


Abstract
The glucagon-like peptide-1 (GLP-1) receptor is a key regulator of insulin secretion and a major therapeutic target for treatment of diabetes. However, GLP-1 receptor function is complex, with multiple endogenous peptides that can interact with the receptor, including full-length (1–37) and truncated (7–37) forms of GLP-1 that can each exist in an amidated form and the related peptide oxyntomodulin. We have investigated two GLP-1 receptor allosteric modulators, Novo Nordisk compound 2 (6,7-dichloro2-methylsulfonyl-3-tert-butylaminoquinoxaline) and quercetin, and their ability to modify binding and signaling (cAMP formation, intracellular Ca2+ mobilization, and extracellular signal-regulated kinase 1/2 phosphorylation) of each of the naturally occurring endogenous peptide agonists, as well as the clinically used peptide mimetic exendin-4. We identified and quantified stimulus bias across multiple endogenous peptides, with response profiles for truncated GLP-1 peptides distinct from those of either the full-length GLP-1 peptides or oxyntomodulin, the first demonstration of such behavior at the GLP-1 receptor. Compound 2 selectively augmented cAMP signaling but did so in a peptide-agonist dependent manner having greatest effect on oxyntomodulin, weaker effect on truncated GLP-1 peptides, and negligible effect on other peptide responses; these effects were principally driven by parallel changes in peptide agonist affinity. In contrast, quercetin selectively modulated calcium signaling but with effects only on truncated GLP-1 peptides or exendin and not oxyntomodulin or full-length peptides. These data have significant implications for how GLP-1 receptor targeted drugs are screened and developed, whereas the allosterically driven, agonist-selective, stimulus bias highlights the potential for distinct clinical efficacy depending on the properties of individual drugs.
Introduction
Type II (non–insulin-dependent) diabetes mellitus (DM) is a major disease of the Western world with many complications, including renal failure and vascular conditions leading to heart attack and stroke (Wild et al., 2004). Type II DM is characterized by a decrease in peripheral tissue response to insulin in association with impaired β cell function, which results in an increase in fasting glycemia (DeFronzo, 1992). Although antihyperglycemic drugs such as metformins, sulfonylureas, or thiazolidinediones may be prescribed to promote insulin secretion or enhance insulin sensitivity (Mitri and Hamdy, 2009), these drugs do not target all of the symptoms of type II DM. In recent years, drugs that enhance the activity of the glucagon-like peptide 1 receptor (GLP-1R) have been of particular interest to the pharmaceutical industry, in that activation of this receptor addresses most of the manifestations of the condition.
The GLP-1R is a family B peptide hormone G protein-coupled receptor (GPCR) primarily expressed in pancreatic β cells and responds to at least four distinct endogenous GLP-1 variants as well as to the related peptide oxyntomodulin and exogenous mimetic peptides such as exendin-4. The four secreted forms of GLP-1 include a full-length peptide GLP-1(1–37) and a truncated form GLP-1(7–37), each of which also has an amidated counterpart: GLP-1(1–36)NH2 and GLP-1(7–36)NH2, respectively (Estall and Drucker, 2006; Baggio and Drucker, 2007). Although levels of GLP-1 are reduced in patients with type II DM, the receptor retains insulinotropic properties (Toft-Nielsen et al., 2001). However, the promise of this receptor as a target in the development of type II DM is hindered by the rapid degradation of endogenous peptides by dipeptidyl peptidase IV (DPPIV) in vivo (Deacon et al., 1995a; Kieffer et al., 1995). This has in part been overcome by the development of DPPIV-resistant GLP-1 mimetics such as exendin-4 (Göke et al., 1993; Edwards et al., 2001) and liraglutide (Knudsen et al., 2000; Elbrønd et al., 2002) as well as DPPIV inhibitors that prolong the plasma half-life of endogenous GLP-1R peptides (Deacon et al., 1995b). Although these have therapeutic potential (indeed, exendin-4 is currently used clinically), they require frequent intravenous or subcutaneous administration, reducing compliance. In addition, exendin-4 has also been associated with significant adverse side effects in some patients, including pancreatitis (Olansky, 2010), of which the mechanistic basis is unknown. These difficulties have therefore driven the search for the development of small molecule orally active drugs that augment GLP-1R signaling.
Allosteric ligands bind to GPCRs at sites distinct from the orthosteric (endogenous agonist) binding site and can modulate binding and/or signaling pathways of the receptor, as well as potentially acting as agonists themselves. Allosteric modulation has recently gained much traction as a means to overcome the limitations of many orthosterically targeted ligands, because it has the ability to provide novel receptor specificity and selectively control receptor function (Christopoulos and Kenakin, 2002). Little is known about allosteric modulation of the GLP-1R, and few small nonpeptide ligands acting allosterically at the GLP-1R have been reported. A number of small-molecule agonists have recently been identified by Novo-Nordisk; the most potent of these, compound 2, increases the affinity of GLP-1(7–36)NH2 and also displays intrinsic efficacy in cAMP accumulation assays (Knudsen et al., 2007). There is also a preliminary report that the naturally occurring flavonol quercetin may modulate GLP-1R-mediated calcium (Ca2+) signaling by GLP-1(7–36)NH2 (Schann et al., 2009) and evidence that the substituted cyclobutane Boc5 may also act as an agonist of the receptor (Chen et al., 2007).
A major development in GPCR research is the recognition that different ligands can engender unique receptor conformations, giving rise to distinct signaling profiles. This concept of “ligand-induced stimulus bias” is particularly relevant to receptor systems that have multiple endogenous ligands and is further complicated when allosteric ligands are considered. Although allosteric drugs acting at the GLP-1R offer great promise as therapeutics, the consequences of allosteric modulation of the GLP-1R and whether such ligands promote or modify stimulus bias at the receptor have not been fully explored. Furthermore, the natural complexity of the GLP-1R system, encompassing numerous endogenous peptide agonists, provides potential for small-molecule compounds to differentially modulate individual peptide responses, a behavior termed “probe dependence” (Kenakin, 2008). Consequently, we investigated the signaling and binding properties of these putative allosteric modulators in association with the physiologically relevant endogenous agonists of the GLP-1R, as well as the clinically used mimetic exendin-4. We demonstrate, for the first time, that compound 2 and quercetin each have distinct pharmacological profiles, exhibiting selective modulation of specific peptide agonists and engendering stimulus bias at the GLP-1R. These data have significant implications for how GLP-1 receptor targeted drugs are screened and developed, whereas the allosterically driven, agonist-selective, stimulus bias highlights the potential for distinct clinical efficacy depending on the properties of individual drugs.
Materials and Methods
Materials.
Dulbecco's modified Eagle's medium (DMEM) and Fluo-4 acetoxymethyl ester were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Thermo Fisher Scientific (Melbourne, VIC, Australia). AlphaScreen reagents, 125I-exendin(9–39), 96-well UniFilter GF/C filter plates, 384-well ProxiPlates and MicroScint 40 scintillant were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). SureFire extracellular signal-regulated kinases 1 and 2 (ERK1/2) reagents were obtained from TGR Biosciences (Adelaide, SA, Australia). The bicinchoninic acid (BCA) protein assay kit was purchased from Thermo Fisher Scientific. Compound 2 was generated, according to a method published previously (Teng et al., 2007), to a purity of >95%, and compound integrity was confirmed by NMR. GLP-1 and GLP-1 peptide analogs were purchased from American Peptide (Sunnyvale, CA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) or BDH Merck (Melbourne, VIC, Australia) and were of an analytical grade.
Cell Culture.
FlpIn Chinese hamster ovary (CHO) cells stably transfected with human GLP-1R (FlpInCHO-huGLP-1R) were generated using Gateway technology (Invitrogen) as described previously (May et al., 2007a). These cells expressed the human GLP-1R at a density of 123,500 ± 1368 receptors/cell. Cells were maintained in DMEM supplemented with 10% heat-inactivated FBS and were incubated in a a humidified environment at 37°C in 5% CO2. Untransfected CHO-FlpIn cells were used to control for receptor-independent effects. Additional control experiments were performed using COS-7 cells expressing the human CTa calcitonin receptor (Morfis et al., 2008).
Radioligand Binding Assay.
Membrane preparations of FlpInCHO-huGLP-1R were prepared as described previously (Avlani et al., 2004). Protein concentration was determined using the BCA protein assay kit (Thermo Fisher Scientific) according to the manufacturer's instructions, with bovine serum albumin (BSA) as standard. Competition binding assays were performed in 96-well plates using 20 μg of membrane expressing GLP-1R. Membranes were incubated in HEPES buffer [1 mM HEPES, 10 mM MgCl2, 100 mM NaCl, and 1% (w/v) BSA, pH 7.4] containing 0.5 nM 125I-exendin(9–39) and increasing concentration of unlabeled ligand for 1 h. For interaction studies, competition of 125I-exendin(9–39) binding by each orthosteric agonist was performed in the presence of increasing concentrations of either compound 2 or quercetin. For all experiments, nonspecific binding was defined by 1 μM GLP-1(7–36)NH2. Incubation was terminated by rapid filtration through Whatman (Clifton, NJ) GF/C filters (presoaked in 0.03% (v/v) polyethylenimine for a min of 2 h) using a 96-well harvester (Tomtec, Hamden, CT). Filters were washed three times with 0.9% NaCl (w/v) and 0.3% BSA (w/v) and allowed to dry before addition of 30 μl of scintillant and determination of radioactivity by scintillation counting.
cAMP Accumulation Assay.
FlpInCHO-huGLP-1R cells were seeded at a density of 5 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2. Growth media was replaced with stimulation buffer [phenol-free DMEM containing 0.1% (w/v) BSA and 1 mM 3-isobutyl-1-methylxanthine] and incubated for a further 1 h at 37°C in 5% CO2. Cells were stimulated with increasing concentrations of peptide ligand alone, allosteric ligand alone, or simultaneously with increasing concentrations of allosteric ligand and peptide, and incubated for 30 min at 37°C in 5% CO2. Selected additional experiments were also performed 5 and 10 min after ligand stimulation to account for the potential for kinetic differences between different signaling assays to contribute to apparent stimulus bias. The reaction was terminated by rapid removal of the ligand-containing buffer and addition of 50 μl of ice-cold 100% ethanol. After ethanol evaporation, 75 μl of lysis buffer [0.1% (w/v) BSA, 0.3% (v/v) Tween 20, and 5 mM HEPES, pH 7.4] was added, and 10 μl of lysate was transferred to a 384-well ProxiPlate (PerkinElmer Life and Analytical Sciences). Five microliters of acceptor bead mix (1.0% AlphaScreen cAMP acceptor beads diluted in lysis buffer) and 15 μl of donor bead mix [0.3% AlphaScreen cAMP donor beads, 0.025% AlphaScreen cAMP biotinylated cAMP (133 units/μl) diluted in lysis buffer, and preincubated for a minimum of 30 min] were added in reduced lighting conditions. Plates were incubated at room temperature overnight before measurement of the fluorescence using a Fusion-Alpha plate reader (PerkinElmer Life and Analytical Sciences) with standard AlphaScreen settings. All values were converted to concentration of cAMP using a cAMP standard curve performed in parallel.
ERK1/2 Phosphorylation Assay.
FlpInCHO-huGLP-1R cells were seeded at a density of 5 × 104 cells/well into 96-well culture plates, and receptor-mediated ERK1/2 phosphorylation was determined by using the AlphaScreen ERK SureFire protocol as described previously (May et al., 2007a). Initial ERK1/2 phosphorylation time course experiments were performed over 1 h to determine the time at which ERK1/2 phosphorylation was maximal after stimulation by agonists. Cells were stimulated with peptide ligand and/or simultaneously with increasing concentrations of allosteric ligand for the time required to generate a maximal ERK1/2 phosphorylation response (10 min). Data were normalized to the maximal 3% FBS response, determined at 7 min (peak FBS response).
Intracellular Ca2+ Mobilization Assay.
FlpInCHO-huGLP-1R cells were seeded at a density of 5 × 104 cells/well into 96-well culture plates, and receptor-mediated intracellular Ca2+ mobilization was determined as described previously (Werry et al., 2005). Because of their autofluorescence at high concentrations, increasing concentrations of allosteric modulators were added 30 min before addition of peptide agonist in the FlexStation (Molecular Devices, Palo Alto, CA) to establish a basal fluorescence signal. Fluorescence was determined immediately after peptide addition, with an excitation wavelength set to 485 nm and an emission wavelength set to 520 nm, and readings were taken every 1.36 s for 120 s. Peak magnitude was calculated using five-point smoothing, followed by correction against basal fluorescence. The peak value was used to create concentration-response curves. Data were normalized to the maximal response elicited by 100 μM ATP.
Data Analysis.
All data obtained were analyzed in Prism 5.02 (GraphPad Software Inc., San Diego, CA). Concentration response signaling data were analyzed using a three-parameter logistic equation as described previously (May et al., 2007a).
 where Bottom represents the y value in the absence of ligand(s), Top represents the maximal stimulation in the presence of ligand/s, [A] is the molar concentration of ligand, and EC50 represents the molar concentration of ligand required to generate a response halfway between Top and Bottom. Likewise, this equation was used in inhibition binding, replacing EC50 with IC50. In this case, Bottom defines the specific binding of the radioligand that is equivalent to nonspecific ligand binding, whereas Top defines radioligand binding in the absence of a competing ligand. In a similar manner, the IC50 value represents the molar concentration of ligand required to generate a response halfway between Top and Bottom.
where Bottom represents the y value in the absence of ligand(s), Top represents the maximal stimulation in the presence of ligand/s, [A] is the molar concentration of ligand, and EC50 represents the molar concentration of ligand required to generate a response halfway between Top and Bottom. Likewise, this equation was used in inhibition binding, replacing EC50 with IC50. In this case, Bottom defines the specific binding of the radioligand that is equivalent to nonspecific ligand binding, whereas Top defines radioligand binding in the absence of a competing ligand. In a similar manner, the IC50 value represents the molar concentration of ligand required to generate a response halfway between Top and Bottom.
Allosteric modulator inhibition binding data were subsequently analyzed according to an allosteric ternary complex model (May et al., 2007a) to determine ligand cooperativity. In this case, nondepletion of ligands was assumed (Avlani et al., 2008):
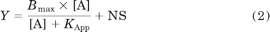 where
where
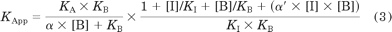 and where Y represents radioligand binding, Bmax denotes maximal binding site density, and NS denotes the fraction of nonspecific binding. [A] and KA denote the concentration of radioligand and equilibrium dissociation constant for the radioligand, respectively. [B] and KB denote the concentration of allosteric ligand and equilibrium dissociation constant for the allosteric ligand, respectively. [I] and KI denote the concentration of peptide agonist used in competition with the radioligand and the equilibrium dissociation constant for the peptide agonist, respectively. α and α′ represent cooperativity factors, which are measures of the magnitude and direction of the allosteric interaction between the modulator and the radioligand, or the peptide agonist, respectively. Values of α > 1 are indicative of a modulator-mediated increase in binding activity, whereas values of α between 0 and 1 are indicative of a modulator-mediated decrease in binding affinity.
and where Y represents radioligand binding, Bmax denotes maximal binding site density, and NS denotes the fraction of nonspecific binding. [A] and KA denote the concentration of radioligand and equilibrium dissociation constant for the radioligand, respectively. [B] and KB denote the concentration of allosteric ligand and equilibrium dissociation constant for the allosteric ligand, respectively. [I] and KI denote the concentration of peptide agonist used in competition with the radioligand and the equilibrium dissociation constant for the peptide agonist, respectively. α and α′ represent cooperativity factors, which are measures of the magnitude and direction of the allosteric interaction between the modulator and the radioligand, or the peptide agonist, respectively. Values of α > 1 are indicative of a modulator-mediated increase in binding activity, whereas values of α between 0 and 1 are indicative of a modulator-mediated decrease in binding affinity.
cAMP interaction data were also analyzed with an operational model of allosterism:
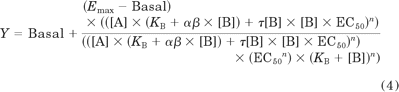 where Emax is the maximal possible response of the system (not the agonist), basal is the basal level of response in the absence of agonist, KB denotes the functional equilibrium dissociation constant of the agonist (B), τ is an index of the coupling efficiency (or efficacy) of the agonist and is defined as the total concentration of receptors divided by the concentration of agonist-receptor complex that yields half the maximum system response (Emax), and n is the slope of the transducer function that links occupancy to response. αβ is the combined affinity-efficacy parameter that measures the magnitude and direction of the functional interaction between the modulator and peptide agonist.
where Emax is the maximal possible response of the system (not the agonist), basal is the basal level of response in the absence of agonist, KB denotes the functional equilibrium dissociation constant of the agonist (B), τ is an index of the coupling efficiency (or efficacy) of the agonist and is defined as the total concentration of receptors divided by the concentration of agonist-receptor complex that yields half the maximum system response (Emax), and n is the slope of the transducer function that links occupancy to response. αβ is the combined affinity-efficacy parameter that measures the magnitude and direction of the functional interaction between the modulator and peptide agonist.
To quantify signaling bias, which may be manifested either as selective affinity (KA) and/or efficacy (τ) of an agonist for a given pathway, agonist concentration-response curves data were analyzed with an operational model (Gregory et al., 2010), but modified to directly estimate the ratio of τ/KA, in a manner similar to that described by Figueroa et al. (2009), for each pathway:
 where all other parameters are as defined for eq. 4.
where all other parameters are as defined for eq. 4.
All estimated parameters are expressed as logarithms (mean ± S.E.M.); where relevant, statistical analysis was performed by one-way analysis of variance and Dunnett's post test using GraphPad Prism 5.02, and statistical significance was accepted at p < 0.05.
Results
Compound 2 and Quercetin Selectively Modulate the Binding Affinity of Antagonists and Agonists of the GLP-1R.
To establish the ability of the putative small molecule GLP-1R modulators, compound 2 and quercetin, to modify peptide binding affinity, equilibrium binding studies were performed between each of these modulators and the radiolabeled orthosteric antagonist 125I-exendin(9–39). Analysis of the data using an allosteric ternary complex model revealed negative cooperativity between compound 2 and the antagonist, with the level of inhibition reaching a limit consistent with the predicted allosteric mode of inhibition (Table 1, Fig. 1B). Quercetin also displayed weak negative cooperativity with 125I-exendin(9–39) (Table 1, Fig. 1B). Affinity estimates (pKB) for compound 2 and quercetin were 7.58 ± 0.22 and 6.79 ± 0.41, respectively.
Differential effects of putative allosteric modulators used in this study on binding properties of peptide agonists of the human GLP-1R
Data were fit with a one-site competition plus allosteric modulator model as defined in eqs. 2 and 3 or by a three-parameter logistic model to yield binding parameters. pKi is the negative logarithm of the estimated affinity of the peptide agonist for the receptor. pKB is the negative logarithm of the estimated affinity of the allosteric compounds. Logα is the logarithm of the of the cooperativity factor governing the allosteric interaction between the peptide ligand and modulator. Antilogarithms are shown in parentheses. All values are mean ± S.E.M. from three to six independent experiments performed in duplicate.
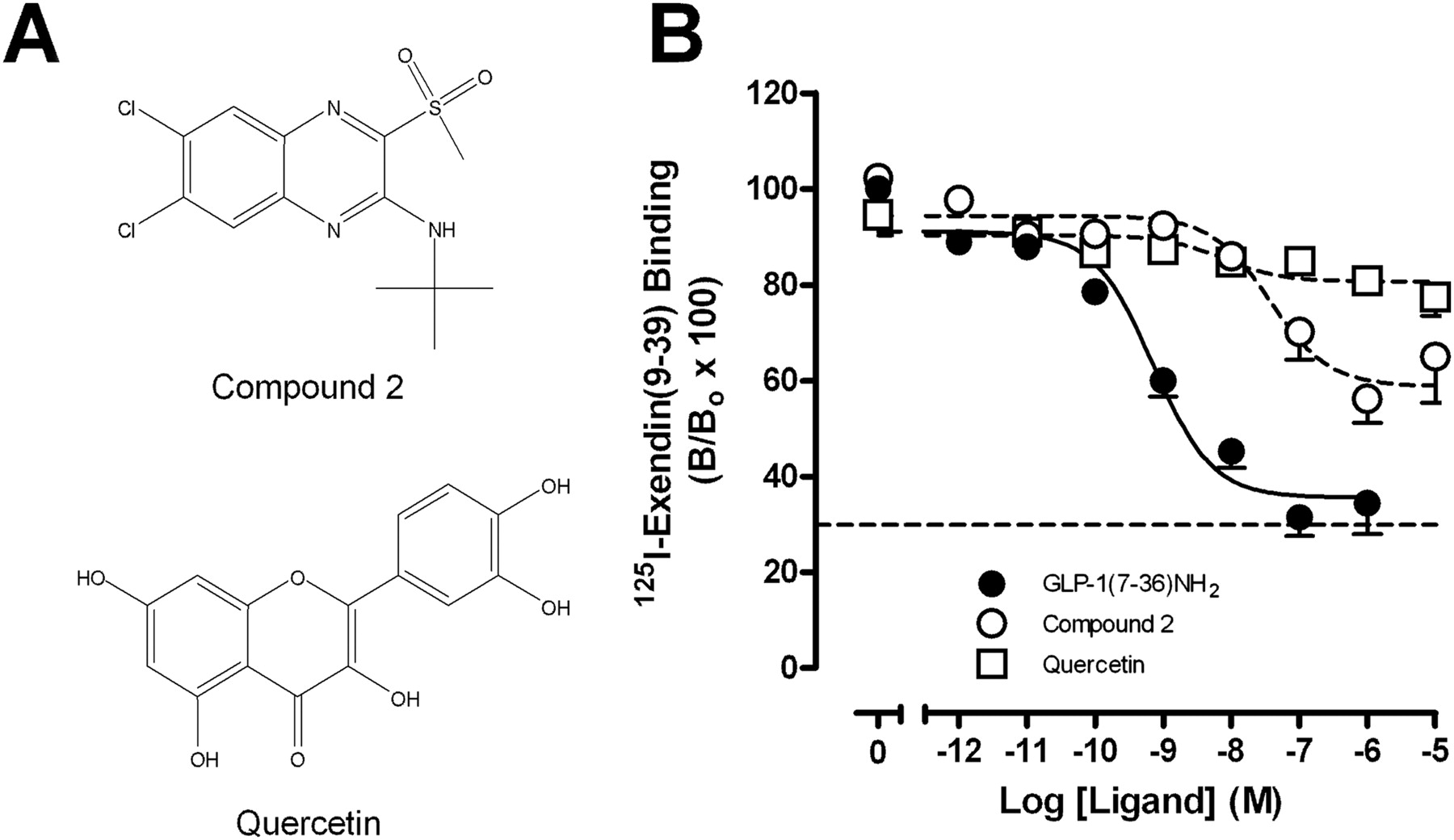
Structure and binding interactions elicited by allosteric modulators of the human GLP-1R. A, structures of the human GLP-1R small-molecule allosteric modulators used in this study. B, characterization of the inhibition binding profiles of compound 2 and quercetin at the human GLP-1R in relation to the endogenous peptide agonist GLP-1(7–36)NH2 using 125I-exendin(9–39) as the radioligand and membranes prepared from FlpInCHO cells stably expressing the human GLP-1R. Data are normalized to total binding and are analyzed with an allosteric modulator titration curve as defined in eqs. 2 and 3, assuming nondepletion (compound 2 and quercetin) or a competitive inhibition model [GLP-1(7–36)NH2]. All values are mean ± S.E.M. of 6 to 12 independent experiments conducted in duplicate. Nonspecific binding, measured in the presence of 10−6 M exendin-4, ranged from 25 to 30% of total binding (dotted line in B). B, bound radioligand; Bo, binding in the absence of peptide ligand (total binding).
To establish the ability of compound 2 to modulate orthosteric agonist affinity, competition binding studies were performed with GLP-1(7–36)NH2, GLP-1(7–37), GLP-1(1–36)NH2, GLP-1(1–37), oxyntomodulin, or exendin-4 in the absence or presence of increasing concentrations of compound 2 (Fig. 2, Table 1, Supplemental Fig. S1) or quercetin (Supplemental Fig. S2, Table 1). These assays demonstrated that compound 2 displays probe dependence (Leach et al., 2007), whereby the cooperativity between the orthosteric and allosteric binding sites is dependent on the orthosteric ligand present in the system. Compound 2 caused a concentration-dependent increase in the affinity of GLP-1(7–36)NH2 (Fig. 2A, Table 1) and GLP-1(7–37) (Supplemental Fig. S1A, Table 1). Compound 2 also displayed robust positive cooperativity for oxyntomodulin to a greater extent than either GLP-1(7–36)NH2 or GLP-1(7–37) (Fig. 2D, Table 1). In contrast, compound 2 minimally altered the binding of GLP-1(1–36)NH2, GLP-1(1–37) and exendin-4, therefore displaying neutral cooperativity with these agonists (Fig. 2, B and C, Supplemental Fig. S1B, Table 1). No significant modulation in affinity of any peptide agonist was observed in interaction binding studies with quercetin (Supplemental Fig. S2, Table 1).
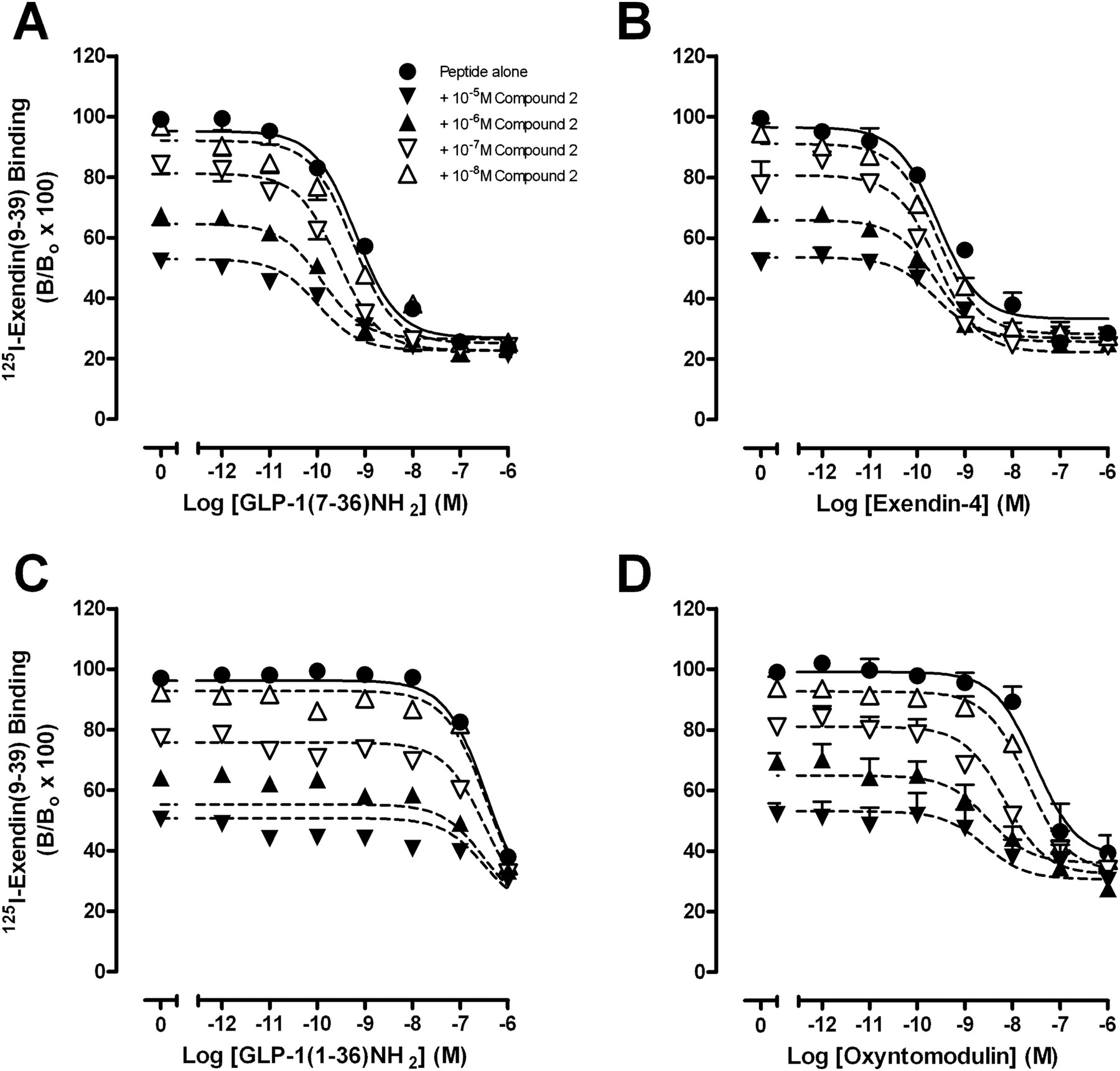
Characterization of the inhibition binding of varying concentrations of compound 2 in the presence of GLP-1(7–36)NH2 (A), exendin-4 (B), GLP-1(1–36)NH2 (C), or oxyntomodulin using membranes prepared from FlpInCHO cells stably expressing the human GLP-1R (D). Data are normalized to total radioligand binding and are analyzed with a one-site competition plus allosteric modulator curve as defined in eqs. 2 and 3, assuming nondepletion. All values are mean ± S.E.M. of four to six independent experiments conducted in duplicate. Nonspecific binding, measured in the presence of 10−6 M exendin-4, ranged from 25 to 30% of total binding. B, bound radioligand; Bo, binding in the absence of peptide ligand (total binding).
Compound 2 but Not Quercetin Selectively Augments cAMP Responses in an Agonist-Specific Manner at the GLP-1R.
The GLP-1R preferentially couples to the Gαs pathway, activating adenylate cyclase and augmenting intracellular levels of cAMP, which in turn play a direct role in the secretion of insulin (Drucker et al., 1987). Compound 2 stimulated a robust increase in cAMP production displaying low potency partial agonism in the absence of an orthosteric ligand (Fig. 3E, Table 2). In contrast, quercetin did not act as an agonist at the GLP-1R for cAMP (Supplemental Fig. S3G), whereas no response was seen in untransfected CHO-FlpIn cells for any of the ligands studied.

Characterization of the interaction between compound 2 and GLP-1(7–36)NH2 (A), exendin-4 (B), GLP-1(1–36)NH2 (C), or oxyntomodulin (D) in a cAMP accumulation assay using FlpInCHO cells stably expressing the human GLP-1R. E, compound 2 alone. Data are normalized to maximal peptide response and analyzed with an operational model of allosterism as defined in eq. 4. All values are mean ± S.E.M. of three to eight independent experiments conducted in duplicate.
Differential effects of compound 2 on peptide agonists of the human GLP-1R in cAMP accumulation in FlpInCHO cells stably expressing the human GLP-1R
Data were analyzed with an operational model of allosterism as defined in eq. 4. pEC50 values represent the negative logarithm of the concentration of agonist that produces half the maximal response. Emax represents the maximal response normalized to that of GLP-1(7–36)NH2. Logαβ values represent the logarithm of the product of binding (α) and activation (β) cooperativity factors between compound 2 and the peptide agonists. Antilogarithms are shown in parentheses. All values are mean ± S.E.M. of three to six independent experiments, conducted in duplicate. Data were analyzed with one-way analysis of variance and Dunnett's post test.
To investigate the potential for allosteric effects on peptide agonist-mediated cAMP signaling, interaction studies between the small molecule modulators and peptide ligands were performed. Coaddition of each of the peptides with compound 2 resulted in an observed elevation in cAMP at low concentrations of peptide agonist as a result of the intrinsic efficacy of compound 2 (Fig. 3). Analysis of the interaction between compound 2 and each of the peptides with the allosteric operational model revealed combined affinity-efficacy (αβ) estimates (Table 2) that were consistent with the affinity cooperativity estimates from the binding studies (Table 1), suggesting that effects of compound 2 on peptide-mediated cAMP responses were driven principally by changes in affinity. Thus, where peptides exhibited neutral cooperativity with compound 2 in binding, as seen for exendin-4, GLP-1(1–36)NH2, and GLP-1(1–37), the αβ estimates for compound 2 were not significantly different from 1 (Fig. 3, B and C, Supplemental Fig. S4B, Table 2), whereas oxyntomodulin displayed greatest combined cooperativity with compound 2 (Fig. 3D, Table 2), and GLP-1(7–36)NH2 and GLP-1(7–37) only modest cooperativity (Fig. 3A, Supplemental Fig. S4A, Table 2). For the latter two peptides, this level of cooperativity was insufficient to elicit a significant change in potency, consistent with simulation of the interaction, based on the affinity cooperativity estimate, which predicts a log unit shift of only 0.06 in potency at 10 μM compound 2 (Supplemental Fig. S5). Additional experiments after either 5 or 10 min of agonist stimulation revealed equivalent profiles of compound 2 interaction (data not shown).
Quercetin did not influence the production of cAMP for any of the peptides used in this study (Supplemental Fig. S3). This is consistent with the binding data, where the presence of quercetin did not alter the binding profile of any peptide (Supplemental Fig. S2).
Quercetin but Not Compound 2 Selectively Modifies Intracellular Ca2+ Responses via the GLP-1R in an Agonist-Specific Manner.
Given that allosteric ligands can bind simultaneously with orthosteric ligands and promote unique changes in receptor conformation, the resulting conformations may engender “stimulus-bias” across different signaling pathways in the same cellular background (Urban et al., 2007). In addition to Gαs coupling, the GLP-1R couples to Gαq proteins, resulting in mobilization of intracellular Ca2+ (Hällbrink et al., 2001). To investigate whether compound 2 or quercetin could impose stimulus bias on the actions of the orthosteric peptide agonists, we performed functional interaction assays for mobilization of intracellular Ca2+. Neither compound 2 nor quercetin displayed any intrinsic efficacy for the GLP-1R in this pathway (Supplemental Fig. S6E, Fig. 4E). To assess the roles of these allosteric modulators, concentration-response curves were established for the peptide ligands in the presence and absence of increasing concentrations of compound 2 or quercetin. In contrast to the cAMP data, quercetin caused biphasic changes in peptide agonist potency and efficacy for GLP-1(7–36)NH2, GLP-1(7–37), and exendin-4 (Fig. 4, A–C). Weak inhibition of peptide efficacy was observed between 10 nM and 1 μM quercetin, whereas augmentation of peptide efficacy was observed between 30 and 50 μM quercetin. However, no significant modulation of efficacy was observed for oxyntomodulin, which itself is only a partial agonist for this signaling pathway (Fig. 4D). In contrast to quercetin, compound 2 had no effect on peptide agonist-mediated Ca2+ responses (Supplemental Fig. S6). Loss of Ca2+ signal with the addition of 10 μM compound 2 was observed with all peptides studied; however, there was also a parallel reduction in the observed ATP response, indicating that this effect was an experimental artifact at this concentration of compound 2. In contrast, all ATP responses in the presence of increasing concentrations of quercetin remained robust and consistent, as did calcitonin-mediated Ca2+ signaling in CTa receptor expressing COS-7 cells, consistent with quercetin's effects on high-affinity agonists being mediated at the level of the GLP-1 receptor. Neither GLP-1(1–36)NH2 nor GLP-1(1–37) displayed any agonism in this signaling pathway at the tested concentration range (1 pM–1 μM) in either the absence or presence of compound 2 or quercetin (Supplemental Fig. S6E, Fig. 4E).

Characterization of the interaction between quercetin and GLP-1(7–36)NH2 (A), exendin-4 (B), GLP-1(7–37) (C), or oxyntomodulin in an intracellular Ca2+ mobilization assay using FlpInCHO cells stably expressing the human GLP-1R (D). E, GLP-1(1–36)NH2, GLP-1(1–37), or quercetin alone. Data are normalized to the maximal response elicited by 100 μM ATP and analyzed with a three-parameter logistic curve as defined in eq. 1. All values are mean ± S.E.M. of four to eight independent experiments conducted in duplicate. Statistical significance of changes in Emax in the presence of quercetin in comparison to the Emax of the peptide alone were determined by one-way analysis of variance and Dunnett's post test and are indicated with an asterisk (*, p < 0.05).
Neither Compound 2 nor Quercetin Modulates Agonist-Mediated ERK1/2 Phosphorylation via the GLP-1R.
ERK1/2 phosphorylation is often used as a general marker of convergent activation of multiple pathways, including G protein-independent signaling such as arrestin scaffolding (Lee et al., 2008). Although we observed that both compound 2 and quercetin could engender stimulus bias for a subset of peptides, we failed to observe any modulation of agonist responses in ERK1/2 phosphorylation in the presence of compound 2 or quercetin (Fig. 5, Supplemental Fig. S7). However, compound 2 displayed weak partial agonism for phosphorylation of ERK1/2 with a pEC50 of 5.76 ± 0.06 and a maximum of 51% of the GLP-1(7–36)NH2 response (Fig. 5G). In contrast, quercetin displayed no intrinsic agonism in any pathway tested.

Characterization of the interaction between compound 2 and GLP-1(7–36)NH2 (A), exendin-4 (B), GLP-1(7–37) (C) GLP-1(1–36)NH2 (D), oxyntomodulin (E), or GLP-1(1–37) (F) in an ERK 1/2 phosphorylation assay using FlpInCHO cells stably expressing the human GLP-1R. G, compound 2 alone. Data are normalized to maximal peptide response and analyzed with a three-parameter logistic curve as defined in eq. 1. All values are mean ± S.E.M. of three independent experiments conducted in duplicate.
Discussion
Despite the increasing prevalence of “biologicals” (high molecular weight natural or modified peptides/proteins) for the clinical treatment of disease, low molecular weight, orally active compounds are still pursued as the idealized therapeutic drug. Such drugs have traditionally been developed to mimic the properties of the natural ligand of the receptor by targeting the orthosteric binding site. Although this approach has been broadly successful for many GPCRs, including adrenergic receptors and histamine receptors (Black, 1989), there are many cases in which it has been problematic. Consequently, there has been increasing interest in the development of allosteric and/or bitopic drugs as a way of targeting these receptors (Valant et al., 2009). However, allosteric interactions are often complex, with the potential for modulation of affinity and efficacy, independently or concomitantly, as well as potential for the allosteric ligand to exhibit its own intrinsic efficacy; efficacy modulation and intrinsic efficacy may also be manifested in a pathway-dependent manner (May et al., 2007b). Furthermore, the nature of the allosteric interaction is probe-dependent (May et al., 2007b). In many physiological systems, this behavior is largely irrelevant, because there is principally one endogenous ligand for the receptor. However, there are numerous examples in which individual receptors can respond physiologically or pathophysiologically to multiple endogenous ligands, including the melanocortin receptors α-melanocyte stimulating hormone, adrenocorticotropin, agouti-related peptide (Tao, 2010), the parathyroid hormone receptor 1 (Gardella and Jüppner, 2000), and GLP-1Rs. In this study, we have demonstrated that allosteric modulation of the GLP-1R is complex, with pathway-dependent modulation of receptor response that is reliant upon both the peptide ligand and the allosteric modulator. Furthermore, it highlights the fact that distinct peptide ligands can exhibit stimulus bias at the GLP-1R and that allosteric modulators can impose further bias on this activity. Although some caution should be applied when interpreting data from a transfected cell background rather than a native GLP-1R-expressing cell line, collectively, these data stress the need for broad elucidation of mechanism of action when developing allosteric compounds.
The GLP-1R is pleiotropically coupled, eliciting signals via diverse pathways, including, prominently, the formation of cAMP (Baggio and Drucker, 2007). The signaling downstream of GLP-1R activation leading to the release of insulin is well studied and is critically dependent upon the activation of Gαs and formation of cAMP with subsequent effects via protein kinase A and exchange protein directly activated by cAMP, including direct inhibition of KATP channels, cell depolarization, and influx of extracellular Ca2+ (Baggio and Drucker, 2007). Nonetheless, there is also a role for mobilization of intracellular Ca2+ in augmentation of response (Baggio and Drucker, 2007), which is also manifested with Gαq-coupled receptors such as the muscarinic M3 acetylcholine receptor (Gautam et al., 2008). Sustained effects on gene transcription and the preservation of β-cell mass involve multiple signaling pathways, both cAMP-dependent and -independent; the latter include activation of mitogen-activated protein kinases such as ERK1/2. However, for many therapeutically important effects, including the modulation of appetite, the underlying GLP-1R-mediated signaling is not yet fully elucidated. Nonetheless, it is clear that the physiological response is a composite of the interplay between the various signaling pathways activated by individual ligands.
Evaluation of signaling across three pathways (cAMP production, elevation of intracellular Ca2+, and ERK1/2 phosphorylation) demonstrated, as expected, that all of the peptide agonists coupled the receptor most strongly to Gαs-mediated cAMP production. Each of the high-affinity agonists, GLP-1(7–36)NH2, GLP-1(7–37), and exendin-4 exhibited a similar profile of activation (Table 3), consistent with other functional and physiological studies with these peptides (Göke et al., 1993; Baggio and Drucker, 2007). In contrast, GLP-1(1–36)NH2, GLP-1(1–37), and oxyntomodulin exhibited significant bias relative to GLP-1(7–36)NH2. Oxyntomodulin exhibited less preference for cAMP relative to ERK1/2, but similar preference for cAMP relative to Ca2+, indicating that physiological responses to oxyntomodulin via the GLP-1R could differ from those elicited by GLP-1(7–36)NH2 (Table 3). Oxyntomodulin, like GLP-1(7–36)NH2 and GLP-1(7–37), is elevated postprandially, reaches higher plasma levels than those of GLP-1(1–36)NH2 and GLP-1(1–37), and can reach very high levels in some conditions, including celiac disease in children (Le Quellec et al., 1998). However, it is generally thought to play only a limited role therapeutically because of its lower affinity for the GLP-1R. Although oxyntomodulin can bind to both the glucagon and GLP-1R, most of its physiological responses seem to be mediated via the GLP-1R, as demonstrated in GLP-1R-knockout mice (Estall and Drucker, 2006). Oxyntomodulin, however, has a physiological profile distinct from that of GLP-1(7–36)NH2 (Druce and Bloom, 2006; Maida et al., 2008), which is not fully consistent with a purely affinity-driven reduction in signaling but is consistent with the observations of stimulus bias in this study.
Efficacy of agonists of the GLP-1R in FlpInCHO cells stably expressing the human GLP-1R
τ is an index of the coupling efficiency (or efficacy) of the agonist and is defined as the total concentration of receptors divided by the concentration of agonist-receptor complex that yields half the maximum system response (Emax). KA is the affinity of the agonist. Ratios were determined using eq. 5. Log[τ/KA]normalized is expressed relative to the Log τ/KB values for GLP-1(7–36)NH2 and is a measure of the relative strength of coupling of each individual agonist for an individual pathway relative to that of the control agonist. Log[stimulus bias] is the ratio of the efficacy of each agonist for the two pathways relative to the values for GLP-1(7–36)NH2 and is a measure of the degree of stimulus bias exhibited by individual agonists across the pathways, relative to that of the control agonist. Data are mean ± S.E.M. of three to eight individual experiments.
The flavonol quercetin lacked intrinsic activity but selectively modulated intracellular Ca2+ responses for the high-affinity agonists GLP-1(7–36)NH2, GLP-1(7–37), and exendin-4, causing weak inhibition at low concentrations and significant augmentation of response at concentrations above 10 μM, but had no effect on oxyntomodulin response. Thus, quercetin imparts stimulus bias with respect to Ca2+ signaling, but in a peptide-agonist dependent manner, which is consistent with a receptor-dependent mode of action. The selective bias for the high-affinity peptides is consistent with the overlap in signaling pathway activation profile exhibited by the peptides, which suggests that these peptides induce similar receptor conformations.
In contrast, analysis of the actions of compound 2 revealed intriguing differences in behavior of peptide agonists and also in modulation of peptide agonist function. Compound 2 displayed intrinsic efficacy for cAMP accumulation and ERK1/2 phosphorylation; while more efficacious in the former, it demonstrated less bias between these pathways than the peptide agonists of the receptor (Table 3). A formal assessment of relative signaling via mobilization of intracellular Ca2+ was not possible because of the nonspecific loss of response seen at higher compound 2 concentrations in the CHO cell background. Such distinction in receptor activation by compound 2 versus peptide agonists is not surprising, because it engages the receptor via different interactions and could be expected to engender unique conformation(s). This type of behavior has recently been observed at the M4 muscarinic acetylcholine receptor, where the allosteric agonist 3-amino-5-chloro-6-methoxy-4-methyl-thieno(2,3-b)pyridine-2-carboxylic acid cyclopropylamide (LY2033298) activates the receptor by conformational shifts that are partially distinct from those used by orthosteric agonists (Nawaratne et al., 2010).
In binding assays, compound 2 displayed significant probe dependence for modulation of affinity, negative cooperativity with the antagonist radioligand 125I-exendin(9–39), strong positive cooperativity with oxyntomodulin, weaker positive cooperativity with GLP-1(7–36)NH2 and GLP-1(7–37), and essentially neutral cooperativity with exendin-4 and full-length GLP-1 peptides. The differential modulation of affinity for peptide agonists versus antagonists is not necessarily surprising because compound 2 is an allosteric agonist and thus would be expected to favor binding to activated states of the receptor (Hall, 2000). However, the profile for modulation of peptide agonists reveals unanticipated effects that could translate into unexpected responses in preclinical and/or clinical evaluation of drug efficacy. In this regard, there were a number of important findings: the first was a markedly greater modulation of oxyntomodulin affinity relative to that for GLP-1(7–36)NH2 and GLP-1(7–37), which manifested in the cAMP assay as significant augmentation of oxyntomodulin potency but minimal augmentation of truncated GLP-1 peptide signaling; this response could be expected to allow drugs with this type of profile to elicit actions via oxyntomodulin that would not normally be seen with circulating levels of the peptide [40–60 pM physiologically and >500 pM in some pathophysiological states (Le Quellec et al., 1998)]. A second important observation was that GLP-1(7–36)NH2 and GLP-1(7–37) had a distinct interaction with compound 2 from that of exendin-4. As discussed above, exendin-4 and the truncated forms of GLP-1 are considered functionally equivalent; however, the positive cooperativity between compound 2 and truncated GLP-1 peptides versus neutral cooperativity with exendin-4 implies that the conformational state(s) adopted by exendin-4 is indeed different from that induced/occupied by GLP-1(7–36)NH2 and GLP-1(7–37); this may lead to divergence in signaling outcomes between the endogenous peptides, and exendin-4 may be detected if more broad analysis of signaling and/or receptor regulation is undertaken. Such differences in receptor interaction could underlie unexpected side effect profiles in susceptible populations.
Like quercetin, compound 2 modulated peptide signaling from only one of the three pathways assayed in the current study. Although quercetin modulated GLP-1R-mediated intracellular Ca2+, compound 2 modulated GLP-1R-mediated cAMP, suggesting that both modulators engender distinct forms of stimulus bias. Although efficacy cooperativity may be influenced by the propensity of an allosteric ligand to activate an individual pathway (Hall, 2000), simulation of compound 2 modulation of response based on affinity cooperativity, in the absence of intrinsic efficacy, predicts that there should be increased potency for each pathway, at least for oxyntomodulin. The absence of this effect indicates that compound 2 is generating true stimulus bias toward production of cAMP relative to the other pathways measured. Because allosteric interactions are due to conformational changes in the receptor as a result of co-occupancy of two ligands and conformational differences are the driver for biased signaling, broad understanding of receptor signaling is critical to understanding the success/failure of allosteric drugs during clinical development. It also highlights the importance of determining the function of putative allosteric modulators in both binding and signaling assays, and that discrimination of allosteric properties of small molecules depends on screening in multiple pathways, regardless of coupling strength. Likewise, for endogenous receptor systems that involve the interplay of multiple natural ligands, probe dependence of allosteric drugs is a major factor that needs consideration during discovery and development; indeed, this could also extend to otherwise inert metabolic products of the ligands.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was funded in part by the National Health and Medical Research Council (NHMRC) of Australia [Program Grant 519461]; and by an NHMRC of Australia Principal Research Fellowship (to P.M.S.) and a Senior Research Fellowship (to A.C.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.065664.
-
ABBREVIATIONS:
- DM
- diabetes mellitus
- GLP-1R
- glucagon-like peptide 1 receptor
- GPCR
- G protein-coupled receptor
- DPPIV
- dipeptidyl peptidase IV
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- ERK1/2
- extracellular signal-regulated kinases 1 and 2
- BCA
- bicinchoninic acid
- CHO
- Chinese hamster ovary
- BSA
- bovine serum albumin
- LY2033298
- 3-amino-5-chloro-6-methoxy-4-methyl-thieno(2,3-b)pyridine-2-carboxylic acid cyclopropylamide
- compound 2
- (6,7-dichloro2-methylsulfonyl-3-tert-butylaminoquinoxaline
- GLP-1
- glucagon-like peptide 1.

- Received April 16, 2010.
- Accepted June 14, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}