MP Biomedicals (Solon, OH), and the detergent Nonidet P-40 was from Fluka Chemical Corp. (Ronkonkoma, NY).
Cell Culture and Cell Lines.
The 293T clone of human embryonic kidney (HEK) fibroblasts were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 4.5 mg/ml glucose, 2 mM l-glutamine, penicillin, and streptomycin. Stably transfected HEK populations and stable cell lines were generated by transfection of 2 to 5 μg of plasmid DNA with Effectene (QIAGEN, Valencia, CA), according to the manufacturer's protocol, followed by 2-week selection in growth medium supplemented with 0.2 mg/ml hygromycin B (Invitrogen, Carlsbad, CA) for epitope-tagged MOR-1 constructs or 0.2 mg/ml Zeocin (Invitrogen) for epitope-tagged σ1 constructs. Stable expression of MOR-1 and σ1 was confirmed by ligand-binding saturation assays and Western blot for the latter.
[35S]GTPγS Binding Assays.
Assays and membrane preparations were performed as described previously (Bolan et al., 2004). Cell culture medium was changed 24 h before harvesting cells. Membranes were prepared from 70 to 80% confluent monolayers of evenly distributed cells. Membrane preparations from cell lines were immediately frozen on dry ice and stored at −80°C. Multiple freeze-thaw cycles were strictly avoided. Brain membrane [35S]guanosine-5′-O-(3-thio)triphosphate ([35S]GTPγS) binding assays were performed with freshly prepared CD-1 mouse brain membrane homogenate. DAMGO stimulation of μ opioid receptors and DPDPE stimulation of δ opioid receptors were performed similarly. Muscarinic acetylcholine receptor stimulation was performed with methacholine. Fresh BD1047 solutions were prepared immediately before each experiment. [35S]GTPγS binding reactions were performed in the presence and absence of the indicated agonist for 60 min at 30°C in the assay buffer (50 mM Tris-HCl, pH 7.4, 3 mM MgCl2, 0.2 mM EGTA, and 10 mM NaCl) containing 0.05 nM [35S]GTPγS and 30 μM GDP, as reported previously (Bolan et al., 2004). Twenty-five micrograms of membrane protein was used per tube. After the incubation, the mixture was filtered through glass-fiber filters (Whatman Schleicher and Schuell, Keene, NH) and washed three times with 3 ml of ice-cold 50 mM Tris-HCl, pH 7.4, on a semiautomatic cell harvester (Brandel Inc., Gaithersburg, MD). Filters were transferred into vials, and radioactivity was determined using 3 ml of Liquiscent (National Diagnostics, Atlanta, GA) in a Tri-Carb 2900TR scintillation counter (PerkinElmer Life and Analytical Sciences, Waltham, MA). Basal binding was determined in the presence of GDP and the absence of drug.
Receptor Binding Assays.
Ligand binding competition assays were performed on isolated membranes as described previously (Bolan et al., 2004). [3H]DAMGO and [3H]diprenorphine saturation and competition binding assays were performed at 25°C for 60 min in potassium phosphate buffer (50 mM, pH 7.4) containing 5 mM magnesium sulfate. Specific binding was defined as the difference between total binding and nonspecific binding, determined in the presence of levallorphan (10 μM). Protein concentrations were determined as described previously using bovine serum albumin as the standard.
Plasmid Constructions.
The murine σ1 receptor with a carboxyl- terminal hemagglutinin (HA) tag was cloned into the NotI site of pcDNA3.1/zeo(+) (Invitrogen). The murine MOR-1 with a triple amino-terminal Flag (DYKDDDD) epitope tag was generated by cloning into the p3XFlag expression vector (Sigma-Aldrich) as described previously (Pan et al., 2002).
Immunoprecipitation and Immunoblot Analysis.
Cells were harvested in ice-cold phosphate-buffered saline solution containing 2 mM EDTA, washed twice, and lysed for 30 min at 4°C in a solubilization buffer containing 0.5% Nonidet P-40, 50 mM Tris-HCl, pH 7.4, and 150 mM NaCl supplemented with a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). The detergent-soluble fraction was obtained after centrifugation at 4°C for 10 min at 20,000g. The clarified Nonidet P-40 soluble cell lysate was used for immunoprecipitation with either 5 μg of agarose-coupled rabbit anti-HA polyclonal antiserum (Santa Cruz Biotechnology, Santa Cruz, CA) or 2 μg of agarose-coupled anti-FLAG(M2) mouse monoclonal antibody (Sigma-Aldrich). Immunoprecipitated fractions and whole-cell lysates (∼10 μg of protein) were separated on 10% SDS-polyacrylamide gels and transferred electrophoretically to nitrocellulose. Membranes were immunoblotted with either a horseradish peroxidase-conjugated rabbit anti-HA antibody (Santa Cruz Biotechnology) or a mouse anti-FLAG antibody (Sigma Aldrich). Immunoreactive proteins were visualized using the enhanced chemiluminescence detection system (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK).
RNA Interference Knockdown of σ1.
BE(2)-C cells were transfected with nonspecific control or human σ1 receptor-selective siRNA (Santa Cruz Biotechnologies). Oligofectamine (Invitrogen) was used as the siRNA transfection reagent, according to manufacturer's suggested protocol. Cells were harvested and membranes prepared, as described above, 96 h after transfection.
Data and Statistical Analysis.
Pairs of data were analyzed by Student's t test, whereas multiple comparisons were evaluated by ANOVA. Unless indicated otherwise, all experiments were replicated at least three times independently. Mean EC50 and Bmax values were determined by nonlinear regression analysis of dose-response data using the curve-fitting program in Prism 5 (GraphPad Software Inc., San Diego, CA).
Results
Interaction of σ Ligands with Opiate Binding Sites.
Because σ drugs potently modulate opiate analgesia in mice and rats (Chien and Pasternak, 1994; Mei and Pasternak, 2002), we first determined whether σ ligands directly affected opioid receptor binding. σ1 Binding is stereoselective. Although (−)pentazocine labels μ and κ opioid binding sites with high affinity, it has only modest affinity for σ sites. Conversely, (+)pentazocine selectivity labels σ1 receptors (Ki = 1.8 nM) approximately 500-fold more potently than μ opioid sites (Ki > 700 nM) and more than 30-fold more potently than κ1 binding sites (Chien et al., 1997). Likewise, the σ ligand BD1047 shows poor affinity for opioid binding sites (Ki > 1000 nM) while labeling σ1 sites with very high affinity (Ki = 0.9 nM) (Matsumoto et al., 1995). Inclusion of BD1047 at concentrations up to 100 nM in binding assays with the μ agonist failed to inhibit [3H]DAMGO binding to μ receptors. Furthermore, inclusion of a fixed concentration of BD1047, corresponding to the concentration used in the functional assays (10 nM), did not influence the KD or Bmax of [3H]DAMGO binding in saturation studies performed with BE(2)-C neuroblastoma cell membranes (Fig. 1). Thus, neither (+)pentazocine nor BD1047 influence opioid binding directly despite their ability to modulate opioid actions in vivo.

Effect of σ-selective ligands on μ opioid receptor binding. Radioligand binding saturation assays were performed using BE(2)-C membranes, as described under Materials and Methods. Increasing concentrations of [3H]DAMGO were added in the absence (●) or presence of BD1047 (10 nM; ○). Data are presented as the mean of two independent determinations. The Bmax and KD of [3H]DAMGO alone (0.054 pmol/mg and 0.55 nM, respectively) and with 10 nM BD1047 (0.062 pmol/mg and 0.8 nM, respectively) showed no significant differences, as determined by nonlinear regression analysis (Prism; GraphPad Software Inc.).
σ Modulation of μ Opioid-Stimulated [35S]GTPγS Binding in Brain Membranes.
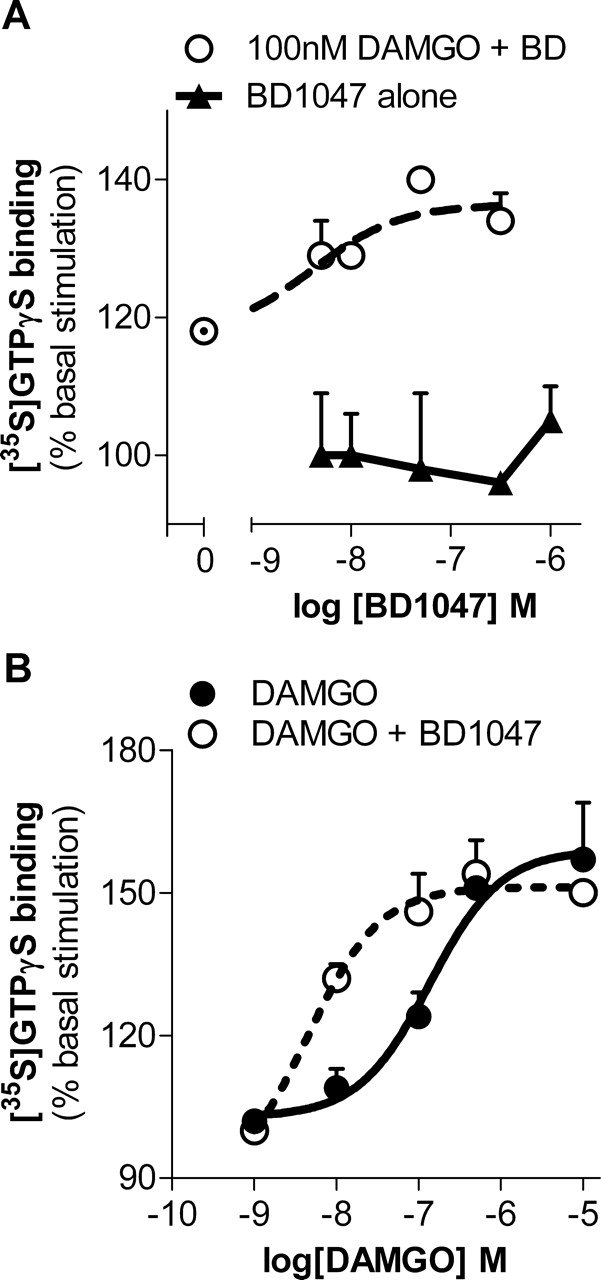
The lack of effect of σ ligands on the receptor binding of opioids raised the question of whether their ability to modulate opioid analgesia reflected a direct effect on receptor activity or downstream signaling. To assess whether there was a direct influence on opioid receptor activation, we examined the effects of σ ligands on opioid-induced stimulation of [35S]GTPγS binding. This assay assesses receptor activation by measuring guanine nucleotide exchange, an early event in GPCR-mediated signaling (Sim et al., 1996). BD1047 alone had no effect on [35S]GTPγS binding (Fig. 2A). As anticipated, the μ opioid DAMGO (100 nM) modestly stimulated [35S]GTPγS binding by approximately 20%. The stimulation of a fixed concentration of DAMGO was potentiated in a dose-dependent manner by BD1047 (EC50 = 4 nM), almost doubling the DAMGO-induced [35S]GTPγS binding at BD1047's maximal effective concentration of approximately 100 nM (Fig. 2A). It is interesting that the EC50 for BD1047 is similar to its affinity for σ1 binding sites (Ki = 0.9 nM) (Matsumoto et al., 1995).

Potentiation of μ opioid signaling in brain membranes by σ-selective ligand. [35S]GTPγS binding assays were performed with fresh mouse brain membrane preparations as described under Materials and Methods. A, [35S]GTPγS binding was measured in mouse brain membrane preparations dosed with BD1047 (5–500 nM) alone. Data are presented as the percentage of stimulation over basal levels. DAMGO (100 nM) was incubated with increasing concentrations of BD1047. DAMGO combined with BD1047 stimulated [35S]GTPγS binding by up to 36 ± 3% greater than basal levels, with a logEC50 of −8.38 ± 0.35. B, dose-response curves of DAMGO stimulation of [35S]GTPγS binding by DAMGO alone (●) or DAMGO with BD1047 (10 nM; ○) revealed a logEC50 of −6.86 ± 0.21 of DAMGO alone and −8.34 ± 0.23 when combined with BD1047 (10 nM; P = 0.05, determined by two-way ANOVA). Bmax values of DAMGO alone and combined with BD1047 are 159 ± 6 and 151 ± 3% basal stimulation, respectively, and are not significantly different.
DAMGO alone stimulated [35S]GTPγS binding in a dose-dependent manner (EC50 = 137 nM), with a maximal stimulation (Bmax) of 59 ± 6% over baseline values. Inclusion of a fixed concentration of BD1047 (10 nM) potentiated DAMGO-induced stimulation of [35S]GTPγS binding, shifting the DAMGO dose-response curve more than 25-fold to the left (EC50 = 5 nM) with no appreciable change in maximal binding (51 ± 3% over baseline values) (Fig. 2B).
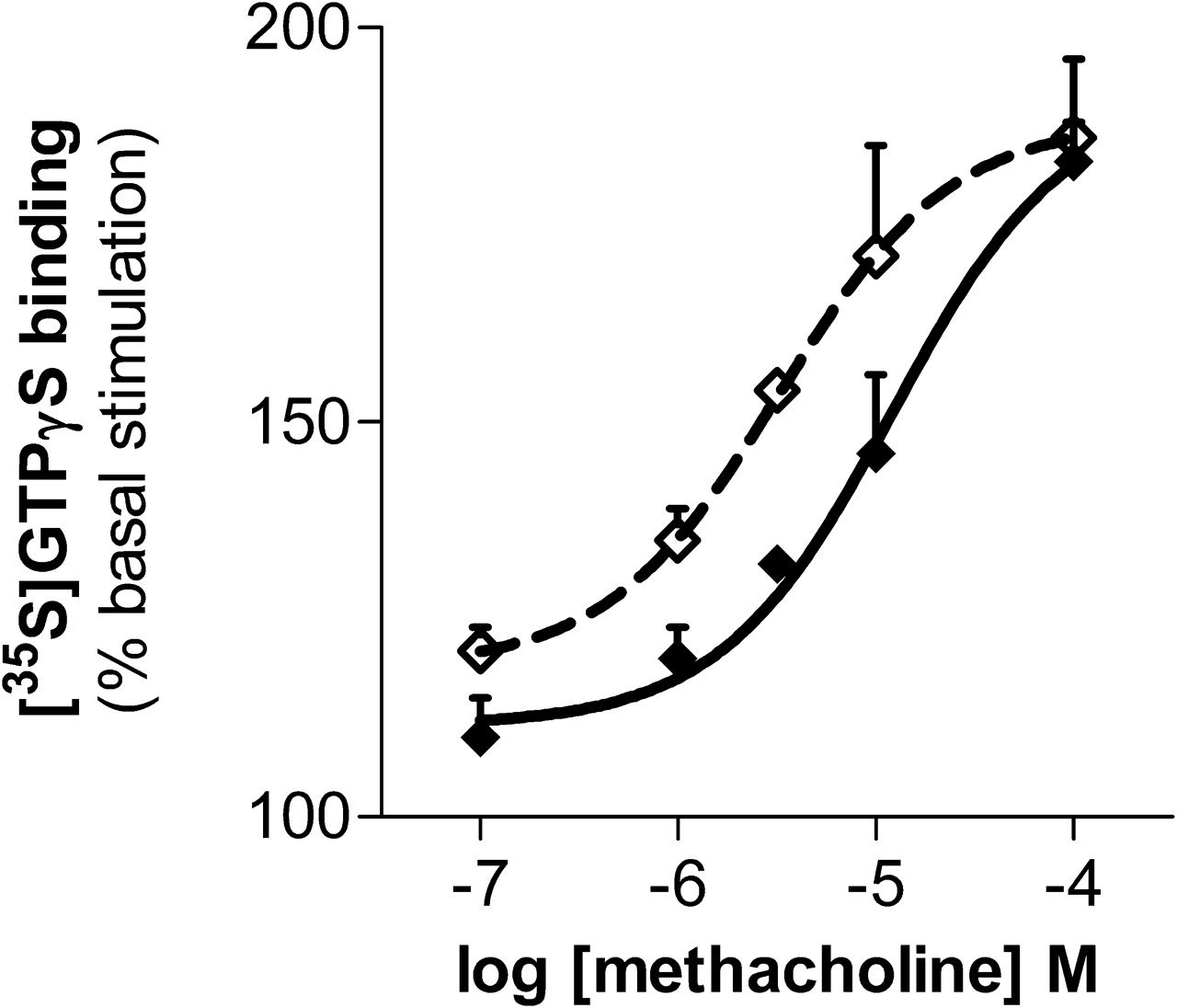
The effects were not limited to opioid-stimulated [35S]GTPγS binding. In brain homogenates, methacholine stimulated [35S]GTPγS binding in a dose-dependent manner (Fig. 3). Inclusion of BD1047 shifted the dose-response curve to the left, documenting the ability of σ1 systems to influence nonopioid G-protein-coupled receptors.

Potentiation of muscarinic acetylcholine receptor signaling by σ-selective ligand. Methacholine-induced stimulation of [35S]GTPγS binding was assessed in mouse brain membranes alone and in combination with BD1047 (10 nM). Assays were performed with fresh brain membrane preparations. The logEC50 of methacholine alone is −4.91 ± 0.22 and −5.49 ± 0.26 when combined with 10 nM BD1047 (P < 0.001). Bmax values are 191 ± 12 and 188 ± 8% basal stimulation, respectively, and are not significantly different.
σ Ligand Modulation of μ and δ Opioid-Stimulated [35S]GTPγS Binding in BE(2)-C Neuroblastoma Cells.
Mouse brain comprises a heterogeneous population of cell types and receptor systems. We next examined a more homogenous system, the human BE(2)-C neuroblastoma cell line that natively expresses μ opioid receptors (Standifer et al., 1994). As in the brain, BD1047 alone had no effect on [35S]GTPγS binding (Fig. 4A). However, BD1047 enhanced in a dose-dependent manner the stimulation of a fixed concentration of DAMGO on [35S]GTPγS binding. DAMGO at 100 nM stimulated [35S]GTPγS binding 33 ± 11% over basal levels alone. BD1047 increased this stimulation in a dose-dependent manner, raising the maximal response over basal activity approximately 3-fold (100 ± 10%) with potency (EC50 = 4 nM) very similar to that seen in the brain (EC50 5 nM) (Fig. 4A).
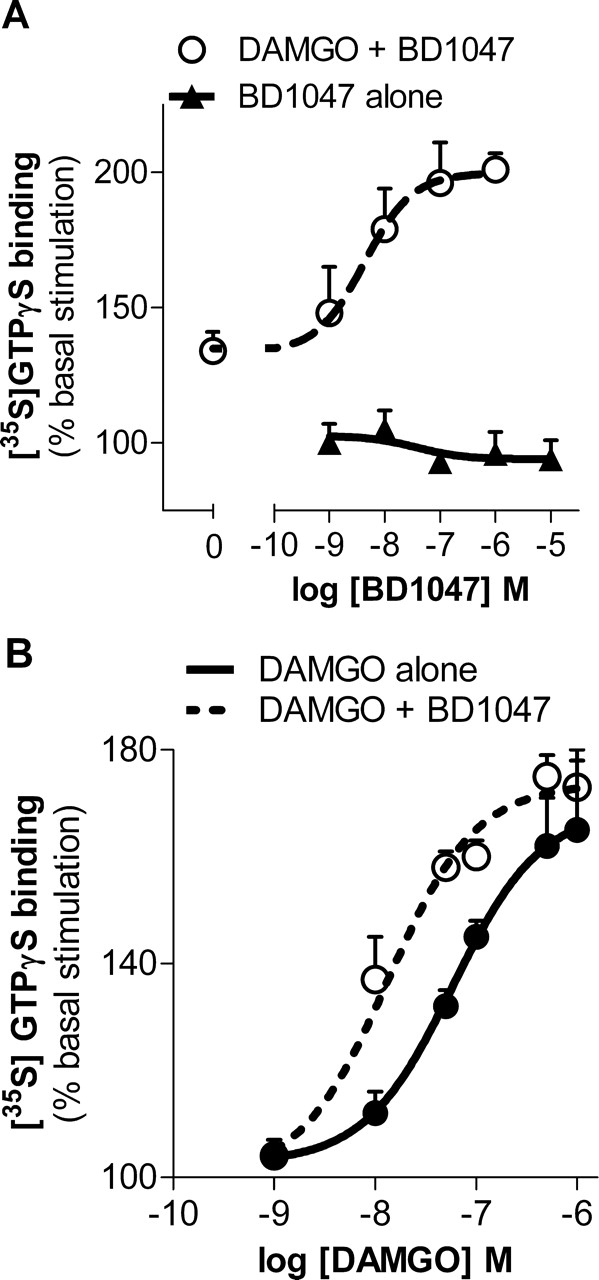
Potentiation of μ opioid receptor signaling in membranes from human neuroblastoma cell line, BE(2)-C, by σ selective ligand. A, stimulation of [35S]GTPγS binding was measured in BE(2)-C cell membrane preparations dosed with BD1047 (1–1000 nM) alone. Data are presented as the percentage of stimulation over basal levels. BD1047 alone did not stimulate [35S]GTPγS binding. DAMGO (100 nM) increased [35S]GTPγS binding by 33 ± 11% over basal levels. BD1047 increased DAMGO stimulation to 100 ± 10% over basal levels at 1 μM with a logEC50 of −8.37 ± 0.42 (P = 0.01, determined by one-way ANOVA). B, DAMGO dose-response curves of [35S]GTPγS binding by DAMGO alone (●) or combined with BD1047 (10 nM; ○). The logEC50 of DAMGO alone (−7.23 ± 0.19) was lowered more than 4-fold (logEC50 −7.87 ± 0.19) when combined with BD1047 (10 nM); P = 0.0002, determined by two-way ANOVA). Bmax values (174 ± 3 and 169 ± 6% basal stimulation, respectively) were not significantly different.
As in the brain, BD1047 altered the potency, not the efficacy, of DAMGO. Administered alone, DAMGO increased [35S]GTPγS binding in a dose-dependent manner with an EC50 of 59 nM. Inclusion of a fixed concentration of BD1047 (10 nM) shifted the DAMGO dose-response curve more than 4-fold to the left, yielding an EC50 of only 13 nM (Fig. 4B). Despite this increase in DAMGO potency, BD1047 had no appreciable effect on the maximal stimulation, a measure of efficacy, which was 69 ± 6% greater than basal levels alone and 74 ± 3% in the presence of BD1047.
Although the selectivity of this antagonist strongly implicated a role for σ1 receptors in these actions, we next confirmed this using siRNA to down-regulate σ1 receptors in BE(2)-C cells. σ1 siRNA down-regulated the level of σ1 receptors in this paradigm by approximately 50%, as measured by Western blots (Fig. 5, A and B). In the σ1 siRNA-treated cells, DAMGO stimulated [35S]GTPγS binding significantly more than in controls (Fig. 5D), firmly establishing a role for σ1 receptors in opioid stimulation of [35S]GTPγS binding.

Potentiation of μ opioid receptor signaling in BE(2)-C cells by siRNA down-regulation of σ1 receptors. A, immunoblot of membrane proteins extracted from BE(2)-C cells transfected with nonspecific control or human σ1 receptor selective siRNA. Immunoblot represents σ1 proteins levels in BE(2)-C cells that were harvested 96 h after transfection. B, histogram represents the quantification of siRNA-mediated σ1 receptor knockdown from three determinations. Control siRNA did not significantly alter σ1 levels (103 ± 5% of basal). σ1 Was knocked down to 49 ± 18% of basal levels by σ1-selective siRNA. C, confirmation of equivalent protein loading by Coomassie stain of SDS-Tris-glycine gel run in parallel to immunoblot shown in A. D, comparison of DAMGO stimulation of [35S]GTPγS binding measured in BE(2)-C membrane preparations from σ1 receptor siRNA and control siRNA-transfected cells. Data are presented as the percentage of stimulation over basal levels and are from three independent determinations (P values were determined by two-tailed, unpaired t test).
The ability of BD1047 to enhance agonist-induced stimulation of [35S]GTPγS binding was not limited to μ opioid receptors. BE(2)-C cells also natively express δ opioid receptors. In these cells, the δ opioid peptide DPDPE stimulated [35S]GTPγS binding in a dose-dependent manner (EC50 = 209 nM), with a maximal stimulation of 89 ± 6% over basal levels (Fig. 6). Inclusion of a fixed concentration of BD1047 (10 nM) increased the potency of DPDPE, shifting its response curve to the left more than 15-fold and decreasing its EC50 value to 12 nM. As with the μ-selective opioids, the maximal response in the presence of BD1047 (76 ± 4% over basal) did not differ significantly from that of DPDPE alone.

Potentiation of δ opioid receptor signaling in BE(2)-C cell membranes by σ-selective ligand. Dose-response curves measuring stimulation of [35S]GTPγS binding by increasing concentrations of δ opioid receptor ligand, DPDPE, alone (●, solid line) or combined with 10 nM BD1047 (○, broken line). The logEC50 of DPDPE alone is −6.68 ± 0.13 and −7.91 ± 0.23 when combined with 10 nM BD1047 (P = 0.002, determined by two-way ANOVA). Bmax values are 189 ± 6 and 176 ± 4%, respectively, and are not significantly different.
In vivo, we observed opposing actions by the σ1 agonist (+)pentazocine and σ1 antagonists (Chien and Pasternak, 1994; Mei and Pasternak, 2002). Whereas a σ1 antagonist potentiated opioid analgesia, (+)pentazocine diminished the opioid analgesic activity. We examined the effect of (+)pentazocine on opioid agonist-induced stimulation of [35S]GTPγS binding. Despite using doses of (+)pentazocine up to 1 μM, we were unable to observe an effect of (+)pentazocine on DAMGO stimulation of [35S]GTPγS. This inactivity of (+)pentazocine suggests that the σ1 receptors in the BE(2)-C cell line may already be in an “agonist” conformation, limiting the activity of σ1 agonist ligands in this in vitro assay to σ antagonists.
σ Ligand Modulation of μ Opioid-Stimulated [35S]GTPγS Binding in HEK Cells Stably Expressing MOR-1.
σ1 Receptors modulate μ opioid receptors in a transfected cell line in a manner similar to that observed in both brain and neuroblastoma cells. Endogenous σ1 receptors are expressed in all cell lines that have been examined, including HEK cells, as shown by the binding of [3H](+)pentazocine (Fig. 7A). However, HEK cells have no opioid receptor binding, as confirmed by the absence of binding by the opioid antagonist [3H]diprenorphine and the opioid agonist [3H]DAMGO (data not shown). To ensure that the effects we observed in brain and BE(2)-C cells reflected an interaction with MOR-1, we explored the effects of σ1 antagonists in HEK cells stably expressing transfected MOR-1.

σ-Selective ligand potentiation of opiate signaling in a heterologous expression system. HEK293T was stably transfected with MOR-1 cDNA. Stably transfected cell populations and clones were tested for ligand binding and stimulation of [35S]GTPγS binding, as described under Materials and Methods. The data presented are from a representative stably transfected clone. A, saturation study of [3H](+)pentazocine binding in HEK293T cells. The mean number of endogenous [3H](+)pentazocine binding sites is 0.116 pmol/mg membrane, with a KD of 2.6 nM. These mean values were generated from two independent experiments performed in triplicate. B, saturation study of [3H]DAMGO binding in membranes from MOR-1-transfected HEK293T cells (■). [3H]DAMGO binding was undetectable in parental cell membranes. The mean number of [3H]DAMGO binding sites in the MOR-1 transfected cell line is 0.164 pmol/mg membrane. The KD is 0.76 nM. These mean values were generated from two independent experiments performed in triplicate. C, BD1047-induced stimulation of [35S]GTPγS binding. D, DAMGO-induced stimulation of [35S]GTPγS binding (●, solid line) alone or combined with 10 nM BD1047 (○, broken line). The logEC50 of DAMGO alone is −7.34 ± 0.26 and −8.24 ± 0.31 when combined with 10 nM BD1047 (P < 0.001). Bmax values are 170 ± 8 and 168 ± 14% basal stimulation, respectively, and are not significantly different.
σ Agonists and antagonists alone did not influence [35S]GTPγS binding in either nontransfected HEK or Chinese hamster ovary cells (Table 1). Likewise, opioids do not stimulate [35S]GTPγS binding in nontransfected HEK cells, which do not natively express opioid receptors. After transfection with MOR-1, we isolated a stably expressing clone of HEK cells that expressed μ opioid binding sites (Fig. 7B). As with brain and BE(2)-C membranes, BD1047 alone did not stimulate [35S]GTPγS binding in the MOR-1-expressing HEK transfected cells (Fig. 7C). However, DAMGO stimulated [35S]GTPγS binding in the MOR-1-transfected cells in a dose-dependent manner with an EC50 of 45 nM. Inclusion of a fixed concentration of BD1047 (10 nM) potentiated DAMGO-induced stimulation of [35S]GTPγS binding, shifting the EC50 value more than 7-fold to the left to 6 nM (Fig. 7D). As in the other systems, the effects of BD1047 were limited to the potency of DAMGO, with no appreciable changes in the maximal effects. The ability of DAMGO to stimulate [35S]GTPγS binding only in HEK cells transfected with MOR-1 and not in nontransfected cells confirmed the role of MOR-1 in these actions.
[35S]GTPγS binding assay with a panel of σ ligands
The effects of σ agonists and antagonists alone (10 μM) on [35S]GTPγS binding were determined in CHO cells stably transfected with MOR-1. Results are the means ± S.E.M. of three independent determinations and are given as the percentage of basal stimulation levels. No significant stimulation of [35S]GTPγS binding was observed. Similar results were observed with HEK293T cells.
Physical Association of σ1 and Opioid Receptors.
The [35S]GTPγS binding studies strongly suggested a direct interaction between σ1 and opioid receptors. To determine whether this reflected a physical association between these two proteins within a complex, we performed coimmunoprecipitation experiments using epitope-tagged receptors. The FLAG-epitope was attached to the N terminus of MOR-1, whereas an HA tag was attached to the C terminus of the σ1 receptor, and both were transfected into HEK cells.
Immunoblotting with an HA antibody revealed bands only after a pull-down from lysates of cells transfected with the HA-tagged σ1 receptor (Fig. 8A, middle), confirming the specificity of the pull-down. Silver staining shows an enrichment of a band consistent with σ1 receptors only from σ1-HA-transfected cells, again supporting the specificity of the approach (Fig. 8A, right).

Coimmunoprecipitation of σ1 receptor and MOR-1. Coimmunoprecipitation experiments were performed using detergent-soluble cell membranes prepared from HEK293T Flag-MOR-1 and σ1-HA stably transfected cells. Immunoprecipitated samples were resolved by 10% SDS-polyacrylamide gel electrophoresis. All apparent molecular masses are represented in kilodaltons. A, immunoprecipitation (IP) by agarose bead-coupled HA antibody pulled down Flag-MOR-1, detected by immunoblotting (IB) with an anti-Flag antibody, only in lysates from cells coexpressing both Flag-MOR-1 and σ1-HA (left). Bands are visible at 90, 70, and 45 kDa and are indicated by closed arrows (left). Middle (control) shows similar quantities of immunoprecipitated σ1-HA (30 kDa) with the HA antibody. Right, a representative silver-stained gel of HA-immunoprecipitated samples is shown. Bands representative of σ1 receptors are indicated by open arrows. B, IP by agarose bead coupled Flag antibody pulled down σ1-HA (30 kDa), detected by IB with an anti-HA antibody, only in membranes from cells coexpressing both Flag-MOR-1 and σ1-HA (left). Similar quantities of immunoprecipitated Flag-MOR-1 were detected by immunoblotting with a Flag antibody (middle). The bands at 55 and 25 kDa correspond to heavy- and light-chain IgG, respectively (middle). C, pre-IP (“input”) detergent-soluble membranes were resolved by SDS-polyacrylamide gel electrophoresis as IP loading controls.
In the inverse controls, we saw a similar pattern with the FLAG pull-down of the FLAG-tagged MOR-1 (Fig. 8B, middle). We observed several specific bands with a major band at approximately 45 kDa and less dense bands at approximately 70 and 90 kDa only in cells transfected with the FLAG-tagged MOR-1. These bands probably reflect opioid receptors at different stages of maturation within the cell and variable post-translational modifications. The ∼45-kDa band probably corresponds to the nonglycosylated, unmodified MOR-1 protein, whereas the band at approximately 70 kDa may correspond to the glycosylated MOR-1, and the 90-kDa band may correspond to oligomeric forms of MOR-1 (Jordan and Devi, 1999) or other MOR-1/protein complexes.
To determine whether MOR-1 and the σ1 receptors were physically associated, we pulled down the σ1 proteins with an HA antibody and immunoblotted with a FLAG antibody against MOR-1 (Fig. 8A, left). In this study, we observed three detectable forms of Flag-MOR that were seen only in the cells expressing both MOR-1 and σ1 proteins, confirming the specificity of the association. The major 45-kDa species and minor species at approximately 70 and 90 kDa were indistinguishable from the control blots against MOR-1. It would seem that the 45-kDa band represents a nonglycosylated form of the receptor. In the middle, the HA-band is not present in membranes from the Flag-MOR-1-transfected cells, whereas on the right, the protein loading is shown.
In the inverse pull-down experiment, we immunoprecipitated FLAG-MOR-1. When we then performed an HA-immunoblot, we observed a band only in the cells coexpressing both proteins (Fig. 8B, left). In the middle, the Flag-band is not present in membranes from the FLAG-MOR-1-transfected cells, whereas on the right, the protein loading is shown. Thus, MOR-1 and σ1 receptors coimmunoprecipitate, demonstrating a physical association between them.
To ensure that the associations were not spuriously generated during the solubilization and immunoprecipitation of the complexes, we also performed control studies in which membranes from HEK cells transfected with FLAG-tagged MOR-1 and membranes from HEK cells transfected with HA-tagged σ1 were mixed before the solubilization and immunoprecipitation. Because none of the cells in this control expressed both tagged receptors, any evidence of coimmunoprecipitation would imply that the association occurred during or after the solubilization step. However, this mixing experiment showed no evidence of coimmunoprecipitation of the two tagged proteins (data not shown).
Discussion
A number of years ago, we reported that σ ligands modulate opioid analgesia without influencing other opioid actions, such as the inhibition of gastrointestinal transit (Chien and Pasternak, 1993, 1994, 1995; Mei and Pasternak, 2007). The cloning of the σ1 receptor opened a number of investigations at the molecular level that had not been possible previously. σ1 receptors clearly do not fit, on the basis of structure, within any of the other established receptor classes, including the G-protein-coupled receptor family. Indeed, there is debate about whether they fulfill all of the criteria of a classic receptor. σ1 Receptors were initially classified within the opioid receptor family based on their affinity for the (+)isomers of several benzomorphans, and their actions were believed to be associated with opioid action, but they have now been linked to wide range of diverse biological phenomena extending beyond G-protein-coupled receptors, including voltage-gated potassium channels (Lupardus et al., 2000) and NMDA receptors (Monnet et al., 1996; Bermack and Debonnel, 2005). The potential complexity of σ receptors has been further increased with suggestions of as-yet-unidentified σ receptor subtypes (Ueda et al., 2001; Bermack and Debonnel, 2005).
Using whole mouse brain membranes, neuroblastoma cell lines naturally expressing opioid receptor activity, and a heterologous expression system, we confirmed the ability of σ1 receptors to influence opioid receptor activity through the formation of a functional complex in which the μ opioid receptor and the σ1 receptor are physically associated. Despite the physical association of the two receptors, σ ligands do not influence the binding of opioids to the μ receptor. Yet they clearly modulate opioid receptor signaling. Administered alone, σ ligands do not stimulate [35S]GTPγS binding. Yet the selective σ antagonist BD1047 markedly potentiated DAMGO-induced signaling without altering [3H]DAMGO binding, shifting the dose-response curve for opioid-induced stimulation of [35S]GTPγS binding 3- to 10-fold to the left without altering the maximal response. We observed very similar effects in brain, neuroblastoma, and transfected HEK cell membranes. We also have observed similar actions with another σ1 antagonist, haloperidol (data not shown). Finally, down-regulation of σ1 receptors using siRNA has an effect similar to that of the antagonists, confirming a role of σ1 receptors in these actions.
The σ1 receptor does not fit within any of the established receptor families. Yet drugs with opposing actions (i.e., agonists and antagonists) have been identified and used pharmacologically. The classification of these drugs remains somewhat tentative and has been based, in large part, on the similar effects of antagonists and treatments that down-regulate the expression level of the receptors (King et al., 1997). Although both agonists and antagonists influenced opioid analgesia in vivo, we only observed activity of the antagonists in our membrane systems. The reasons for this are not clear. However, one possibility is that the σ1 receptors in our membranes are already in an agonist conformation. If so, the addition of a σ1 agonist would not be expected to exert an additional effect, whereas antagonists would retain their activity. This possibility is supported by preliminary studies in our laboratory looking at the conformation of the C tail of the receptor using circular dichroism. Compared with the protein in the absence of any ligand, antagonists, but not agonists, induced a conformational shift in the protein (M. Islam, F.J. Kim, and G.W. Pasternak, unpublished observations). Thus, the native conformation of the C terminus in the absence of any ligand seems to be in an “agonist state” in this model, consistent with the above proposal. However, the conformation of the receptor in vivo may be under the influence of other factors, leading to differing ratios of agonist/antagonist conformations. It is interesting that the potentiation of opioid analgesia in vivo by the σ1 antagonist differed among mouse strains and for different classes of opioids (Chien and Pasternak, 1993, 1994, 1995), which would be consistent with differing ratios of agonist/antagonist conformations of the σ1 receptor in these strains.
We had initially anticipated that the σ1 antagonists would increase the affinity of the opioid for the receptor, but this was not the case. Because the affinity of the opioid is not influenced by the σ ligand, the enhanced activation of G-proteins seems to be due to an increase in the intrinsic activity of the opioid because the maximal response can be achieved at lower receptor occupancy. It also is consistent with an increase in spare receptors in that a maximal response can be seen with the occupation of a smaller fraction of receptors. The similar response with two other G-protein-coupled receptors, the δ opioid receptor and the muscarinic acetylcholine receptor, raises the possibility that these σ receptor actions may extend to other GPCR systems.
How σ1 receptors influence intrinsic activity without affecting the binding of the opioid agonist is not known. Opioid receptors exist within a large complex composed of a wide range of proteins. Indeed, early estimates of the size of the opioid receptor complex as early as the 1970s suggested a macromolecular assembly of 300 to 500 kDa, far larger than the predicted size of the opioid receptor (G.W. Pasternak, unpublished observations). Our ability to coimmunoprecipitate σ1 and MOR-1 implies the presence of both proteins within the receptor complex, suggesting that σ receptors may directly modulate the transduction pathways within the complex. However, this does not necessarily imply a direct attachment of the σ1 receptor to the opioid receptor. It is equally possible that they may share unidentified protein partners, leading to their association within the complex without a direct attachment.
The demonstration of similar actions with other G-protein coupled receptors raises the question of whether σ1 receptors have a more general role among G-protein-coupled receptors. Although we have examined only a very small sample of receptors, we were able to demonstrate interactions for all that were studied. However, the differences in the regional distribution of σ1 receptors within the brain may reflect a more selective association of σ1 receptors with G-protein-coupled receptors. Furthermore, σ1 receptors have a far wider range of actions beyond G-protein-coupled receptors. Others have shown an important role of σ1 receptors in both Kv1.4 potassium channels (Lupardus et al., 2000; Aydar et al., 2002) and NMDA receptors (Monnet et al., 1996; Martina et al., 2007). These interactions with monovalent and divalent ion channels and with G-protein-coupled receptors raise more questions on the mechanisms of action and overall role of σ1 receptors. Thus, modulation of σ1 receptors, either through drugs or its expression, would be expected to have a wide range of potential actions. σ Receptors reportedly are enriched in cholesterol-rich lipid microdomains (Crawford et al., 2002; Hayashi and Su, 2003; Gebreselassie and Bowen, 2004), which are believed to mediate the assembly of signaling complexes. They have high levels of expression in brain and other selected organs but are particularly prominent in tumor cells, where they have a role in growth and differentiation (F. J. Kim and G. W. Pasternak, unpublished observations). These observations point out the many questions still unanswered regarding the molecular mechanisms underlying σ receptor biology. Identifying σ receptor-mediated pathways and precisely defining functional domains and molecular mechanisms underlying σ function is indispensable to clearly defining the physiological roles of this protein.
Acknowledgments
We thank Loriann Mazzo and Jin Xu for their assistance.
Footnotes
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grants DA06241, DA02615, DA00220, DA07274]; the National Institutes of Health National Cancer Institute [Grant CA008748]; the National Genetics Foundation; Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research; and The Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.057083.
-
ABBREVIATIONS:
- (±)SKF-10047
- (±)-N-allyl-normetazocine
- BD1047
- N-[2-(3,4-dichlorophenyl)ethyl]-N-methyl-2-(dimethylamino)ethylamine
- DAMGO
- [d-Ala2,N-MePhe4,Gly5(ol)]enkephalin
- DPDPE
- [d-Pen2,d-Pen5] enkephalin
- MOR-1
- μ opioid receptor 1
- [35S]GTPγS
- guanosine 5′-O-(3-[35S]thio)triphosphate
- NMDA
- N-methyl-d-aspartate
- HEK
- human embryonic kidney
- GPCR
- G-protein-coupled receptor
- ANOVA
- analysis of variance
- HA
- hemagglutinin
- siRNA
- small interfering RNA.
- Received April 17, 2009.
- Accepted December 28, 2009.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}