


Abstract
Small molecules targeting allosteric pockets of G protein–coupled receptors (GPCRs) have a great therapeutic potential for the treatment of neurologic and other chronic disorders. Here we performed virtual screening for orthosteric and putative allosteric ligands of the human dopamine D3 receptor (D3R) using two optimized crystal-structure–based models: the receptor with an empty binding pocket (D3RAPO), and the receptor complex with dopamine (D3RDopa). Subsequent biochemical and functional characterization revealed 14 novel ligands with a binding affinity of better than 10 μM in the D3RAPO candidate list (56% hit rate), and 8 novel ligands in the D3RDopa list (32% hit rate). Most ligands in the D3RAPO model span both orthosteric and extended pockets and behave as antagonists at D3R, with compound 7 showing the highest potency of dopamine inhibition (IC50 = 7 nM). In contrast, compounds identified by the D3RDopa model are predicted to occupy an allosteric site at the extracellular extension of the pocket, and they all lack the anchoring amino group. Compounds targeting the allosteric site display a variety of functional activity profiles, where behavior of at least two compounds (23 and 26) is consistent with noncompetitive allosteric modulation of dopamine signaling in the extracellular signal-regulated kinase 1 and 2 phosphorylation and β-arrestin recruitment assays. The high affinity and ligand efficiency of the chemically diverse hits identified in this study suggest utility of structure-based screening targeting allosteric sites of GPCRs.
Introduction
More than 800 G protein–coupled receptors (GPCRs) represent the largest and most diverse superfamily in the human genome. These transmembrane proteins regulate many physiologic and pathophysiologic functions, and mediate the action of more than 36% of all therapeutic drugs (Lagerstrom and Schioth, 2008; Rask-Andersen et al., 2011). New opportunities for GPCR ligand discovery have been unlocked by recent breakthroughs in membrane protein crystallography (Cherezov et al., 2007; Rosenbaum et al., 2007; Chun et al., 2012), leading to a rapidly growing structural coverage of the superfamily (Katritch et al., 2013; Venkatakrishnan et al., 2013). The utility of GPCR structures in the identification of novel antagonists and inverse agonist chemotypes has been demonstrated by structure-based virtual screening campaigns, where hit rates as high as 20 to 70% have been observed (Kolb et al., 2009; Carlsson et al., 2010, 2011; Katritch et al., 2010; de Graaf et al., 2011a; Jacobson and Costanzi, 2012; Mysinger et al., 2012). Active state structures of GPCRs have also been successfully used in virtual screening and structure-based rational design of agonists (Vilar et al., 2011; Shoichet and Kobilka, 2012; Tosh et al., 2012; Weiss et al., 2013). However, the utility of crystal structures in the discovery of new allosteric and bitopic (hybrid allosteric-orthosteric) GPCR ligands has yet to be established.
Allosteric ligands carry a number of potential advantages for GPCR drug development, including enhanced subtype and functional selectivity, and the ability to modulate the action of native agonists without disrupting temporal and spatial aspects of endogenous signaling (May et al., 2007; Kenakin and Miller, 2010; Canals et al., 2012; Melancon et al., 2012). In addition, bitopic ligands can synergistically combine the key functional features of allosteric and orthosteric moieties (Jo et al., 2012; Valant et al., 2012). However, discovery of new chemotypes of allosteric and bitopic ligands often requires more elaborate assays (Allen and Roth, 2011) and is hampered by paucity of starting allosteric scaffolds. In this context, the rapidly growing structural characterization of GPCRs and improved virtual screening technologies could provide an attractive path for faster and more efficient discovery of allosteric ligands.
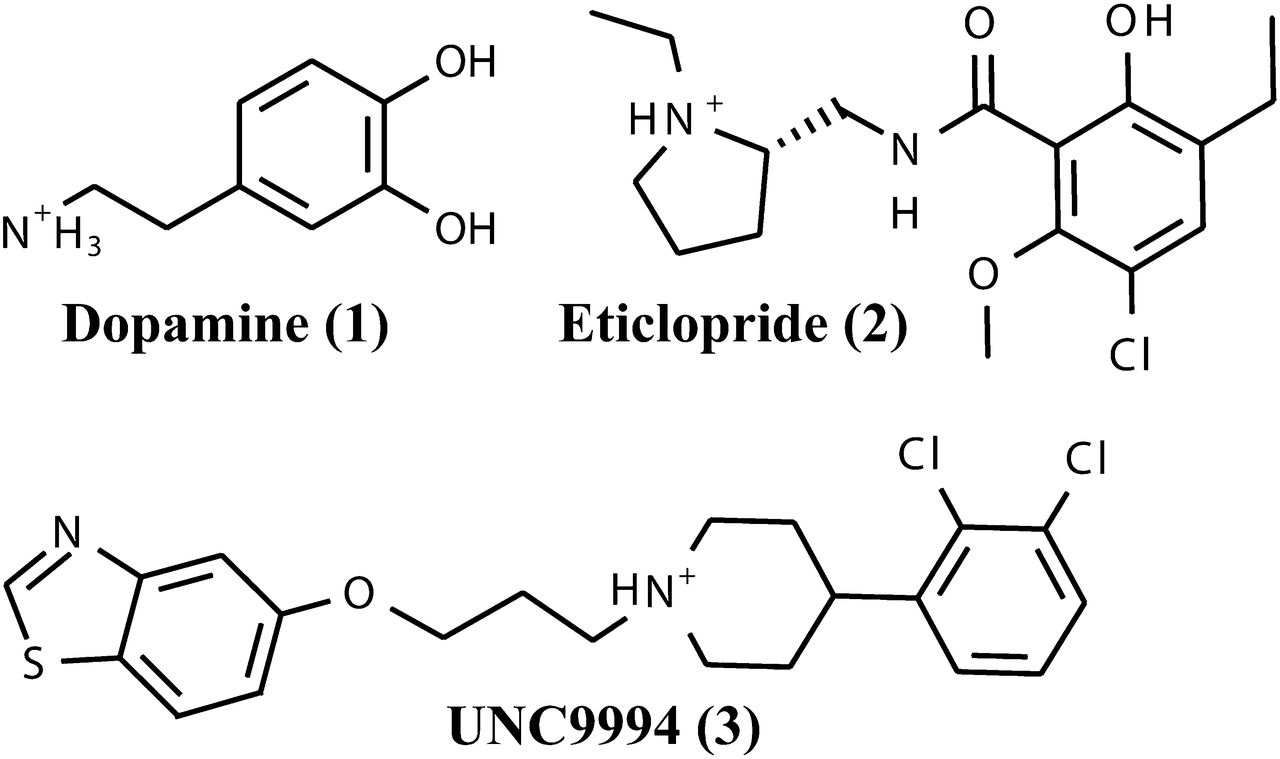
One of the important discovery targets is the dopamine D3 receptor (D3R), a crystal structure of which has been solved recently (PDB ID 3PBL) (Chien et al., 2010). D3R and other so-called D2-like dopamine receptors (Boyd and Mailman, 2012) play an essential role in neurologic processes, including reward and pleasure, cognition, and learning and memory as well as fine motor control, and they are primary clinical targets for the treatment of psychosis, addiction, Parkinson’s disease, and other neuropsychiatric disorders (Pilla et al., 1999; Vorel et al., 2002; Girault and Greengard, 2004; Gilbert et al., 2005; Le Foll et al., 2005; Kienast and Heinz, 2006; Xi et al., 2006; Spiller et al., 2008; Heidbreder and Newman, 2010; Ginovart and Kapur, 2012). The D3R crystal structure reveals the interactions of the antagonist eticlopride (Fig. 1, compound 2) (Hall et al., 1985), which occupies the core orthosteric binding site deep in the transmembrane bundle cavity in atomic detail. The first assessment of the D3R structure in prospective virtual ligand screening (VLS) suggested reasonable hit rates for amine-containing compounds that predominantly occupied the orthosteric binding pocket (Carlsson et al., 2011).

Dopamine receptor agonist dopamine (1), antagonist eticlopride (2), and bitopic antagonist UNC9994 (3).
Analysis of the ligand binding pocket structure in D3R, however, revealed a second, potentially allosteric binding site that extends toward the extracellular loop 2 (ECL2) and was predicted to accommodate aryl amide moieties of D3R-selective antagonists like R22 (Chien et al., 2010). A body of pharmacologic studies shows that these extended polar aryl moieties play a key role in ligand selectivity, including subtype selectivity between closely related D2R and D3R (Heidbreder and Newman, 2010; Newman et al., 2012). Moreover, recent studies also suggest that such extensions of the core ligand scaffold can define β-arrestin–biased signaling of D2R ligands, conferring important therapeutic benefits to these compounds (Allen et al., 2011).
In this study we assess the efficiency of the D3R crystal structure and ligand-optimized structural models in prospective screening for ligands targeting orthosteric and/or putative allosteric binding pockets. The large scale VLS was performed with two types of optimized D3R models: one representing an apo state, D3RAPO, and another one with a prebound dopamine, D3RDopa. The top 25 compounds predicted by each of these two screening models were tested in ligand binding and functional assays. The results suggest that structure-based screening is a promising tool for discovery of not only orthosteric but also allosteric ligands as potential leads for new classes of therapies targeting GPCRs.
Materials and Methods
Receptor Optimization for Apo State and Dopamine-Bound D3R Models
A ligand-guided receptor optimization (LiBERO) procedure for the D3RAPO model was performed as described previously elsewhere (Katritch et al., 2012). The benchmark set included 28 known D3R-selective antagonists with affinities of better than 10 nM from the ChEMBL database (Gaulton et al., 2012). A decoy set was compiled of 300 randomly selected compounds from the Chemdiv drug-like compound library (Chemdiv, San Diego, CA) with a predicted positive charge +1 and molecular mass between 350 and 500 Da, reflecting average properties of the ligand set.
Conformers of the D3R were generated from the crystal structure (PDB ID 3PBL) (Chien et al., 2010) by introducing minor (∼0.1 Å) variations in the protein backbone by normal mode analysis followed by energy-based side chain sampling with one of the 28 ligands from the known antagonist set. Each conformer of the binding pocket was assessed by its VLS performance—that is, the ability to separate D3R ligands from decoys. The area under the curve (AUC) and normalized squared area (NSA_AUC) metrics of VLS performance were calculated from receiver operating characteristic (ROC) curves for each model. In each step, models with the best NSA_AUC values were selected for the next iteration. The iterations of the LiBERO procedure were stopped when the performance of the binding pocket model reached saturation, with no improvement in two consecutive steps. The D3RDopa model was generated from the D3RAPO model by docking dopamine into the binding pocket with flexibility in the pocket side chains and helix V position, as described previously for the β2-adrenergic receptor (β2AR) agonist docking (Katritch et al., 2009). The LiBERO procedure took about 4000 central processing unit (CPU) core-hours and was performed using 64 CPU cores at a high-performance parallel Linux cluster.
Virtual Ligand Screening of Chemical Libraries
Virtual screening of a large library of available compounds was performed using the VLS procedure implemented in ICM-Pro molecular modeling software (Abagyan et al., 2012) as previously described elsewhere (Katritch et al., 2010). The screening was performed independently for two VLS models: 1) optimized apo receptor, D3RAPO, and 2) D3R-dopamine complex, D3RDopa. For both models, the docking procedure was confined to a large rectangular box that included all residues of the orthosteric pocket, extended pocket, and extracellular loops. The screening library of 4.1 million compounds was prepared from the Molsoft ScreenPub database (Molsoft, LLC, San Diego, CA) of unique drug-like compounds available from chemical vendors by automatically removing compounds with reactive groups and molecular mass exceeding 500 Da. On the basis of docking and ICM binding score results, the top 300 compounds in each model hit list were selected and clustered by chemical similarity with 0.3 Tanimoto distance cutoff. Predicted values of ligand efficiency were calculated as LEpred = Score/NHA, where Score is the ICM ligand binding score, and NHA is the number of nonhydrogen atoms in the ligand. From 1 to 10 compounds from each chemical cluster were selected for experimental assays according to their binding scores, LEpred values, chemical properties, and on-shelf availability from vendors, resulting in 50 compounds (25 for each model set). The virtual screening of 4.1 million compounds took about 21,000 CPU core-hours and was performed using 128 CPU cores at a high-performance parallel Linux cluster.
The Tanimoto distance between the hits and known ligands of dopamine receptors was calculated using dynamic linear fingerprints of 1536 bits with chains up to 6 atoms long extended with the atom/bond counts as implemented in ICM-Pro v.3.7-2a (Molsoft, LLC). The set of known ligands with affinity better than 1 μM to any of the dopamine receptor subtypes was derived from the ChEMBL database (Gaulton et al., 2012).
Radioligand Binding Assays
The compounds selected by virtual screening were purchased from available screening collections of chemical vendors, Chembridge (San Diego, CA) and Enamine (Monmouth Junction, NJ). Purity of compounds was equal to or greater than 95% as verified by high-performance liquid chromatography experiments performed by the vendors.
Molecular Biology.
The human wild-type D3R gene was synthesized by DNA2.0 with flanking restrictions sites AscI at the 5′ end and FseI at the 3′ end. The baculovirus expression vector, designated as pFastBac1-830220, was a modified pFastBac1 vector (Invitrogen/Life Technologies, Carlsbad, CA) containing an expression cassette with an hemagglutinin signal sequence followed by a FLAG tag at the N terminus and a PreScission protease site followed by a 10× His tag at the C terminus. The components of the expression cassette were introduced using standard polymerase chain reaction–based site-directed mutagenesis. The expression cassette also contained restriction sites for AscI and FseI, allowing for standard restriction digestion and subsequent ligation of the synthesized wild-type D3R DNA.
Expression and Purification of D3R Protein in Sf9 Cells.
High-titer recombinant baculovirus (>108 viral particles per ml) was obtained using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Briefly, recombinant baculoviruses were generated by transfecting 5 μg of recombinant bacmid containing the target gene sequence into Spodoptera frugiperda (Sf9) cells using 3 μl of FuGENE HD Transfection Reagent (Roche Applied Science, Indianapolis, IN) and Transfection Medium (Expression Systems, Davis, CA). Cell suspensions were incubated for 4 days while shaking at 27°C. P0 viral stocks were isolated after 4 days and used to produce high-titer baculovirus stocks. Viral titers were performed by flow cytometric S2 methods after staining cells with gp64-PE (Expression Systems). Sf9 cells at a cell density of 2–3 × 106 cells/ml were infected with P2 virus at a multiplicity of infection of 5. We added 15 µM cholesterol (Anatrace/Affymetrix, Maumee, OH) in 5% β-methyl cyclodextrin (Sigma-Aldrich, St. Louis, MO) solution to the cells at 24-hours after infection. Cells were harvested by centrifugation at 48 hours after infection and stored at −80°C until use.
Saturation and Competition Binding Assays Using Sf9 Cell Membranes.
For saturation binding assays, cell pellets with expressed D3R were suspended in ice-cold 25 mM HEPES, pH 7.5, as a lysis buffer, containing protease inhibitors (Complete protease inhibitor cocktail tablet; Roche Applied Science) and were homogenized with 20 strokes using a Dounce homogenizer. Crude plasma membranes were isolated by centrifugation at 150,000g for 60 minutes at 4°C, and were further washed 3 times by repeated centrifugation and resuspension in 25 mM HEPES, 1000 mM NaCl, pH 7.5 buffer supplemented with protease inhibitors. Before the ligand binding assays, the membrane pellets were resuspended in the assay buffer: 50 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM CaCl2, and 5 mM MgCl2. [3H]Spiperone (PerkinElmer Life and Analytical Sciences, Waltham, MA) was used as a radioligand. Crude plasma membranes (0.2 μg of total protein per reaction) were incubated for 24 hours at room temperature with serial dilutions of the radioligand (0.04–90 nM). Incubations were rapidly terminated by filtration using a Tomtec Mach III cell harvester (Tomtec, Hamden, CT) through a 96-well GF/B filter plate (MultiScreen Harvest plate; Millipore Corp., Billerica, MA), and rinsed 5 times with 500 μl of ice-cold buffer (50 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM CaCl2, and 5 mM MgCl2). The plates were dried, and 30 μl of OptiPhase-HiSafe III scintillation liquid (PerkinElmer) were added. The bound radioactivity was measured using a Packard TopCount NXT (PerkinElmer). Nonspecific binding was determined in parallel reactions in the presence of an excess of eticlopride (100 μM; Sigma-Aldrich), and specific binding was defined as the difference between total and nonspecific binding. Protein concentrations were determined with a bicinchoninic acid (BCA) protein assay (Pierce/Thermo Scientific, Rockford, IL), using serum albumin as a reference. All incubations were performed in triplicate, and independent experiments were repeated at least 2 times. Equilibrium dissociation constants (Kd) and maximal receptor levels (Bmax) were calculated from the results of saturation experiments using Prism 5 (GraphPad Software, San Diego, CA).
For competition binding assays, membrane preparations from Sf9 cells expressing D3R were incubated at room temperature for 72 hours with different concentrations of the assayed ligand and 15 nM [3H]spiperone (PerkinElmer) in 50 mM HEPES, pH 7.5, 50 mM NaCl, 1 mM CaCl2, and 5 mM MgCl2. Other protocols are identical to the saturation binding assay as described earlier.
Functional Assays
Cell Culture.
Chinese hamster ovary (CHO) FlpIn cells were stably transfected with the human D2(long) dopamine receptor (D2-CHOFlpIn,) or the human D3 dopamine receptor (D3-CHOFlpIn) [cDNA obtained from Missouri S&T cDNA Center (Rolla, MO)]. Cells were grown and maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 20 mM HEPES, 5% fetal bovine serum, and 200 μg/ml hygromycin-B. Cells were maintained at 37°C and 5% CO2 in a humidified incubator. For extracellular signal-regulated kinase 1 and 2 (ERK1/2) phosphorylation assays, cells were seeded into 96-well plates at a density of 50,000 cells/well. After 4 hours, cells were washed twice with phosphate-buffered saline and then serum-starved in DMEM containing 20 mM HEPES for at least 16 hours before assaying. For β-arrestin recruitment experiments, FlpIn CHO cells were transfected with a 2:2:4 ratio of cDNA coding for hemagglutinin-D2L-Rluc8 (a kind gift from Dr. Michelle Glass), GRK2 (a kind gift from Dr. Rob Leurs), and β-arrestin2–yellow fluorescent protein (a kind gift from Marc Caron) (total DNA 8 μg for every 2 million cells). Cells were transfected using linear polyethyleneimine with a molecular mass of 25 kDa (Polysciences, Warrington, PA), as described previously elsewhere (Canals et al., 2012). The day after transfection, cells were trypsinized, resuspended into culture medium, and plated in poly-l-lysine–coated white-bottom 96-well assay plates.
ERK1/2 Phosphorylation.
Receptor activation was followed using the AlphaScreen SureFire assay (TGR BioSciences, Thebarton, Australia), measuring dopamine receptor–mediated downstream phosphorylation of ERK1/2. Dose-response experiments in the absence or presence of ligands were performed at 37°C in a 200 μl total volume of DMEM containing 20 mM HEPES and 0.1% ascorbic acid. Dose-response stimulation or inhibition curves were generated by exposure of the cells to assayed ligands for 30 minutes and then to 10 nM dopamine (EC80 concentration) for 5 minutes. Stimulation of cells was terminated by the removal of the medium and the addition of 100 μl of SureFire lysis buffer to each well. The plate was agitated for 1–2 minutes, and 5 µl of lysate was added to each well of a white opaque 384-well Proxiplate. A 1:30:210 v/v dilution of AlphaScreen beads/activation buffer/SureFire reaction buffer in an 8 μl total volume was then transferred to each well of the 384-well Proxiplate in the dark. This plate was then incubated in the dark at 37°C for 1.5 hours after which time the fluorescence signal was measured by a Fusion plate reader (PerkinElmer), using standard AlphaScreen settings.
β-Arrestin Recruitment.
β-Arrestin recruitment to the D3R and D2R was measured by bioluminescence resonance energy transfer (BRET). CHO FLpIn cells were cotransfected with D2R C-terminally tagged with Rluc8 and β-arrestin2-yellow fluorescent protein as described earlier. At 24 hours after replating in white 96-well plates, cells were washed with Hanks’ buffered saline solution and preincubated for 30 minutes with assayed compounds. The Rluc8 substrate coelenterazine H was added at a final concentration of 5 μM and incubated for 10 minutes. Finally, dopamine was added at concentrations ranging from 100 μM to 0.1 nM and incubated for a further 10 minutes before reading. Measurements were made in a LUMIstar plate reader (BMG Labtech, Ortenberg, Germany) and the BRET ratio (emission at 530 nm/emission at 480 nm) was calculated. The net BRET ratio was obtained by subtracting the BRET ratio of cells only expressing D2-Rluc8. Dopamine caused a concentration-dependent increase in β-arrestin2 recruitment with a potency of (pEC50 = 7.39 ± 0.06).
Data Analysis
In the functional ERK1/2 assay and the β-arrestin assay, agonist concentration response curves were fitted to the following four-parameter Hill equation using Prism 5: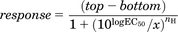 (1)where top represents the maximal asymptote of the concentration response curves, bottom represents the lowest asymptote of the concentration-response curves, logEC50 represents the logarithm of the agonist EC50, x represents the concentration of the agonist, and nH represents the Hill slope. To determine the inhibitory potency of the various test ligands, data were fit to the following equation:
(1)where top represents the maximal asymptote of the concentration response curves, bottom represents the lowest asymptote of the concentration-response curves, logEC50 represents the logarithm of the agonist EC50, x represents the concentration of the agonist, and nH represents the Hill slope. To determine the inhibitory potency of the various test ligands, data were fit to the following equation: (2)where top represents the maximal asymptote of the concentration response curves, bottom represents the lowest asymptote of the concentration response curves, logIC50 represents the logarithm of the antagonist IC50, x represents the concentration of the agonist, and the Hill slope is assumed to be unity. Data shown are the mean ± S.E.M. of at least three separate experiments performed in duplicate.
(2)where top represents the maximal asymptote of the concentration response curves, bottom represents the lowest asymptote of the concentration response curves, logIC50 represents the logarithm of the antagonist IC50, x represents the concentration of the agonist, and the Hill slope is assumed to be unity. Data shown are the mean ± S.E.M. of at least three separate experiments performed in duplicate.
Interaction studies between dopamine and increasing concentrations of the prototypical D2R/D3R antagonists clozapine and haloperidol could be best fit by a Gaddum/Schild equation to describe competitive antagonism: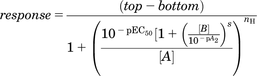 (3)where top represents the maximal asymptote of the concentration response curves, bottom represents the lowest asymptote of the concentration-response curves, pEC50 represents the negative logarithm of the agonist EC50, B is the antagonist, A is the agonist (dopamine), and nH represents the Hill slope. pA2 is the negative logarithm of the concentration in molar that shifts the EC50 by a factor of 2.
(3)where top represents the maximal asymptote of the concentration response curves, bottom represents the lowest asymptote of the concentration-response curves, pEC50 represents the negative logarithm of the agonist EC50, B is the antagonist, A is the agonist (dopamine), and nH represents the Hill slope. pA2 is the negative logarithm of the concentration in molar that shifts the EC50 by a factor of 2.
Data of radioligand binding experiments were analyzed using the nonlinear regression curve fitting program Prism 5 (GraphPad). For the displacement of [3H]spiperone, data were fit using a one site model with a variable Hill slope with the following the equation: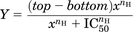 (4)where Y denotes the percent specific binding, top and bottom denote the maximal and minimal asymptotes, respectively, x denotes the inhibitor potency (midpoint location) parameter, and nH denotes the Hill slope factor. Assuming simple competition, IC50 values were converted to Ki values using the Cheng-Prusoff equation.
(4)where Y denotes the percent specific binding, top and bottom denote the maximal and minimal asymptotes, respectively, x denotes the inhibitor potency (midpoint location) parameter, and nH denotes the Hill slope factor. Assuming simple competition, IC50 values were converted to Ki values using the Cheng-Prusoff equation.
Results
Evaluation and Optimization of the Receptor Structure for VLS.
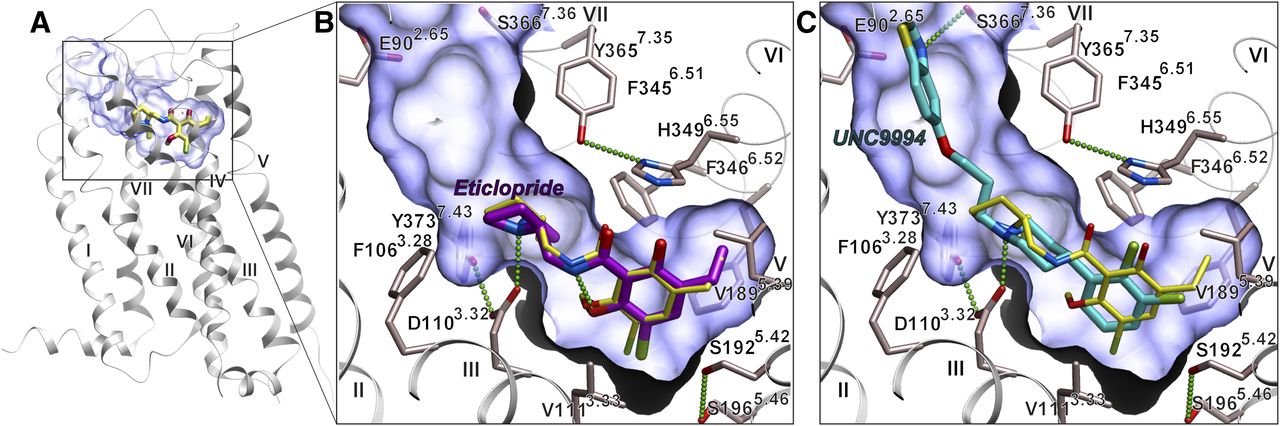
Before initiating VLS studies, the crystal structure of D3R (PDB ID 3PBL) was evaluated for docking of known ligands and its ability to separate ligands from decoys. We used an internal coordinate docking and scoring method (Totrov and Abagyan, 1997, 1999) in the ICM-docking software (Abagyan et al., 2012) for prediction of ligand binding. Docking of eticlopride (Fig. 1, compound 2) into the crystal structure conformation of D3R accurately reproduced the conformation of this ligand in the orthosteric pocket (Figs. 1 and 2). Docking also predicted a binding pose for the high-affinity ligand UNC9994 [5-(3-(4-(2,3-dichlorophenyl)piperidin-1-yl)propoxy)benzo[d]thiazole] (Fig. 1, compound 3) described recently as a β-arrestin–biased D2R/D3R agonist (Allen et al., 2011), which was similar to the extended pose of compound R22 described previously elsewhere (Chien et al., 2010). The top-scoring docking poses of these compounds involve a salt bridge between the basic amino nitrogen and the carboxyl of Asp1103.32, a key anchor site that is conserved in all aminergic receptors (superscript in ×.YY format shows Ballesteros-Weinstein residue numbering for GPCRs) (Ballesteros and Weinstein, 1995). Substituted aromatic rings of eticlopride and the phenylpiperidine scaffold of compound 3 fit within a hydrophobic cavity formed by side chains of helices III, V, VI, and VII as well as Ile183 of ECL2, while the extended tail of the ligand stretches toward the extracellular opening and makes specific polar interactions with the residues of helices II and VII.

Crystal structure of D3R-eticlopride complex (PDB ID 3PBL) (A) and docking poses of eticlopride (B) and a biased bitopic ligand 3 UNC9994 (C). The position of eticlopride in the crystal structure of the complex is shown in all three panels by sticks with yellow carbons. Hydrogen bonds in the predicted ligand complexes are shown by green dotted lines.
To assess performance of the D3R crystal structure model in the docking of other representative D3R ligands, we performed an initial benchmark for a diverse set of D3R-selective high-affinity ligands (Supplemental Fig. 1). The ligand set comprised compounds based on phenylpiperazine and other core scaffolds with a variety of aryl groups as nonorthosteric extensions. Binding modes for these benchmark compounds consistently resembled those of compound 3, with the amino group forming a salt bridge with Asp1103.32 and evidencing additional polar interactions in the extracellular extension of the cavity. However, the crystal structure-based docking yielded relatively low predicted binding scores, which also resulted in only modest separation of ligands from random decoys. To improve the VLS performance of the model, we optimized the D3R pocket conformation using a LiBERO procedure, as described previously elsewhere (Katritch et al., 2012). The ROC curves, calculated for the best models in each iteration (Fig. 3A), showed improvement of VLS performance from initial to the final optimized model. After the fifth iteration, the procedure reached saturation with the area under ROC curve AUC = 89% and the normalized square root AUC metrics NSQ_AUC = 70 (Katritch et al., 2012). Though the optimized model was conformationally close to the crystal structure (root mean square deviation ∼0.9 Å in the binding pocket residues), it substantially improved reproducibility of docking, overall ligand binding scores, and ligand separation from decoys (Fig. 3B). Representative binding poses of the benchmark ligands in this optimized model of apo D3R, which is referred to here as D3RAPO, are shown in Fig. 3, C and D, and Supplemental Fig. 2.

Validation and optimization of the model with known bitopic ligands of D3R. (A) ROC curves are displayed for each iteration of the LiBERO procedure. (B) Binding scores and mean field score are shown as calculated by ICM for ligands (red dots) and decoys (yellow dots). (C and D) Binding poses for two representative bitopic compounds in the optimized D3RAPO model. Compounds are shown by sticks with yellow carbons, and the pocket is shown as a green transparent surface. Hydrogen bonds in the predicted ligand complexes are shown by cyan dotted lines.
Dopamine-Bound Model of the Allosteric Site.
In addition to D3RAPO, a structural model of D3R in complex with dopamine, D3RDopa, was generated to screen for allosteric compounds that can bind along with the endogenous ligand. Flexible receptor docking with dopamine consistently resulted in a binding mode where dopamine largely overlaps with the eticlopride binding pose that was observed in the D3R crystal structure (Chien et al., 2010). In the predicted binding mode, the dopamine molecule engages both the Asp1103.32 anchor site and serine residues Ser1925.42 and Ser1965.46 in helix V (Fig. 4). This is consistent with mutation analysis in dopamine receptors (Woodward et al., 1996), and is also similar to the binding modes of catecholamine ligands in β-adrenergic receptors (de Graaf and Rognan, 2008; Katritch et al., 2009; Rasmussen et al., 2011; Warne et al., 2011). Unlike in β2AR, where the optimal binding of catecholamine agonists required a 1–2 Å inward shift of helix V from its antagonist-bound conformation (Katritch et al., 2009), no dramatic shift in helix V was observed during the flexible docking of dopamine. This difference can be explained by the less constrained amino tail of dopamine as compared with β-adrenergic agonists, and also by the fact that helices V and III in the D3R inactive structure are already ∼3 Å closer than they are in β2AR (Chien et al., 2010).
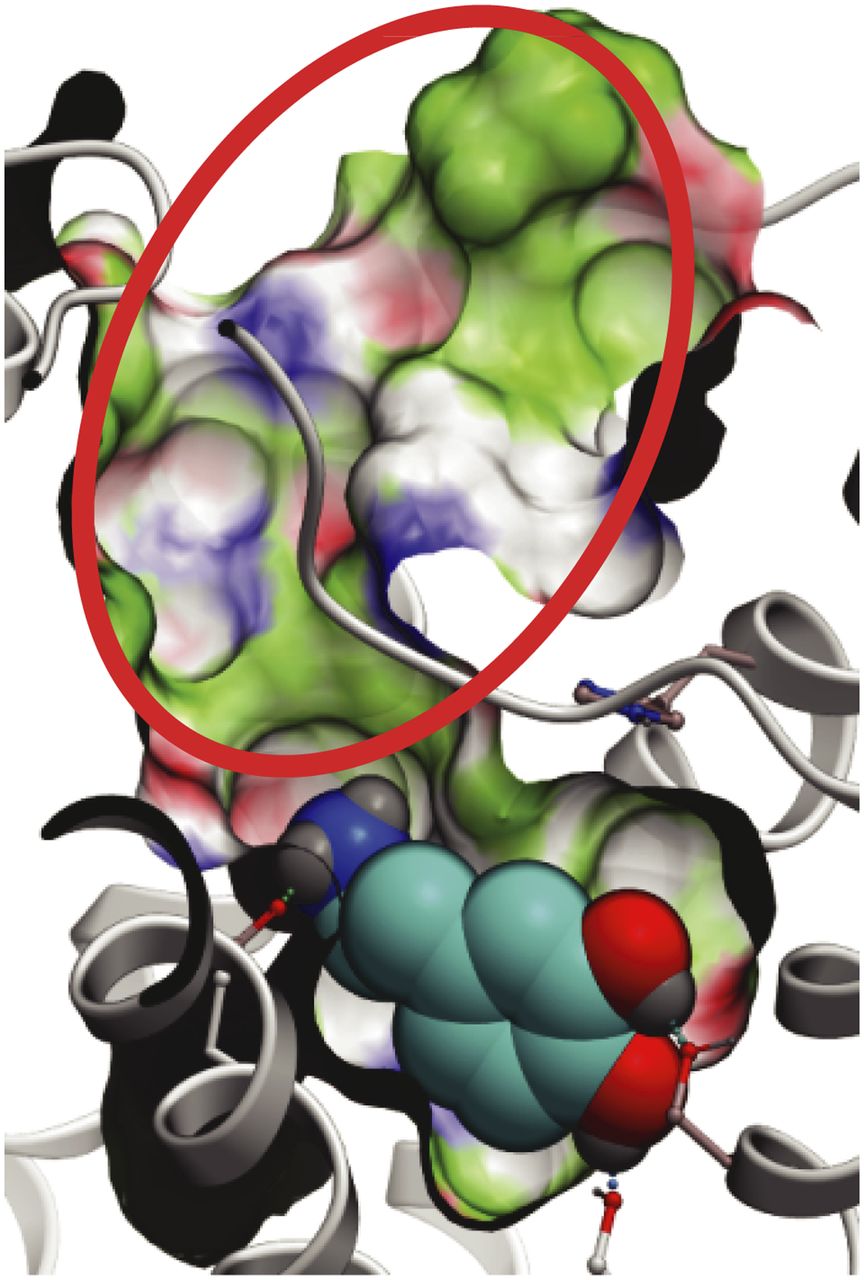
Dopamine-bound model of D3R (D3RDopa) for allosteric compound screening. Dopamine is shown in a space filling representation with cyan carbons. The extended “allosteric” part of the pocket is highlighted by a red circle.
Docking and Virtual Screening of Commercially Available Compounds.
A nonredundant library of 4.1 million commercially available drug-like and lead-like compounds was used for screening with the optimized D3R models as described in Materials and Methods. We performed screening with two distinct crystal structure-based VLS models described above: 1) unliganded receptor, D3RAPO and 2) D3R-dopamine complex, D3RDopa. For each VLS model, the top 150 compounds with the highest ligand binding scores were selected for further redocking and assessment. Both lists of candidate compounds were clustered and pruned to avoid chemical redundancy and close similarity with known D3R ligands. A total of 50 compounds, 25 in the D3RAPO set and 25 in the D3RDopa set, were selected and purchased from chemical vendors (Supplemental Tables 1 and 2). Representative examples of the predicted binding poses for the high-scoring compounds are shown in Fig. 5 and Supplemental Figs. 3 and 4.
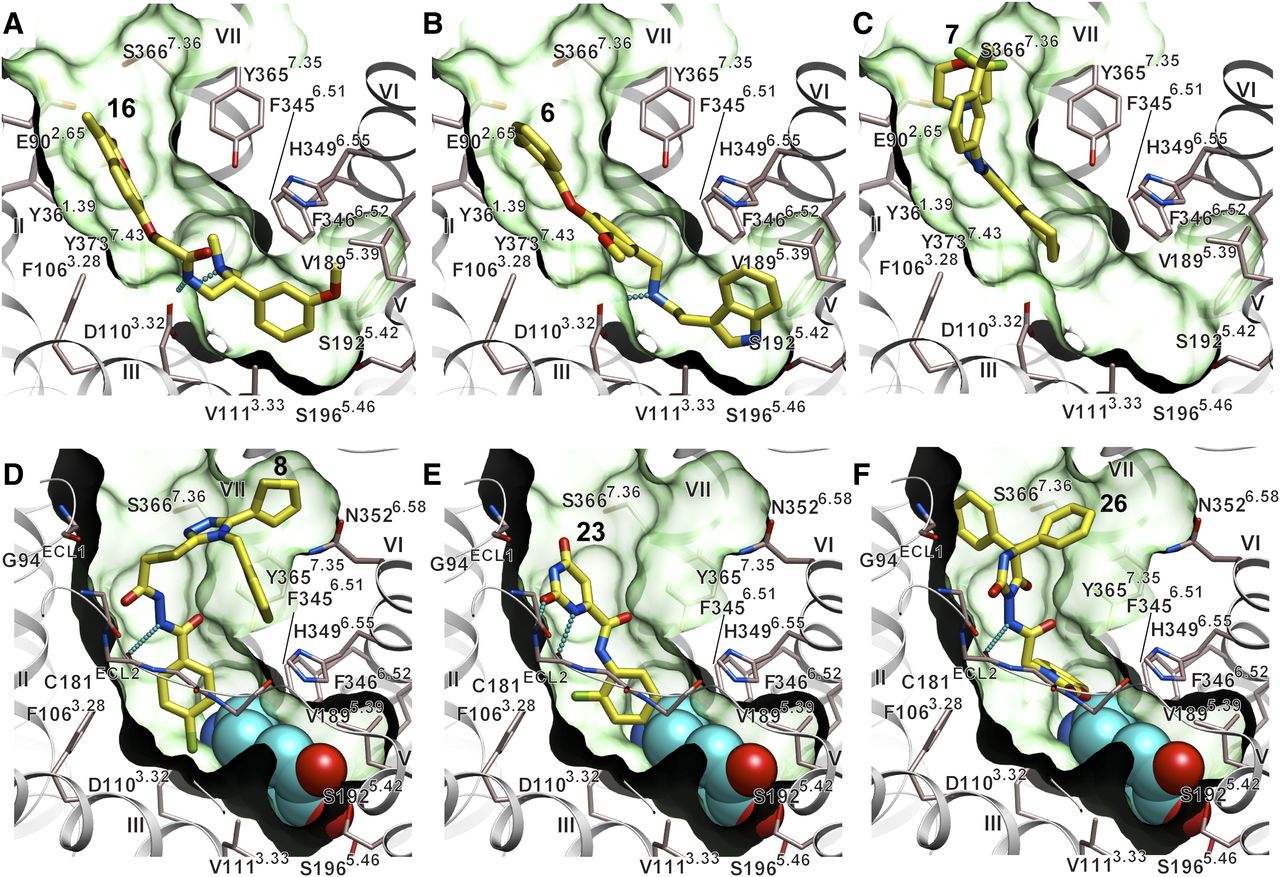
Examples of binding poses for novel D3R ligands, predicted by the ICM-VLS procedure. (A–C) Ligands predicted by VLS with D3RAPO model and (D–F) ligands predicted with D3RDopa model are shown as sticks with yellow carbons. A dopamine molecule bound in the orthosteric pocket of D3RDopa model is shown as spheres with carbon atoms colored cyan. The receptor is shown as a gray ribbon with the key side chains of the pocket shown as thin sticks; the binding pocket is illustrated as a green semitransparent surface.
All high-scoring compounds from the D3RAPO set (Fig. 5, A–C; Supplemental Fig. 3) contained a positively charged amino group, which formed a salt bridge to the conserved Asp1103.32 side chain. Most compounds in this set extend beyond the orthosteric core pocket into the allosteric site in our model, making extensive hydrophobic and polar interactions with side chains of helices I, II, III, and VII as well as with ECL1 and ECL2 residues. One interesting exception in the D3RAPO hit set is presented by compound 7, as its bulky aromatic group apparently does not fit in the orthosteric pocket (Fig. 5C). Instead, compound 7 was predicted to bind in the extended binding pocket, with its amino group forming a salt bridge with a nonconserved Glu902.65 side chain.
In contrast to the D3RAPO set, none of the top 25 candidate hits in the D3RDopa model screening list (Fig. 5, D–F; Supplemental Fig. 4) had a positively charged amine. These compounds were predicted to occupy the allosteric binding pocket, making a variety of interactions with helices I, II, III, and VII as well as ECL1 and ECL2 loops, with some larger compounds (8, 39) reaching ECL3 residues. Most of the candidate hits in the D3RDopa set also reach dopamine and Asp1103.32 at the edge of the orthosteric pocket. Some of the most common predicted interaction motifs include polar interactions with Tyr3657.35 hydroxyl and/or aromatic ring, polar interactions with Ser3667.36 and Glu902.65 side chains, and hydrogen bonding to the backbone amides of Ser1925.42, Cys181 and Ile183 in the ECL2.
Identification of Novel D3R Ligands in Binding Assays.
The top binding compounds from each set identified by the D3R radioligand competition assays are shown in Fig. 6 and Table 1. Novelty of the identified chemotypes was confirmed by their high Tanimoto distance to any of the known ligands of dopamine receptors (Supplemental Table 3; Table 1), which exceeds 0.35 for the D3RDopa set and 0.25 for the D3RAPO set (with exception of compound 19). For the D3RAPO set, we found 14 out of the 25 compounds to bind D3R with Ki < 10 μM (56% hit rate). Ten of them are submicromolar compounds, with the highest affinity Ki = 77 nM measured for ligand 6. For the D3RDopa set, 8 out of the 25 compounds bind D3R with Ki < 10 μM (32% hit rate), with the best affinity for compound 39, Ki = 302 nM. Binding curves for the highest affinity compounds from each set are shown in Fig. 7. Binding curves for the D3RAPO-derived ligands show full competition with [3H]spiperone radioligand binding, which is consistent with their interactions with both orthosteric and extended extracellular sites. The identified ligands in this set are based on a variety of orthosteric scaffolds composed of substituted ring systems and amine groups. The latter include secondary and tertiary amines, including less common for dopamine receptor ligands N-dimethyl amines (12, 16, 29) and pyrrolidine moieties (24). Even higher chemical diversity was found in the ligand moieties that were predicted to bind to the extended pocket, with a variety of heterocycles and conjugated rings making polar contacts with receptor residues in the extracellular region.
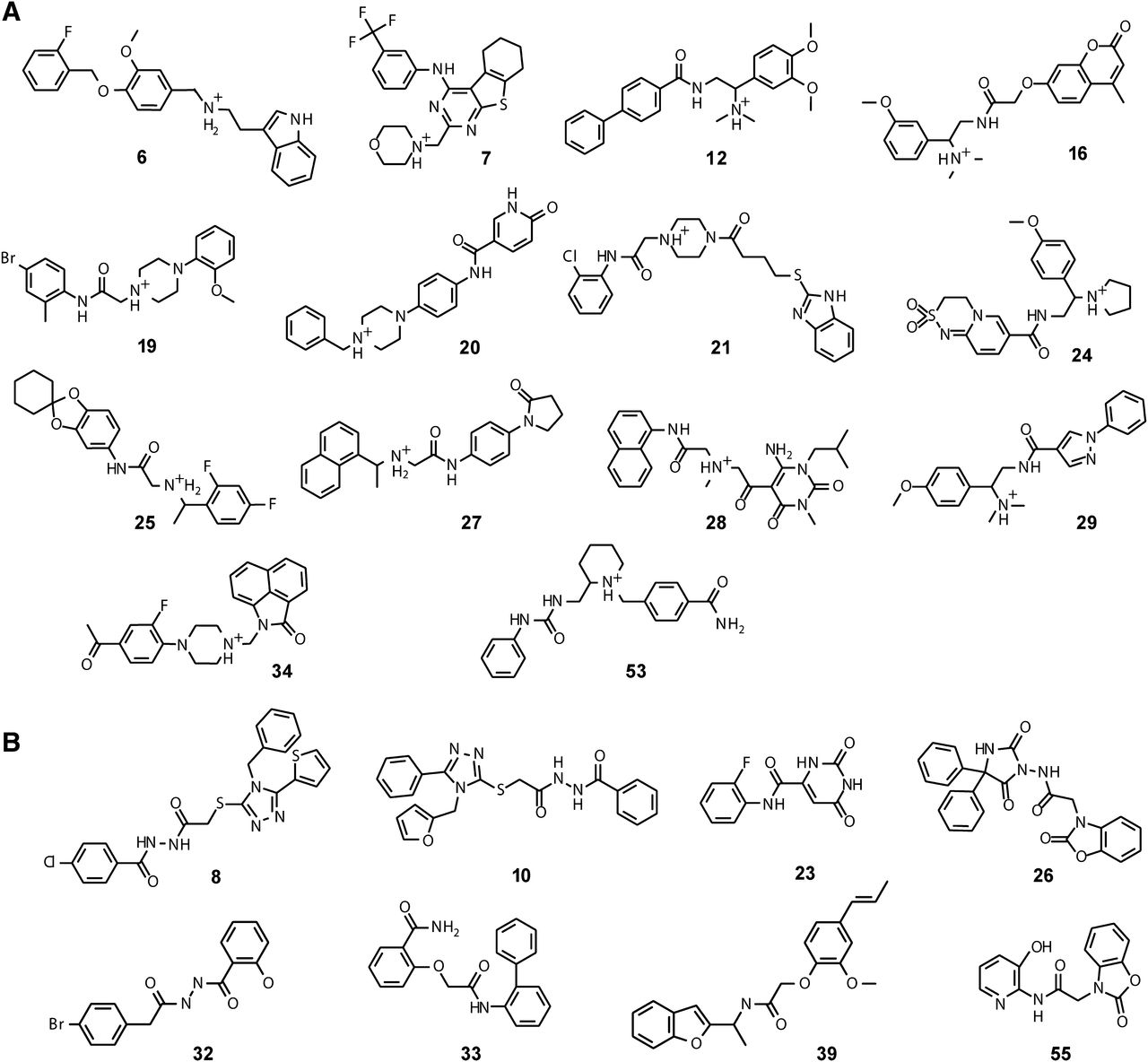
Compounds with D3R binding activity listed in Table 1. (A) Compounds identified from 25 candidates in the D3RAPO VLS set. (B) Compounds identified from 25 candidates in the D3RDopa VLS set.
The binding affinities and functional potencies of compounds identified by virtual screening
Highlighted in bold are binding affinities and potencies in the submicromolar range, and ligand efficiencies 0.3 and higher.

Radioligand binding assays. (A) Competition curves for representative compounds predicted by D3RAPO screening model. (B) Competition curves for representative compounds predicted by D3RDopa screening model.
All compounds derived from the D3RDopa model lacked a positively charged amino group and were not expected to be involved in orthosteric interactions. Nevertheless, our results demonstrate that ligand interactions in the extracellular extension of the pocket can be sufficient for effective binding with submicromolar affinities. Interestingly, binding curves for these compounds show varying levels of inhibition of radioligand [3H]spiperone binding (Fig. 5B), ranging from full inhibition for compound 23 to only ∼40% inhibition for compound 8. This is different from binding curves for the D3RAPO set (Fig. 7A), which all show full inhibition, and are consistent with varying levels of competition between binding poses of D3RDopa ligands and the extended fluorophenyl-alkanone part of [3H]spiperone.
The set of 22 identified ligands of D3R show very high chemical diversity, with their size ranging from 250 ≤ mol. wt. < 350 Da (23, 32, 33, 55) to more bulky compounds with mol. wt. > 450 Da (7, 8, 10, 26, 28). Note that more than half of the new D3R ligands shown in Fig. 6 and Table 1 have a ligand efficiency (LE) ≥ 0.3 kcal/mol per heavy atom, which is considered to be optimal for lead-like compounds (Hopkins et al., 2004). The highest value, LE = 0.45 kcal/mol per heavy atom, was found for compound 23 (D3RDopa set), while several compounds in both sets (6, 19, 29, 32, 39, 55) had LE ≥ 0.33 kcal/mol per heavy atom. These smaller compounds with high ligand efficiency may be especially valuable as lead scaffolds for drug discovery because they provide more room for chemical optimization (Hopkins et al., 2004).
Functional Effects of the New D3R Ligands on Receptor Activation.
To explore the functional features of the newly identified ligands, we tested their ability to modulate dopamine-induced ERK1/2 phosphorylation in D3R and D2R subtypes (see Materials and Methods). As shown in Table 1, all but four of the tested compounds caused a concentration-dependent inhibition of ERK1/2 phosphorylation consistent with their action as either antagonists or as negative allosteric modulators of the dopamine effect (Fig. 8).The highest potency in this assay was observed for compounds 7 (IC50 = 7 nM) and 16 (IC50= 40 nM), and six other ligands had submicromolar potencies. The four exceptions included compounds 6 and 55, which showed no significant modulatory effect in this assay, and compounds 39 and 10, which showed a modest increase in ERK1/2 phosphorylation. This latter action could be consistent with positive allosteric modulation of dopamine-induced ERK1/2 phosphorylation, in line with predicted cobinding of compounds 39 and 10 with dopamine. The majority of compounds displayed similar values of IC50 to the values of affinity (Ki) determined in the radioligand binding assay. For some compounds (for example, 7, 21, and 29) there is an intriguing discrepancy between the two values, which may indicate unusual signal-modulating properties of these ligands. Such discrepancies, however, may simply relate to the different conditions of the two assays such as the different cell backgrounds. None of the D3R ligands in Table 1 displayed agonism in their own right when used at concentrations up to 10 μM (data not shown).

Functional assessment of compounds for modulation of dopamine-induced ERK1/2 phosphorylation mediated by D3R or D2R expressed in CHO cells. Curves of representative compounds are shown. (A and B) Increasing concentrations of ligands yielded by the D3RAPO model were tested for their ability to modulate the effect of 10 nM dopamine at the D3R (A) or D2R (B). (C and D) Increasing concentrations of ligands predicted by the D3RDopa model were tested for their ability to modulate the effect of 10 nM dopamine at the D3R (C) or D2R (D). Responses are normalized to the level of ERK1/2 phosphorylation (pERK1/2) of 10% fetal bovine serum (FBS).
The same ERK1/2 phosphorylation assay was also performed using CHO cells expressing the human D2R subtype (Fig. 8; Table 1). As expected, the majority of compounds showed less potent effects on the D2R as compared with D3R. A 10-fold D3R selectivity was found for compound 16, while several other compounds (7, 28, 23, 26) displayed approximately 5-fold selectivity for D3R.
We further explored the possibility that some of the ligands in the D3RDopa set that were predicted to cobind with dopamine exerted their inhibitory effect on dopamine-stimulated ERK1/2 phosphorylation via negative allosteric modulation of the D3R and D2R. This was tested by measuring dopamine dose-response curves in the presence of increasing concentrations of the ligands, focusing on highly potent compounds 23 and 26. Figure 9 shows that both compounds not only shifted the dopamine dose-response curve toward higher dopamine concentrations, but also caused a dramatic decrease in the maximal response of D2R and D3R expressing CHO cells. Such a pattern was distinct from the action of known competitive antagonists haloperidol (pA2 = 9.76 ± 0.10) and clozapine (pA2 = 7.06 ± 0.12) at the D2R, which each gave a concentration-dependent rightward shift of the dopamine dose-response curve with no decrease in the dopamine maximal response (Fig. 9, C and F). This stark difference suggests that there is a significant allosteric modulation component in the inhibitory effect of compounds 23 and 26 on dopamine signaling.

Assessments of D2R or D3R mediated phosphorylation of ERK1/2 at increasing concentrations of compounds. Dopamine dose response curves are shown for compound 23 (A and D) and 26 (B and E). As a control, response curves are also shown for antagonists clozapine (C) and haloperidol (F), which are known to compete with dopamine. Responses are normalized to the maximal level of ERK1/2 phosphorylation induced by dopamine in the control condition.
Allosteric Ligand Effect on Dopamine-Stimulated Recruitment of β-Arrestin.
To gain further insight into the functional properties of compounds 23 and 26, we investigated their effect upon dopamine-stimulated recruitment of β-arrestin2, which is another measurement of dopamine receptor function (see Materials and Methods). Although dopamine caused a dose-dependent increase in β-arrestin2 recruitment at both D2R and D3R, the magnitude of the effect at D3R was not sufficient to allow useful profiling of these compounds (data not shown); therefore, we focused on D2R for evaluating this effect. As shown in Fig. 10, compounds 23 and 26 caused a strong dose-dependent decrease in Emax with negligible effects upon dopamine potency. For both compounds, the highest concentration used (10 μM) completely abolished the effect of dopamine. In contrast, both the control orthosteric antagonists, clozapine and haloperidol, caused an expected rightward shift in the dose-response curve in this assay. Though haloperidol also decreased dopamine Emax to 50% in this assay, this effect is likely due to the nonequilibrium assay conditions in this case. Specifically, haloperidol has very slow dissociation rate, t1/2 = 40 minutes (Kapur and Seeman, 2000), which exceeds the relatively short 5-minute incubation time for dopamine. Such a nonequilibrium component disappears for fast dissociating clozapine (t1/2 = 0.5 minutes). Consequently, such nonequilibrium effects are unlikely to contribute to the noncompetitive functional profiles of compounds 23 and 26, which have much lower affinity and likely faster dissociation rates. Together, the results of both the ERK1/2 phosphorylation and β-arrestin recruitment assays provide strong evidence for the noncompetitive action of these compounds at the D3R and D2R. In particular, the decrease of dopamine Emax caused by compounds 23 and 26 is consistent with their action as negative allosteric modulators (Kenakin, 2004).

Assessments of dopamine mediated β-arrestin2 recruitment at D2R at increasing concentrations of compounds. Dopamine dose response curves are shown for compound 23 (A) and 26 (B). As controls, response curves are also shown for antagonists clozapine (C) and haloperidol (D), which are known to compete with dopamine. Responses are normalized to the maximal level of stimulation by dopamine in the control condition.
Discussion
Recent applications of structure-based VLS demonstrate high efficiency in the discovery of novel lead-like compounds (Kolb et al., 2009; Carlsson et al., 2010, 2011; Katritch et al., 2010; de Graaf et al., 2011a; Mysinger et al., 2012) targeting orthosteric pockets of GPCRs, where hit compounds largely overlap with the cocrystallized compounds and at least partially reproduce their key receptor interactions. Discovery of allosteric compounds is considered a much more challenging problem, with only a few examples of putative allosteric hits arising from VLS campaigns (de Graaf et al., 2011b; Mysinger et al., 2012). In this study, we specifically aimed at expanding large-scale VLS applications beyond orthosteric sites by 1) developing an optimized D3RAPO model of the full binding pocket with allosteric extension, and 2) developing an alternative model of the pocket with prebound natural ligand dopamine, D3RDopa.
Our results for the D3RAPO model show that a very high 56% hit rate in VLS can be achieved for compounds extending far beyond the dopamine/eticlopride orthosteric site and forming major polar and hydrophobic interactions in the upper part of the pocket, including extracellular loops. One of the most interesting ligands in the D3RAPO set is compound 7 (Figs. 4C and 6), which proved to be the most efficient D3R antagonist in the ERK1/2 phosphorylation assays, with IC50 = 7 nM. The bulky aromatic system of compound 7 was predicted to occupy the extended part of the pocket, with its amino group forming a salt bridge with Glu902.65 at the pocket entrance instead of with the conserved Asp1103.32 anchor. Most ligands in this set behaved as competitive antagonists in ERK1/2 phosphorylation assays, with IC50 values consistent with their binding affinities. This preference for the screening hit compounds toward antagonists was expected, as the D3RAPO screening model was based on the inactive crystal structure of D3R and further optimized with D3R-selective antagonists (Katritch et al., 2012).
Interestingly, a previous VLS study on D3R (Carlsson et al., 2011) observed that the hit rate for crystal structure–based VLS (∼25%) was on par with the hit rate for a homology model built with a closely related β2AR structure. Note, however, that while the homology model in that study was thoroughly optimized using a ligand-optimization procedure, the crystal structure was used for VLS directly without any ligand-guided optimization. Our results here show that ligand-guided optimization of the crystal structure can improve VLS performance and lead to more potent compounds. A similar advantage of ligand-guided optimization in application to crystal structures was also shown previously for VLS on A2A adenosine receptors (Katritch et al., 2010; Carlsson et al., 2011). As ligand-optimization techniques such as LiBERO (Katritch et al., 2010, 2012) or the one described by Carlsson et al. (2011) improve VLS performance for both crystal structures and homology models, higher initial quality of the crystal structure would still confer a substantial advantage in prospective screening over most homology models.
As the most important result of this study, the crystal structure–derived model of dopamine-bound receptor D3RDopa has proved instrumental in the identification of a new class of D3R ligands binding to the allosteric pocket. Although the hit rate was somewhat lower (32%) than in the D3RAPO set, the ligands identified by the D3RDopa model represent unique features and properties. All of the D3RDopa ligands lack a positively charged amino group common for known dopamine ligands and thus cannot form a conserved salt bridge to Asp1103.32 in the orthosteric pocket. Instead, compounds in this new class of D3R ligands are predicted to form hydrogen-bonding interactions with ECL2, particularly peptide-like polar interactions with the backbone amide groups of residues Cys181-Ile183, as well as with a range of polar and aromatic side chains on the extracellular side of the binding cavity. Follow-up structure-function/structure-activity studies will provide further validation of the predicted binding modes and specific interactions for the compounds identified in this study and their derivatives.
The newly identified nonorthosteric ligands showed distinct functional profiles on dopamine-signaling efficiency. Most of these compounds displayed an inhibitory effect on dopamine-induced ERK1/2 phosphorylation, including submicromolar effects of compounds 23, 26, 32, and 33. Other compounds, however, were relatively “silent” in these assays, and compounds 39 and 10 even slightly increased the dopamine effect at D3R and D2R respectively, suggesting a potential positive allosteric modulation, though the latter effect may be too subtle to be of therapeutic interest.
Although our study did not specifically focus on subtype selectivity of the identified ligands, some modest levels of D3R versus D2R selectivity were found for a majority of the compounds, including 10-fold selectivity for 16, and more than 5-fold selectivity for five other compounds. Some of the extended ligands identified here were predicted to reach nonconserved residues in the extracellular opening of the binding pocket. We found that this does not automatically confer them with high selectivity though, probably because interactions with conserved residues or with the ECL2 backbone atoms of the receptor still appear to dominate their contacts. Though VLS in general may be not very effective in screening for subtype-selective compounds (Kolb et al., 2012), these new scaffolds provide a starting platform for further optimization of selectivity by focusing on contacts with nonconserved residues.
Compounds 23 and 26, predicted to bind to the extracellular extension of the binding pocket, were further tested for their potential action as allosteric modulators of dopamine effects at D3R and D2R. Indeed, both compounds showed functional behavior different from prototypical orthosteric antagonists clozapine and haloperidol, and were consistent with noncompetitive antagonism at D2R and D3R. Thus, we demonstrated that both compounds 23 and 26 decrease the maximal efficacy of the endogenous agonist dopamine in the ERK1/2 phosphorylation assay. Similarly, both compounds caused a decrease in the maximal effect of dopamine in the β-arrestin2 recruitment assay at D2R. Thus, the combined data from ERK1/2 phosphorylation, β-arrestin recruitment, and radioligand-binding assays support a noncompetitive or allosteric inhibition of dopamine by compounds 23 and 26, which is consistent with their cobinding with dopamine predicted by computational docking. It is interesting to note that both compounds 23 and 26 caused a more dramatic decrease in the maximal response of dopamine in the β-arrestin recruitment assay as compared with the pERK1/2 phosphorylation assay. There are a number of mechanisms that may underlie such effects. If such compounds act as negative modulators of dopamine efficacy, the less efficiently coupled a signaling pathway is, the greater one would expect the modulatory effect to be. Alternatively, such ligands may display bias in their action and preferentially antagonize the arrestin recruitment pathway as compared with the pERK1/2 pathway, an effect that deserves further assessment in the follow-up studies.
To date only a very few ligands have been characterized as allosteric modulators of dopamine D2-like receptors. One of them is the drug-like ligand SB269,652 [1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide], which has a tertiary amino group and was originally discovered as an antagonist (Taylor et al., 1999). Later assessment has shown that this compound behaves as a negative modulator for D2R and D3R (Silvano et al., 2010). though its allosteric binding mode is not clear. Several analogs of the endogenous neuropeptide Pro-Leu-Gly-NH2 (Fisher et al., 2006; Bhagwanth et al., 2012) were previously reported to have either positive or negative allosteric effects on dopamine signaling. Binding modes of these peptidomimetics are also unknown, though it might be interesting to consider a possibility of their binding in the same region as the allosteric small molecules 23 and 26, amides of which have prominent interactions with the peptide backbone of ECL2 in our models. To the best of our knowledge, this study is the first to systematically search and identify a number of chemically distinct allosteric modulators of the D2R and D3R, and paves the way for the development of a novel class of drugs at dopamine receptors and other therapeutically important GPCRs.
Conclusion
Expanding on the success of recent structure-based VLS applications to class A GPCRs, this study demonstrates highly efficient prospective ligand screening specifically targeting allosteric extensions of the ligand binding cavity of D3R. Our results suggest that the ligand-guided optimization of a receptor can substantially improve the VLS model efficiency without sacrificing chemical diversity of the identified hits. The hit rates of 56% were achieved in identification of novel ligands spanning orthosteric and allosteric sites, with the highest D3R binding affinity Ki = 77 nM and highest antagonism potency IC50 = 7 nM. Most importantly, an additional eight ligands (32% hit rate) were identified for the extended allosteric pocket, using an optimized D3RDopa model of the receptor complex with dopamine bound in its orthosteric site. The most potent D3RDopa set ligands, 23 and 26, demonstrated distinct negative allosteric modulation of dopamine signaling in both ERK1/2 phosphorylation and β-arrestin recruitment assays, which warrants further functional characterization of these allosteric compounds. In the context of current rapidly improving structural coverage of GPCR superfamily, this study suggests that structure-based VLS can become an important tool for exploring not only orthosteric, but also allosteric and bitopic ligands with novel properties potentially beneficial in drug discovery.
Acknowledgments
The authors thank Katya Kadyshevskaya for helping with figure preparation and Angela Walker for helping with manuscript preparation. The authors also thank TGR BioSciences for generously providing the SureFire ERK1/2 kits.
Authorship Contributions
Participated in research design: Katritch, Stevens, Abagyan, Liu.
Conducted experiments: Lane, Chubukov, Liu, Cherezov, Canals.
Contributed new reagents or analytic tools: Katritch, Lane, Stevens, Abagyan.
Performed data analysis: Katritch, Lane, Liu, Chubukov.
Wrote or contributed to the writing of the manuscript: Katritch, Cherezov, Liu, Lane, Canals, Chubukov, Abagyan, Stevens.
Footnotes
- Received June 19, 2013.
- Accepted September 10, 2013.
J.R.L., P.C., and W.L. contributed equally to this work.
The study was supported by National Institutes of Health National Institute of General Medical Sciences Protein Structure Initiative [Grant U54 GM094618] (to R.C.S., R.A., V.C., and V.K.); National Health and Medical Research Council project grant [APP1011920] (to J.R.L.); and The Netherlands Organization for Scientific Research [NWO VENI Grant 863.09.018] (to J.R.L.).
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AUC
- area under the curve
- β2AR
- β2-adrenergic receptor
- BRET
- bioluminescence resonance energy transfer
- CHO
- Chinese hamster ovary
- CPU
- central processing unit
- D3R
- human dopamine D3 receptor
- DMEM
- Dulbecco’s modified Eagle’s medium
- ECL
- extracellular loop
- ERK1/2
- extracellular signal-regulated kinase 1 and 2
- GPCR
- G protein–coupled receptor
- LE
- ligand efficiency
- LiBERO
- ligand-guided receptor optimization
- ROC
- receiver operating characteristic
- SB269,652
- 1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide
- Sf9
- Spodoptera frugiperda cells
- UNC9994
- 5-(3-(4-(2,3-dichlorophenyl)piperidin-1-yl)propoxy)benzo[d]thiazole
- VLS
- virtual ligand screening
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}